Metabolic Interactions of Purine Derivatives with Human ABC Transporter ABCG2: Genetic Testing to Assess Gout Risk

Abstract
:1. Introduction
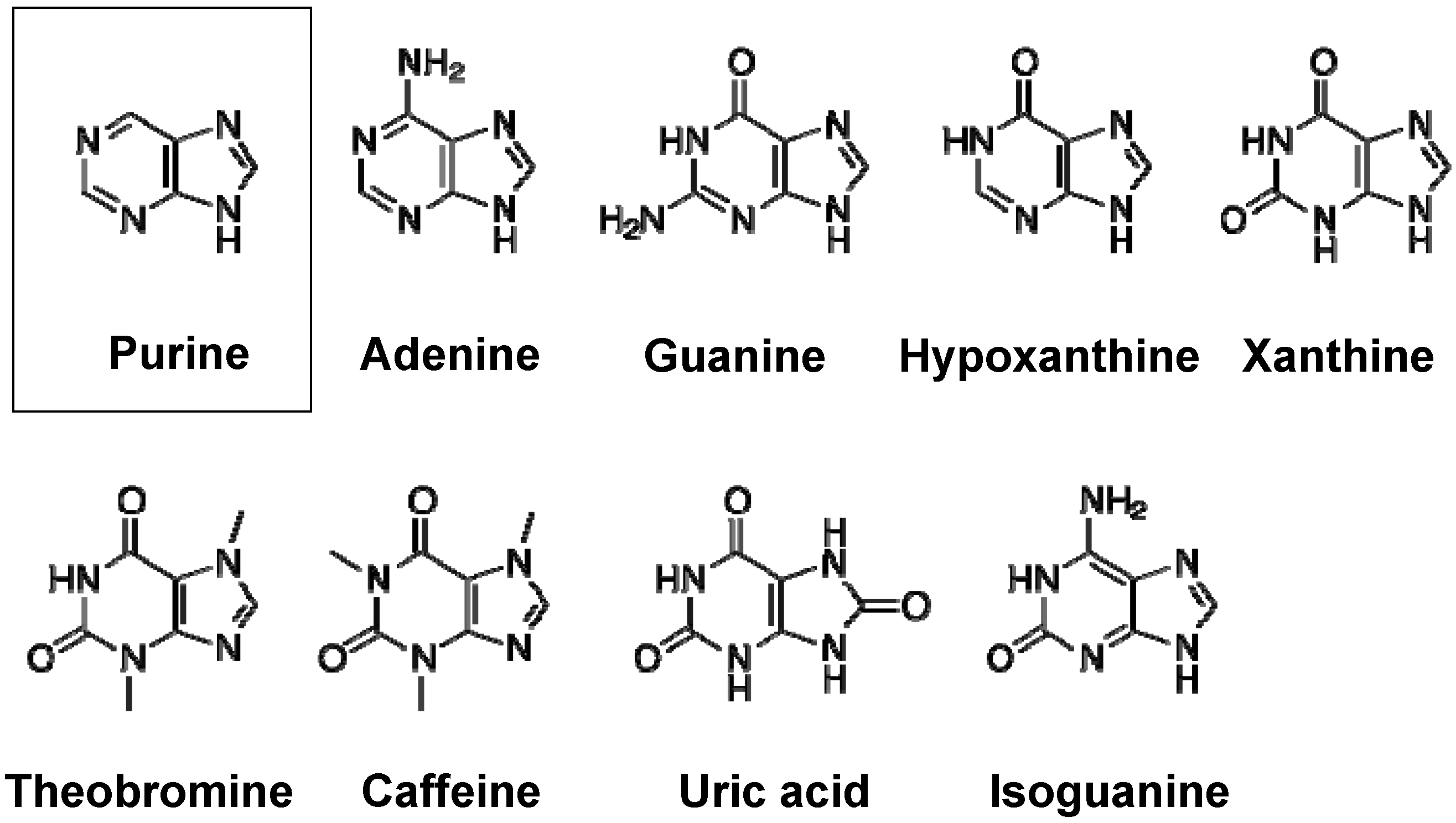
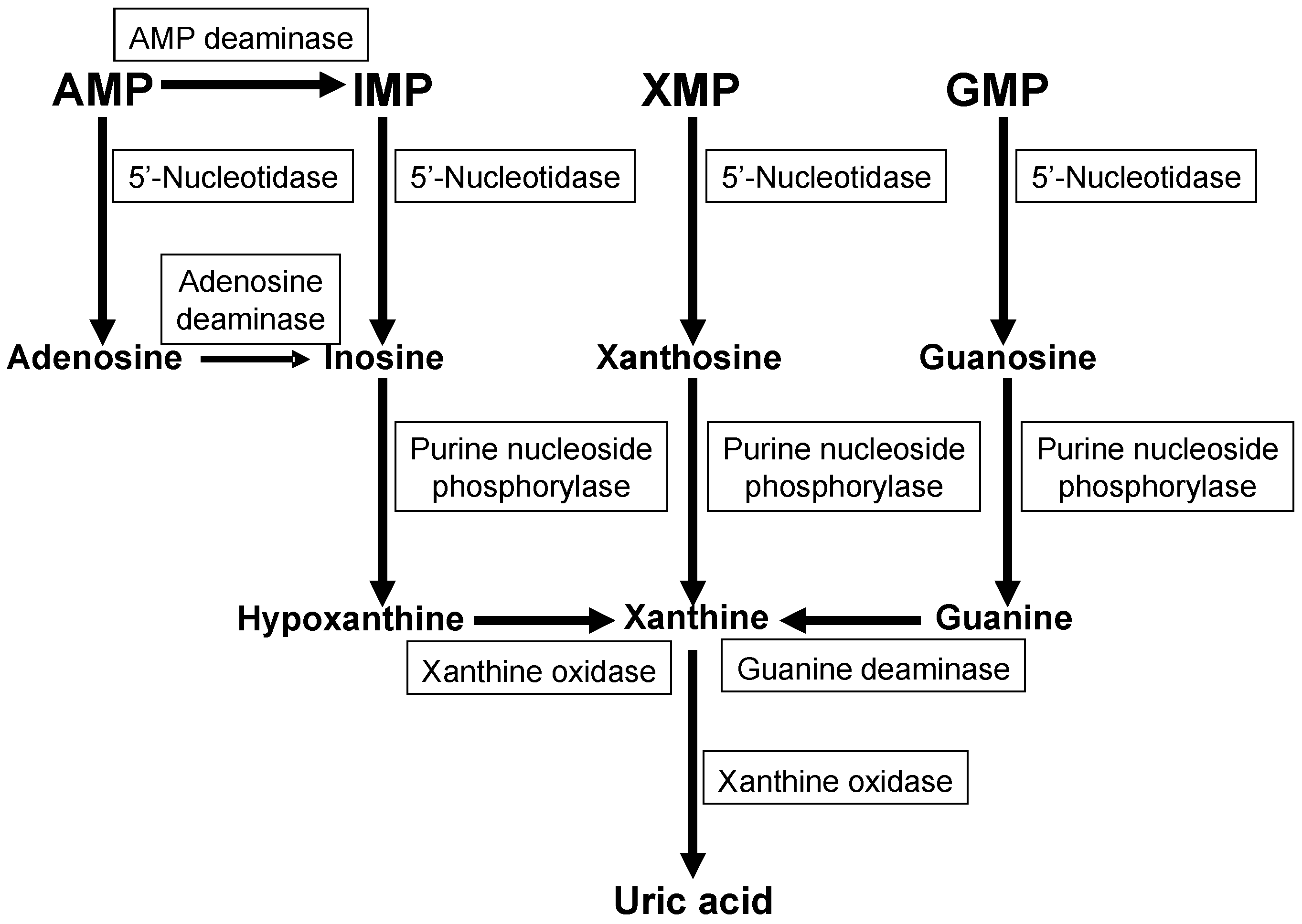
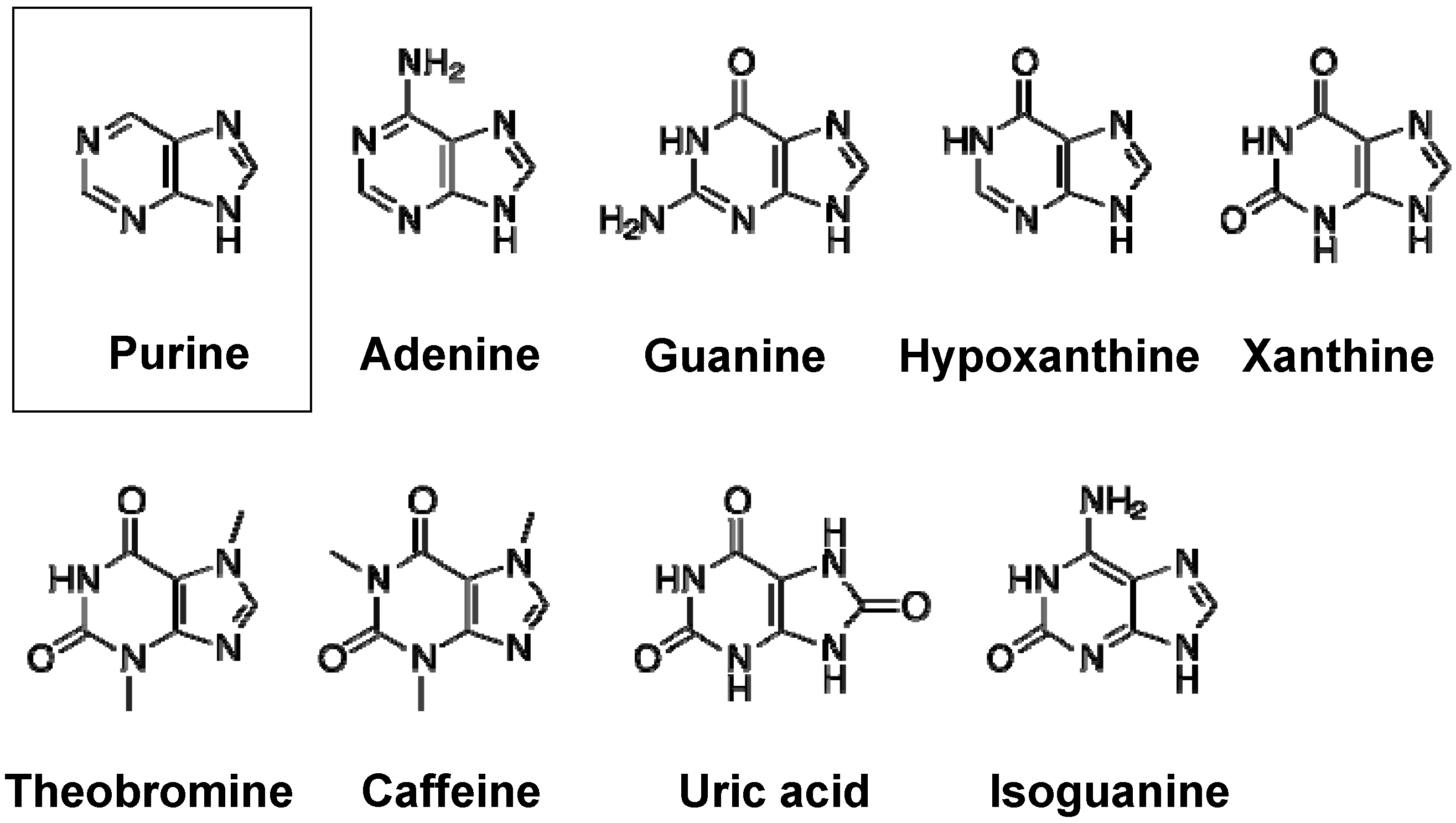
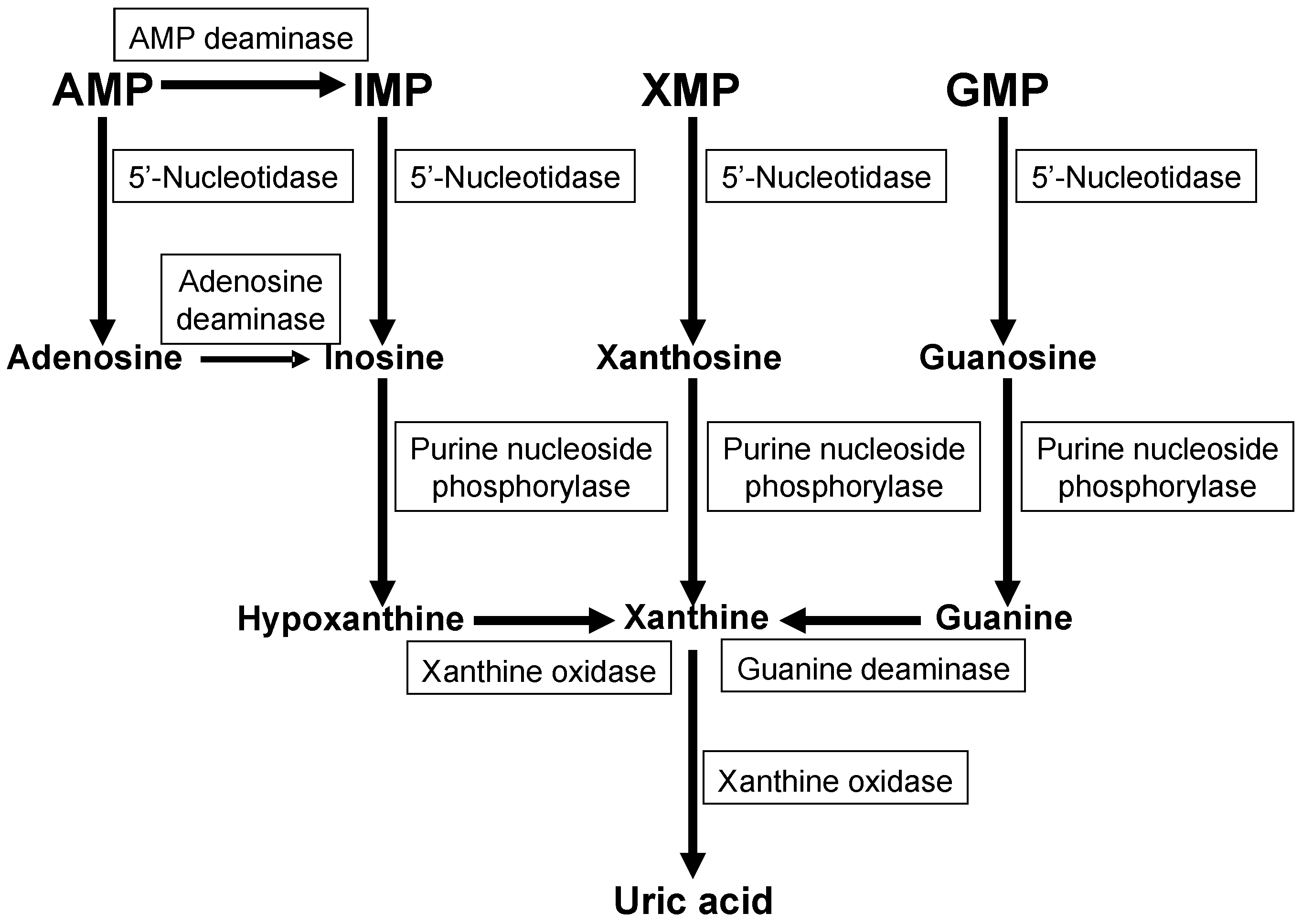
2. Purine Metabolism
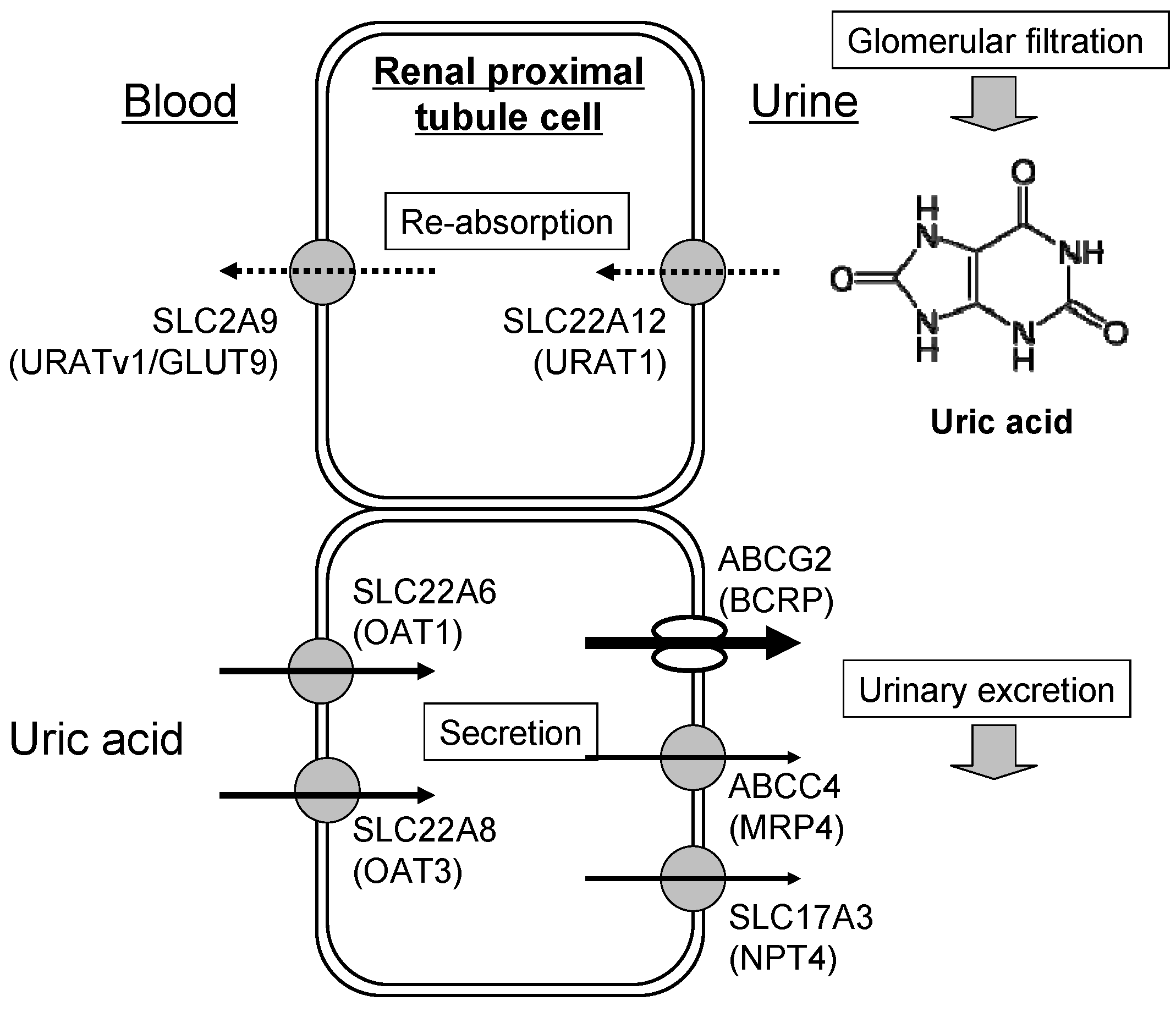
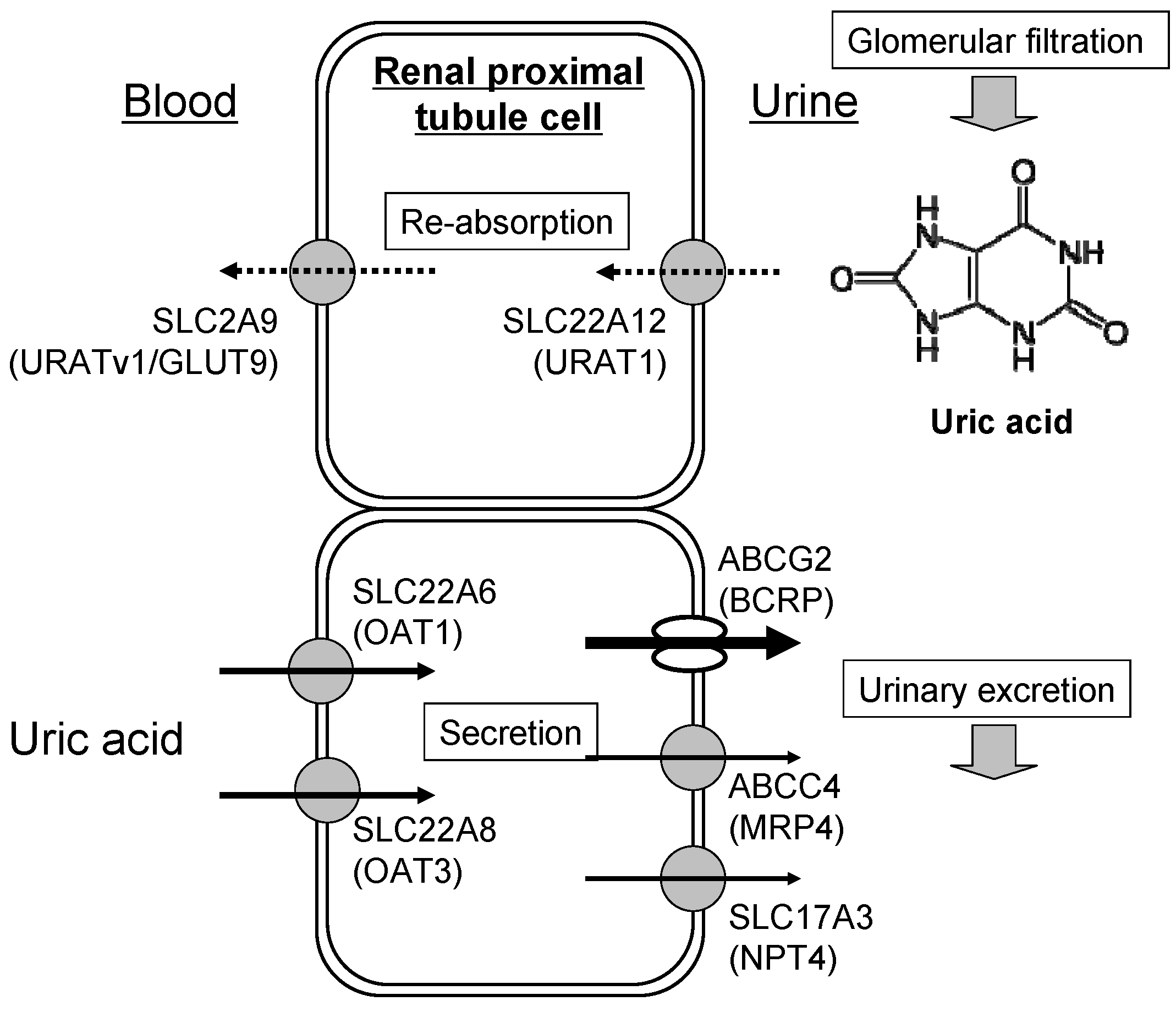
3. Renal Excretion of Uric Acid
4. Cause of Gout and Therapeutics
5. Genetic Analysis of Gout Risk
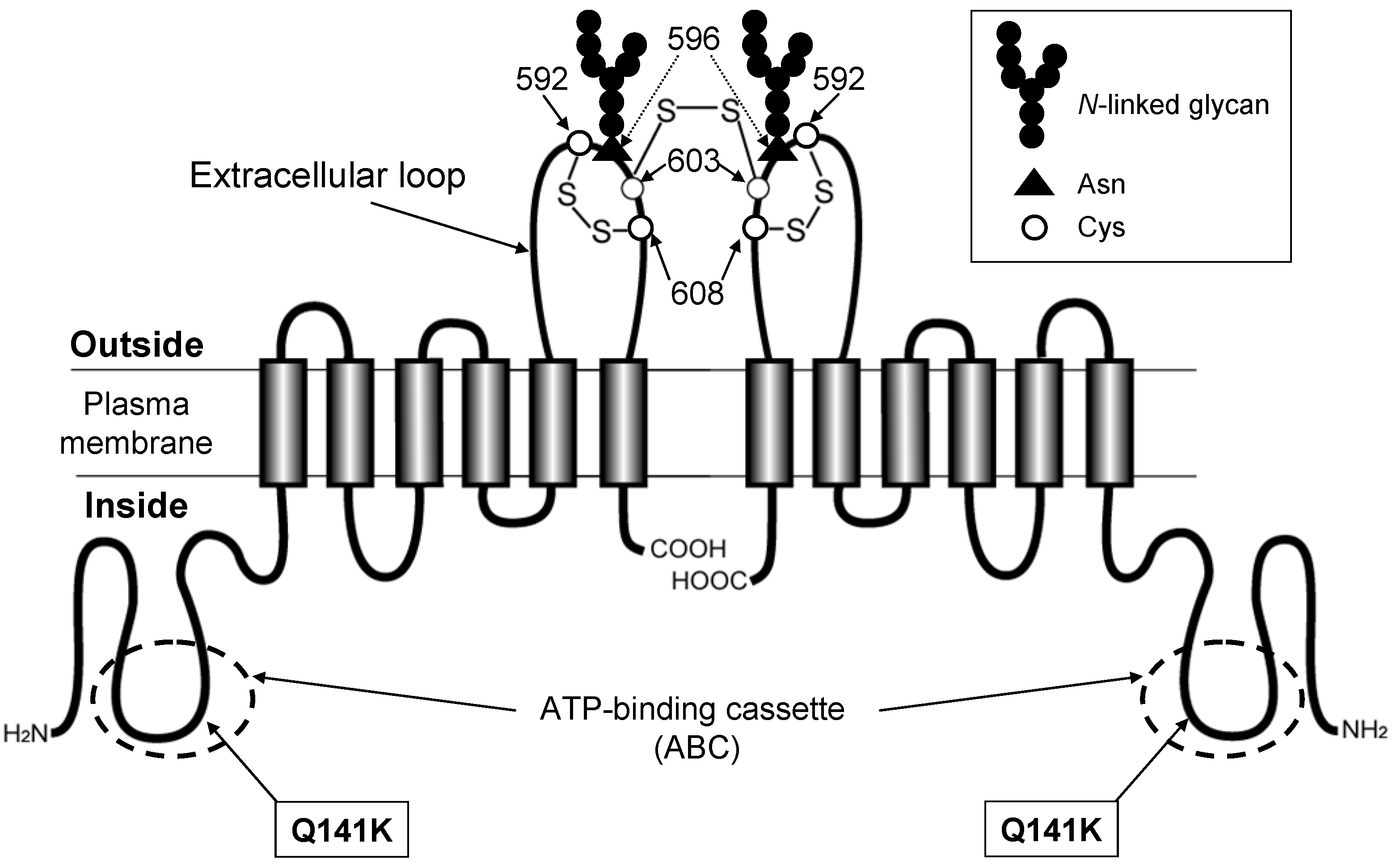
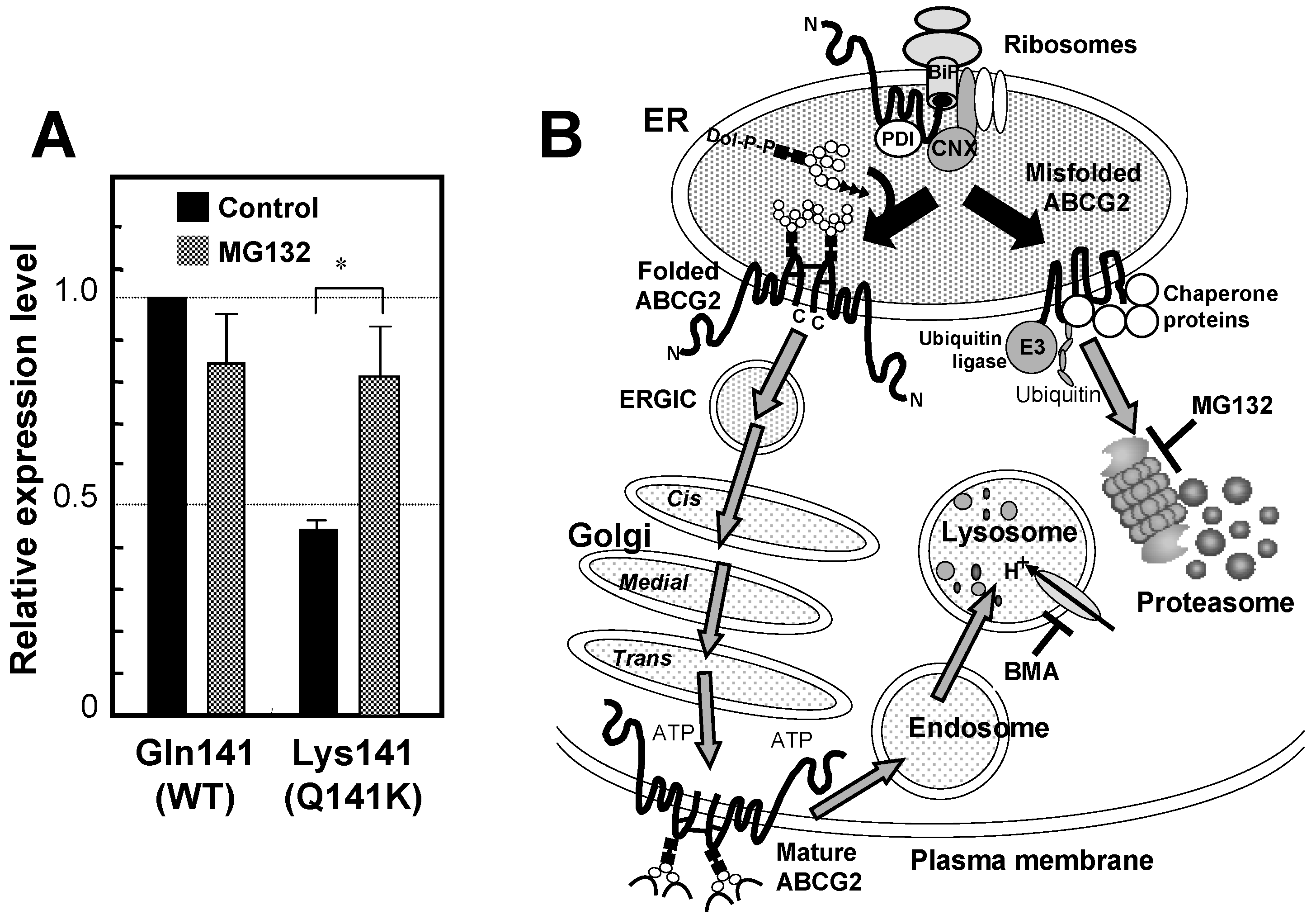
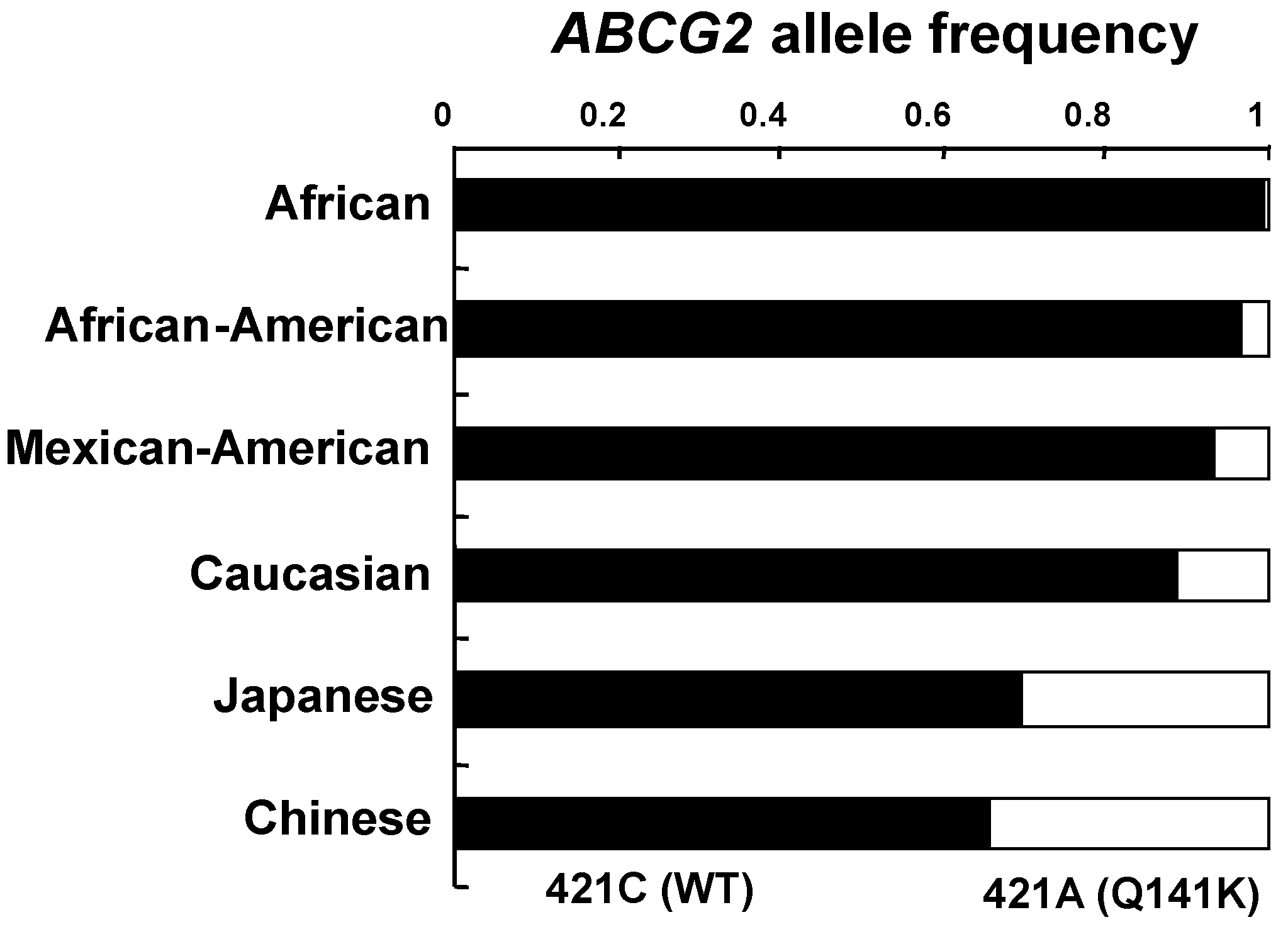
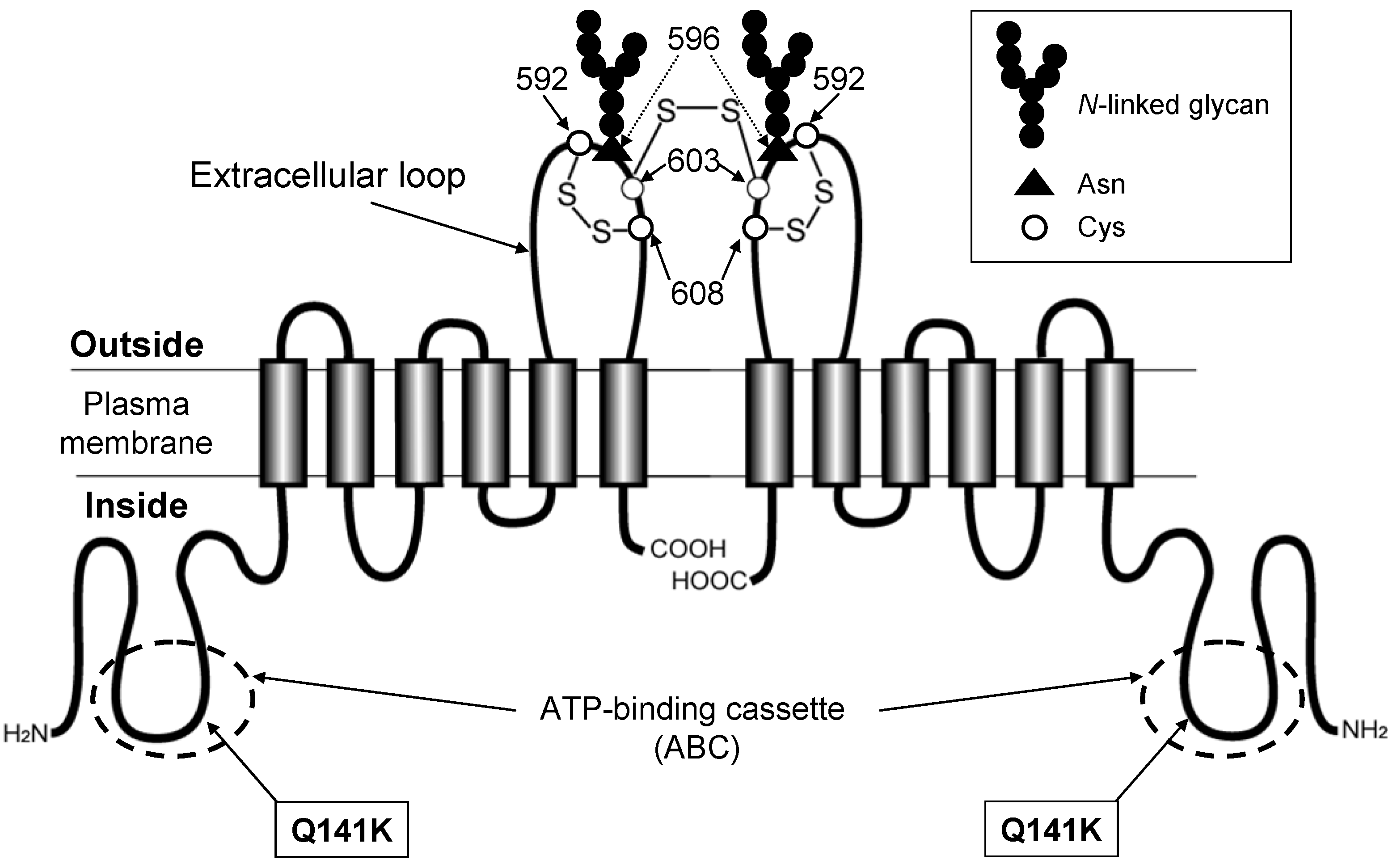
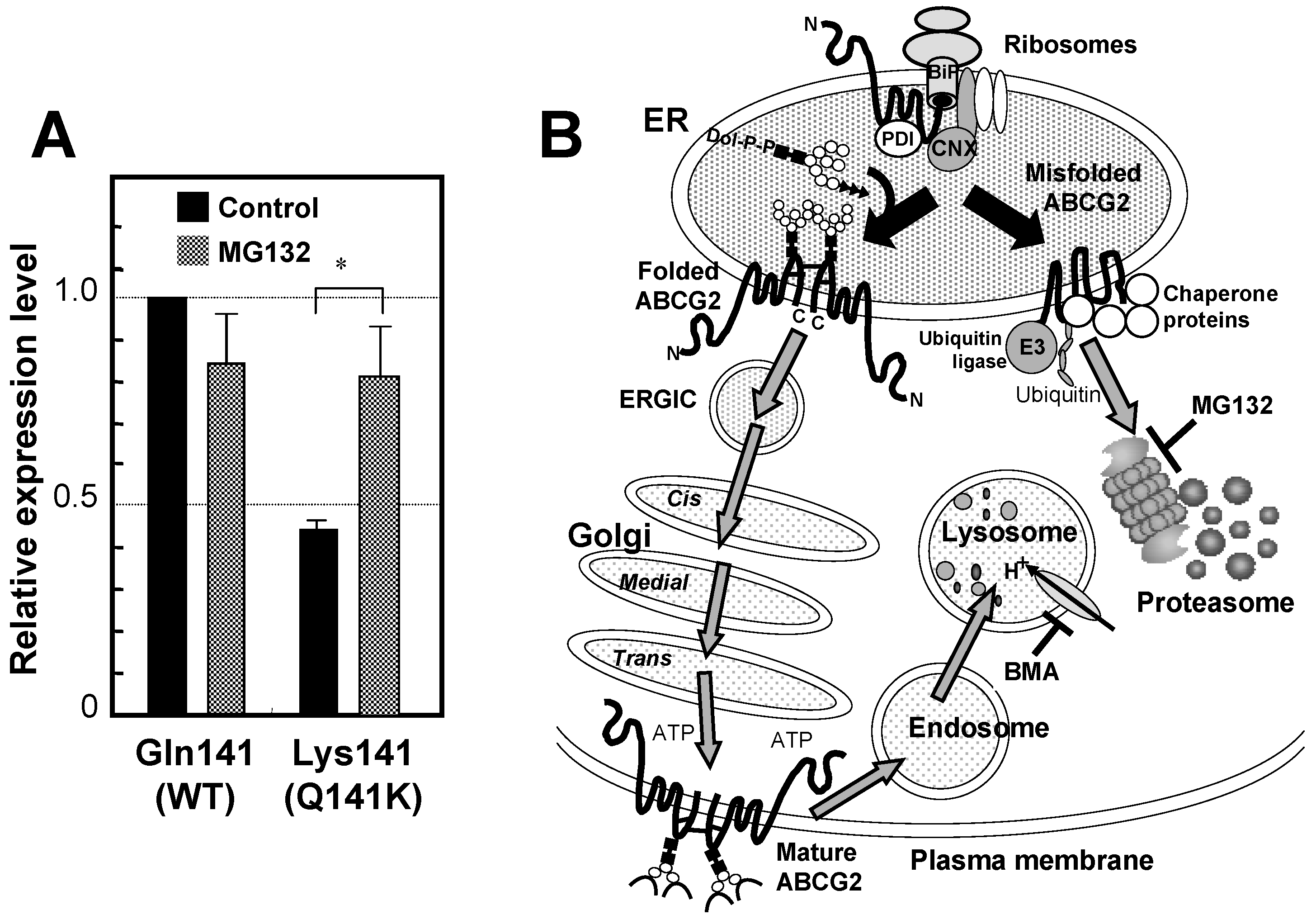
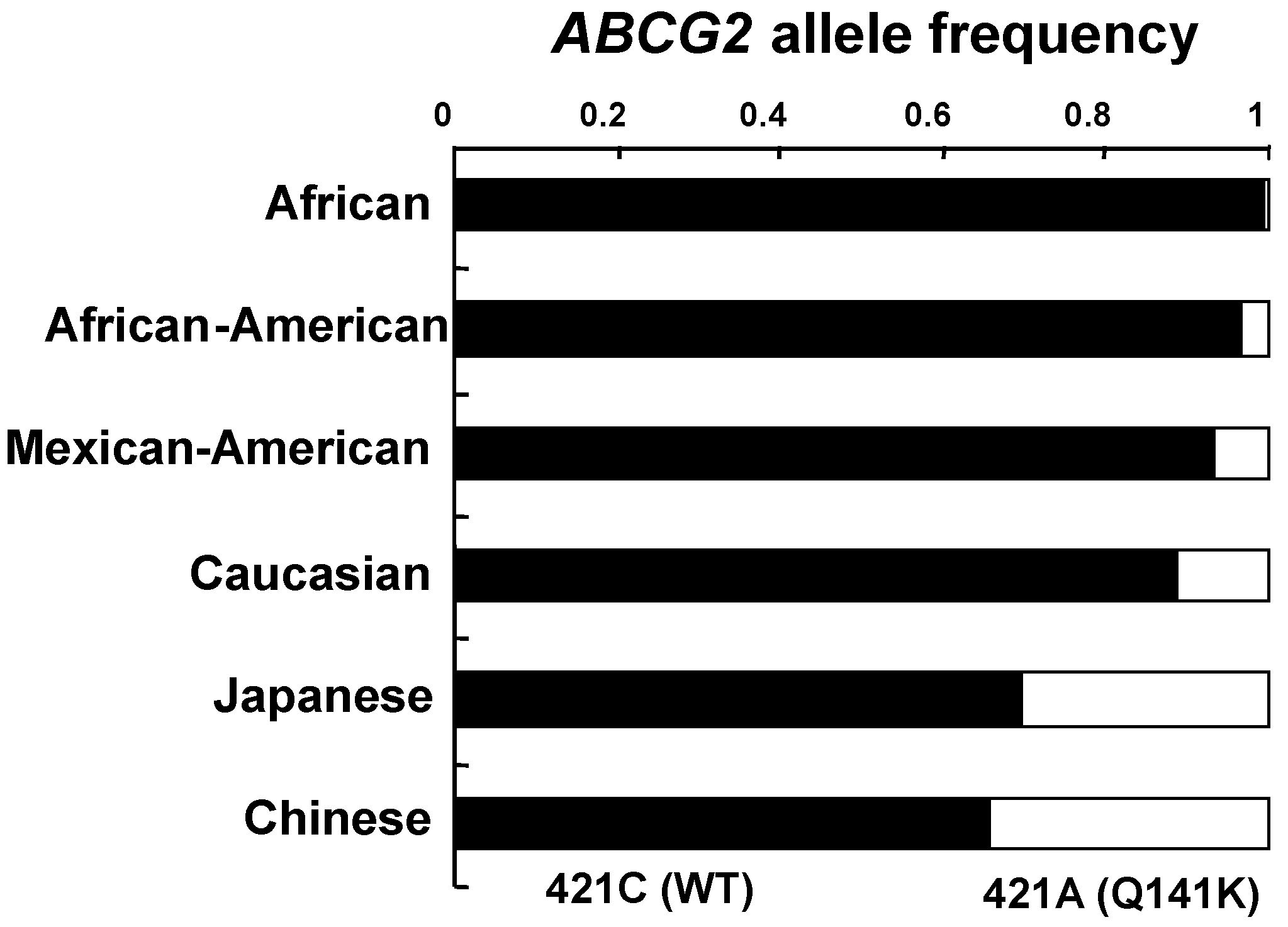
6. Genetic Polymorphisms in ABCG2 Gene
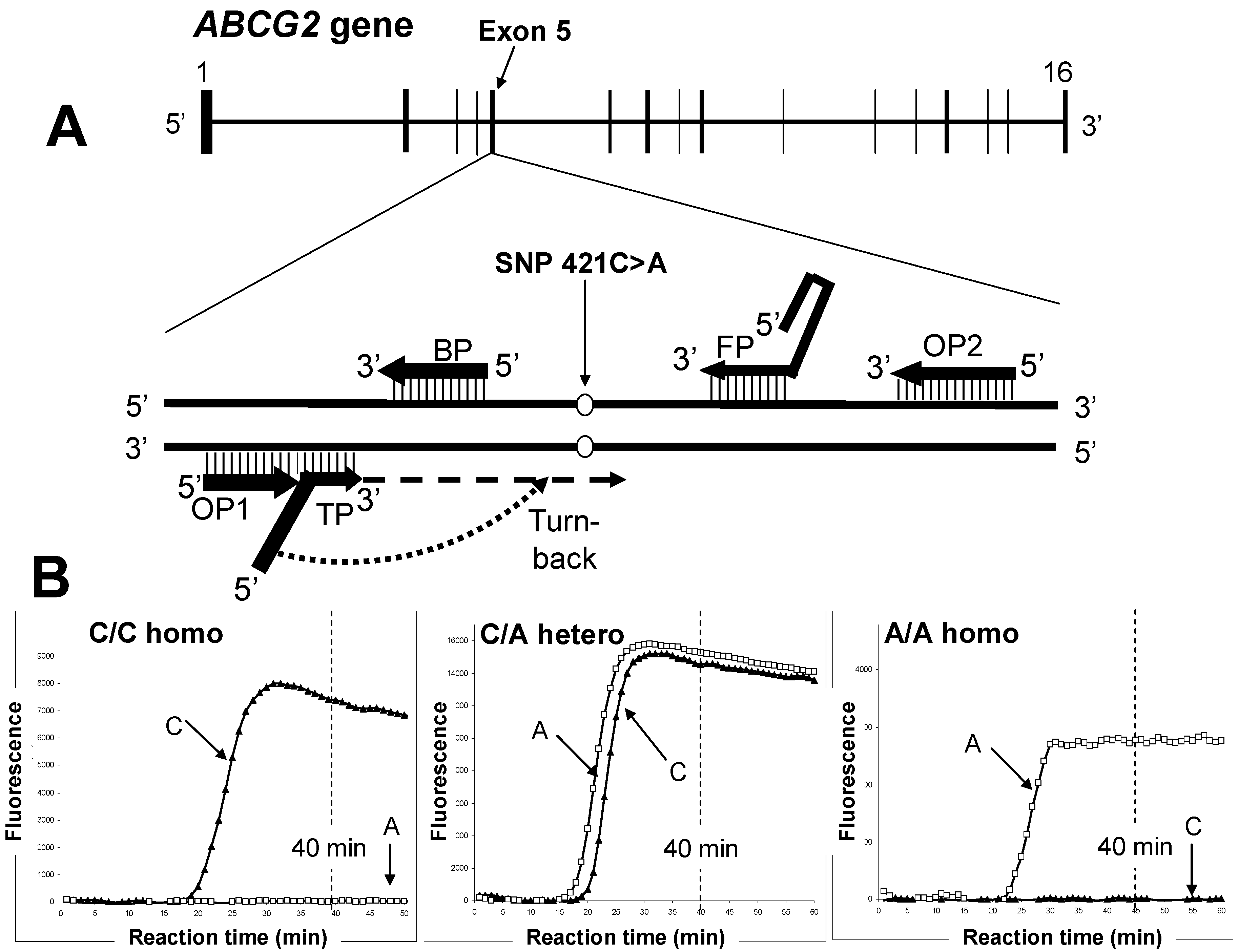
7. Rapid Genotyping to Assess Gout Risk

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
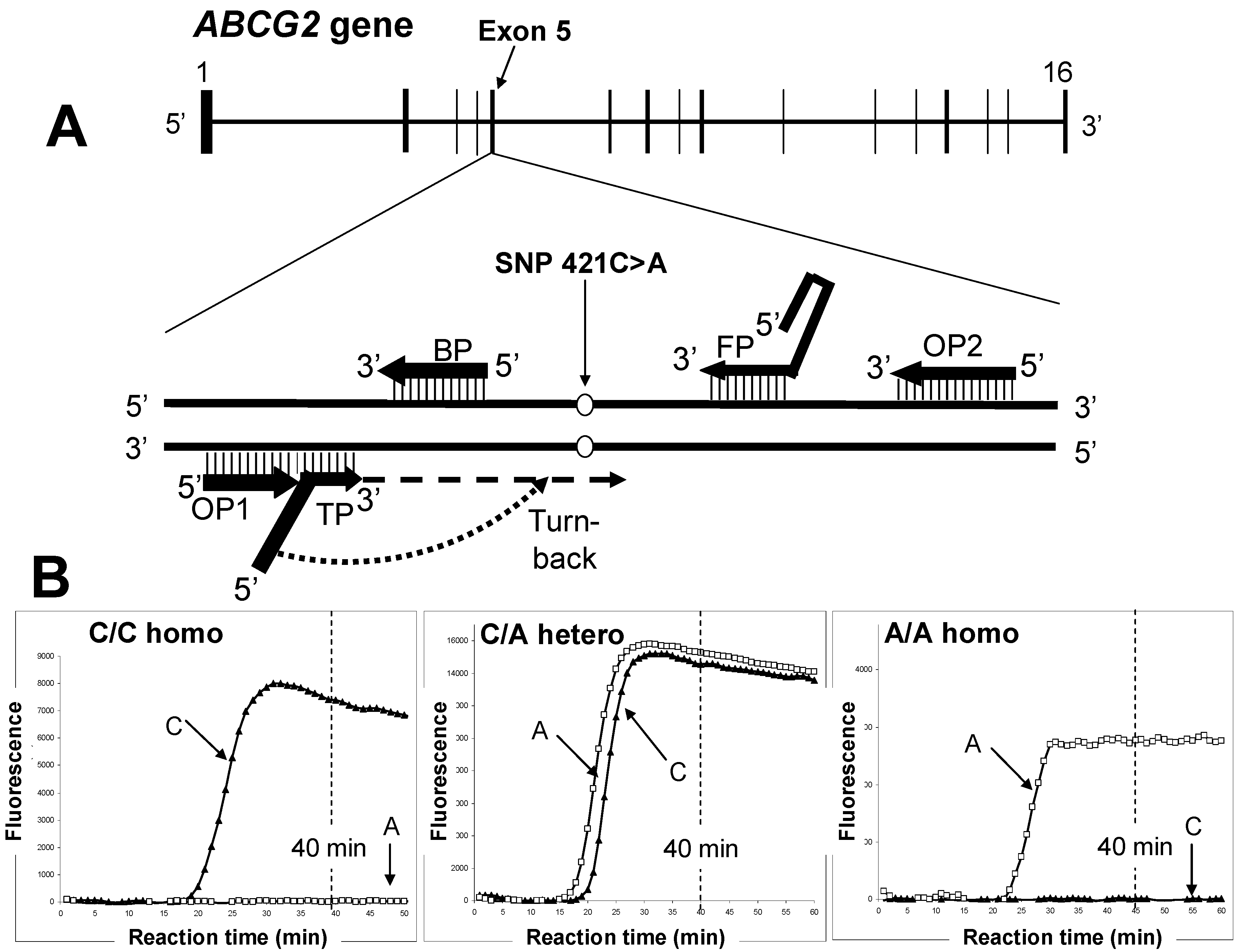{kind=link}
| WT (421C)-detection Primer Set | |
|---|---|
| Primer | Sequence (5'→3') |
| TP |  TAAGTTTTCCTTAAGGATGATGTTGTG TAAGTTTTCCTTAAGGATGATGTTGTG |
| FP | ACCTTCTGTACCCTCAGAAGGTGCCGAAGAGCTGCTGAGAAC |
| BP | ACCGTCAGAGTGCCCAT |
| OP1 | TTATCATTATGTCTCATT |
| OP2 | ATGATTCGTCATAGTTGT |
| SNP (421A)-detecting Primer Set | |
| Primer | Sequence (5'→3') |
| TP | T  TAAGTTTTCTCTTAAGGATGATGTTGTG TAAGTTTTCTCTTAAGGATGATGTTGTG |
| FP | ACCTTCTGTACCCTCAGAAGGTGCCGAAGAGCTGCTGAGAAC |
| BP | ACCGTCAGAGTGCCCAT |
| OP1 | TTATCATTATGTCTCATT |
| OP2 | ATGATTCGTCATAGTTGT |
