Neurotransmitter CART as a New Therapeutic Candidate for Parkinson’s Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Etiology and Pathophysiology of PD
2.1. Genetic Factors
2.2. Mitochondrial Dysfunction and Oxidative Stress
3. CART Is a New Peptide Hormone with Multiple Functions
4. New Functions of CART Related to Mitochondria