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development: Report from the FDA critical path initiative-sponsored workshop. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Ishikawa, T. Multidrug resistance: Genomics of ABC transporters. Nat. Encycl. Hum. Genome 2003, 4, 154–160. [Google Scholar]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S.; et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout. A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1. [Google Scholar] [CrossRef]
- Woodward, O.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar]
- Richette, P.; Bardin, T. Purine-rich foods: An innocent bystander of gout attacks? Ann. Rheum. Dis. 2012, 71, 1435–1436. [Google Scholar] [CrossRef]
- Choi, H.K.; Ford, E.S. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am. J. Med. 2007, 120, 442–447. [Google Scholar] [CrossRef]
- Choi, H.K.; Mount, D.B.; Reginato, A.M. American College of Physicians; American Physiological Society. Pathogenesis of gout. Ann. Intern. Med. 2005, 143, 499–516. [Google Scholar] [CrossRef]
- Iribarren, C.; Folsom, A.R.; Eckfeldt, J.H.; McGovern, P.G.; Nieto, F.J. Correlates of uric acid and its association with asymptomatic carotid atherosclerosis: The ARIC study atherosclerosis risk in communities. Ann. Epidemiol. 1996, 6, 331–340. [Google Scholar] [CrossRef]
- Anzai, N.; Kanai, Y.; Endou, H. New insights into renal transport of urate. Curr. Opin. Rheumatol. 2007, 19, 151–157. [Google Scholar] [CrossRef]
- Enomoto, A.; Endou, H. Roles of organic anion transporters (OATs) and a urate transporter (URAT1) in the pathophysiology of human disease. Clin. Exp. Nephrol. 2005, 9, 195–205. [Google Scholar] [CrossRef]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar]
- Li, S.; Sanna, S.; Maschio, A.; Busonero, F.; Usala, G.; Mulas, A.; Lai, S.; Dei, M.; Orrù, M.; Albai, G.; et al. The GLUT9 Gene is associated with serum uric acid levels in Sardinia and Chianti Cohorts. PLoS Genet. 2007, 3, e194. [Google Scholar] [CrossRef]
- Wallace, C.; Newhouse, S.J.; Braund, P.; Zhang, F.; Tobin, M.; Falchi, M.; Ahmadi, K.; Dobson, R.J.; Marçano, A.C.; Hajat, C.; et al. Genome-wide association study identifies genes for biomarkers of cardiovascular disease: Serum urate and dyslipidemia. Am. J. Hum. Genet. 2008, 82, 139–149. [Google Scholar] [CrossRef]
- Döring, A.; Gieger, C.; Mehta, D.; Gohlke, H.; Prokisch, H.; Coassin, S.; Fischer, G.; Henke, K.; Klopp, N.; Kronenberg, F.; et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat. Genet. 2008, 40, 430–436. [Google Scholar] [CrossRef]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef]
- Matsuo, H.; Chiba, T.; Nagamori, S.; Nakayama, A.; Domoto, H.; Phetdee, K.; Wiriyasermkul, P.; Kikuchi, Y.; Oda, T.; Nishiyama, J.; et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 2008, 83, 744–751. [Google Scholar] [CrossRef]
- Rizwan, A.N.; Burckhardt, G. Organic anion transporters of the SLC22 family: Biopharmaceutical, physiological, and pathological roles. Pharm. Res. 2007, 24, 450–470. [Google Scholar] [CrossRef]
- Van Aubel, R.A.; Smeets, P.H.; van den Heuvel, J.J.; Russel, F.G. Human organic anion transporter MRP4 (ABCC4) is an efflux pump for the purine end metabolite urate with multiple allosteric substrate binding sites. Am. J. Physiol. Renal Physiol. 2005, 288, F327–F333. [Google Scholar]
- Huls, M.; Brown, C.D.; Windass, A.S.; Sayer, R.; van den Heuvel, J.J.; Heemskerk, S.; Russel, F.G.; Masereeuw, R. The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 2008, 73, 220–225. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Schuetz, J.D. Role of ABCG2/BCRP in biology and medicine. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 381–410. [Google Scholar] [CrossRef]
- Choi, H. Epidemiology of crystal arthropathy. Rheum. Dis. Clin. North Am. 2006, 32, 255–273. [Google Scholar] [CrossRef]
- Saag, K.G.; Choi, H. Epidemiology, risk factors, and lifestyle modifications for gout. Arthritis Res. Ther. 2006, 8, S2. [Google Scholar] [CrossRef]
- Becker, M.A.; Schumacher, H.R., Jr.; Wortmann, R.L.; MacDonald, P.A.; Eustace, D.; Palo, W.A.; Streit, J.; Joseph-Ridge, N. Febuxostatcompared with allopurinol in patients with hyperuricemia and gout. N. Engl. J. Med. 2005, 353, 2450–2461. [Google Scholar] [CrossRef]
- Kaneko, K.; Kobayashi, R.; Yasuda, M.; Izumi, Y.; Yamanobe, T.; Shimizu, T. Comparison of matrix proteins in different types of urinary stone by proteomic analysis using liquid chromatography-tandem mass spectrometry. Int. J. Urol. 2012, 19, 765–772. [Google Scholar] [CrossRef]
- Hamada, T.; Ichida, K.; Hosoyamada, M.; Mizuta, E.; Yanagihara, K.; Sonoyama, K.; Sugihara, S.; Igawa, O.; Hosoya, T.; Ohtahara, A.; et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am. J. Hypertens. 2008, 21, 1157–1162. [Google Scholar] [CrossRef]
- Deghan, A.; Köttgen, A.; Yang, Q.; Hwang, S.J.; Kao, W.L.; Rivadeneira, F.; Boerwinkle, E.; Levy, D.; Hofman, A.; Astor, B.C.; et al. Association of three genetic loci with uric acid concentration and risk of gout: A genome-wide association study. Lancet 2008, 372, 1953–1961. [Google Scholar] [CrossRef]
- Kolz, M.; Johnson, T.; Sanna, S.; Teumer, A.; Vitart, V.; Perola, M.; Mangino, M.; Albrecht, E.; Wallace, C.; Farrall, M.; et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009, 5, e1000504. [Google Scholar] [CrossRef] [Green Version]
- Stark, K.; Reinhard, W.; Grassi, M.; Erdmann, J.; Schunkert, H.; Illig, T.; Hengstenberg, C. Common polymorphisms influencing serum uric acid levels contribute to susceptibility to gout, but not to coronary artery disease. PLoS One 2009, 4, e7729. [Google Scholar]
- Zhang, L.; Spencer, K.L.; Voruganti, V.S.; Jorgensen, N.W.; Fornage, M.; Best, L.G.; Brown-Gentry, K.D.; Cole, S.A.; Crawford, D.C.; Deelman, E.; et al. Association of functional polymorphism rs2231142 (Q141K) in the ABCG2 gene with serum uric acid and gout in 4 US populations: The PAGE study. Am. J. Epidemiol. 2013, 177, 923–932. [Google Scholar] [CrossRef]
- Tamura, A.; Wakabayashi, K.; Onishi, Y.; Takeda, M.; Ikegami, Y.; Sawada, S.; Tsuji, M.; Matsuda, Y.; Ishikawa, T. Re-evaluation and functional classification of nonsynonymous single nucleotide polymorphisms of human ABC transporter ABCG2. Cancer Sci. 2007, 98, 231–239. [Google Scholar] [CrossRef]
- Nakagawa, H.; Tamura, A.; Wakabayashi, K.; Hoshijima, K.; Komada, M.; Yoshida, T.; Kometani, S.; Matsubara, T.; Mikuriya, K.; Ishikawa, T. Ubiquitin-mediated proteasomal degradation of non-synonymous SNP variants of human ABC transporter ABCG2. Biochem. J. 2008, 411, 623–631. [Google Scholar] [CrossRef]
- Furukawa, T.; Wakabayashi, K.; Tamura, A.; Nakagawa, H.; Morishima, Y.; Osawa, Y.; Ishikawa, T. Major SNP (Q141K) variant of human ABC transporter ABCG2 undergoes lysosomal and proteosomal degradations. Pharm. Res. 2009, 26, 469–479. [Google Scholar] [CrossRef]
- Wakabayashi-Nakao, K.; Tamura, A.; Furukawa, T.; Nakagawa, H.; Ishikawa, T. Quality control of human ABCG2 protein in the endoplasmeic reticulm: Ubiquitination and proteasomal degradation. Adv. Drug Deliv. Rev. 2009, 61, 66–72. [Google Scholar] [CrossRef]
- Nakagawa, H.; Toyoda, Y.; Wakabayashi-Nakao, K.; Tamaki, H.; Osumi, M.; Ishikawa, T. Ubiquitin-mediated proteasomal degradation of ABC transporters: A new aspect of genetic polymorphisms and clinical impacts. J. Parm. Sci. 2011, 100, 3602–3619. [Google Scholar]
- Ishikawa, T.; Tamura, A.; Saito, H.; Wakabayashi, K.; Nakagawa, H. Pharmacogenomics of the human ABC transporter ABCG2: From functional evaluation to drug molecular design. Naturwissenschaften 2005, 92, 451–463. [Google Scholar] [CrossRef]
- Honjo, Y.; Morisaki, K.; Huff, L.M.; Robey, R.W.; Hung, J.; Dean, M.; Bates, S.E. Single-nucleotide polymorphism (SNP) analysis in the ABC half-transporter ABCG2 (MXR/BCRP/ABCP1). Cancer Biol. Ther. 2002, 1, 696–702. [Google Scholar]
- Iida, A.; Saito, S.; Sekine, A.; Mishima, C.; Kitamura, Y.; Kondo, K.; Harigae, S.; Osawa, S.; Nakamura, Y. Catalog of 605 single-nucleotide polymorphisms (SNPs) among 13 genes encoding human ATP-binding cassette transporters: ABCA4, ABCA7, ABCA8, ABCD1, ABCD3, ABCD4, ABCE1, ABCF1, ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8. J. Hum. Genet. 2002, 47, 285–310. [Google Scholar] [CrossRef]
- Imai, Y.; Nakane, M.; Kage, K.; Tsukahara, S.; Ishikawa, E.; Tsuruo, T.; Miki, Y.; Sugimoto, Y. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol. Cancer Ther. 2002, 1, 611–616. [Google Scholar]
- Zamber, C.P.; Lamba, J.K.; Yasuda, K.; Farnum, J.; Thummel, K.; Schuetz, J.D.; Schuetz, E.G. Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharmacogenetics 2003, 13, 19–28. [Google Scholar] [CrossRef]
- Itoda, M.; Saito, Y.; Shirao, K.; Minami, H.; Ohtsu, A.; Yoshida, T.; Saijo, N.; Suzuki, H.; Sugiyama, Y.; Ozawa, S.; et al. Eight novel single nucleotide polymorphisms in ABCG2/BCRP in Japanese cancer patients administered Irinotecan. Drug Metab. Pharmacokin. 2003, 18, 212–217. [Google Scholar] [CrossRef]
- Bäckström, G.; Taipalensuu, J.; Melhus, H.; Brandstrom, H.; Svensson, A.C.; Artursson, P.; Kindmark, A. Genetic variation in the ATP-binding cassette transporter gene ABCG2 (BCRP) in a Swedish population. Eur. J. Pharm. Sci. 2003, 18, 359–364. [Google Scholar] [CrossRef]
- De Jong, F.A.; Marsh, S.; Mathijssen, R.H.; King, C.; Verweij, J.; Sparreboom, A.; McLeod, H.L. ABCG2 pharmacogenetics: Ethnic differences in allele frequency and assessment of influence on irinotecan disposition. Clin. Cancer Res. 2004, 10, 5889–5894. [Google Scholar] [CrossRef]
- Kobayashi, D.; Ieiri, I.; Hirota, T.; Takane, H.; Maegawa, S.; Kigawa, J.; Suzuki, H.; Nanba, E.; Oshimura, M.; Terakawa, N.; et al. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab. Dispos. 2005, 33, 94–101. [Google Scholar]
- Bosch, T.M.; Kjellberg, L.M.; Bouwers, A.; Koeleman, B.P.C.; Schellens, J.H.M.; Beijnen, J.H.; Smits, P.H.M.; Meijerman, I. Detection of single nucleotide polymorphisms in the ABCG2 gene in a Dutch population. Am. J. Pharmacogenomics 2005, 5, 123–131. [Google Scholar] [CrossRef]
- Basseville, A.; Bates, S.E.; Figg, W.D.; Sparreboom, A. BCRP (ABCG2). In Pharmacogenomics of Human Drug Transporters: Clinical Impacts, 1st ed.; Ishikawa, T., Kim, R.B., König, J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 311–343. [Google Scholar]
- Woodward, O.M.; Tukaye, D.N.; Cui, J.; Greenwell, P.; Constantoulakis, L.M.; Parker, B.S.; Rao, A.; Köttgen, M.; Maloney, P.C.; Guggino, W.B. Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, 5223–5228. [Google Scholar] [CrossRef]
- Ishikawa, T.; Hayashizaki, Y. Clinical SNP detection by the SmartAmp method. Methods Mol. Biol. 2013, 1015, 55–69. [Google Scholar] [CrossRef]
- Matsuo, H.; Ichida, K.; Takada, T.; Nakayama, A.; Nakashima, H.; Nakamura, T.; Kawamura, Y.; Takada, Y.; Yamamoto, K.; Inoue, H.; et al. Common dysfunctional variants in ABCG2 are a major cause of early-onset gout. Sci. Rep. 2013, 3, 2014. [Google Scholar]
- Ishikawa, T.; Ware, J. Future Perspectives. In Pharmacogenomics of Human Drug Transporters: Clinical Impacts, 1st ed.; Ishikawa, T., Kim, R.B., König, J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 401–416. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishikawa, T.; Aw, W.; Kaneko, K. Metabolic Interactions of Purine Derivatives with Human ABC Transporter ABCG2: Genetic Testing to Assess Gout Risk. Pharmaceuticals 2013, 6, 1347-1360. https://doi.org/10.3390/ph6111347
Ishikawa T, Aw W, Kaneko K. Metabolic Interactions of Purine Derivatives with Human ABC Transporter ABCG2: Genetic Testing to Assess Gout Risk. Pharmaceuticals. 2013; 6(11):1347-1360. https://doi.org/10.3390/ph6111347
Chicago/Turabian StyleIshikawa, Toshihisa, Wanping Aw, and Kiyoko Kaneko. 2013. "Metabolic Interactions of Purine Derivatives with Human ABC Transporter ABCG2: Genetic Testing to Assess Gout Risk" Pharmaceuticals 6, no. 11: 1347-1360. https://doi.org/10.3390/ph6111347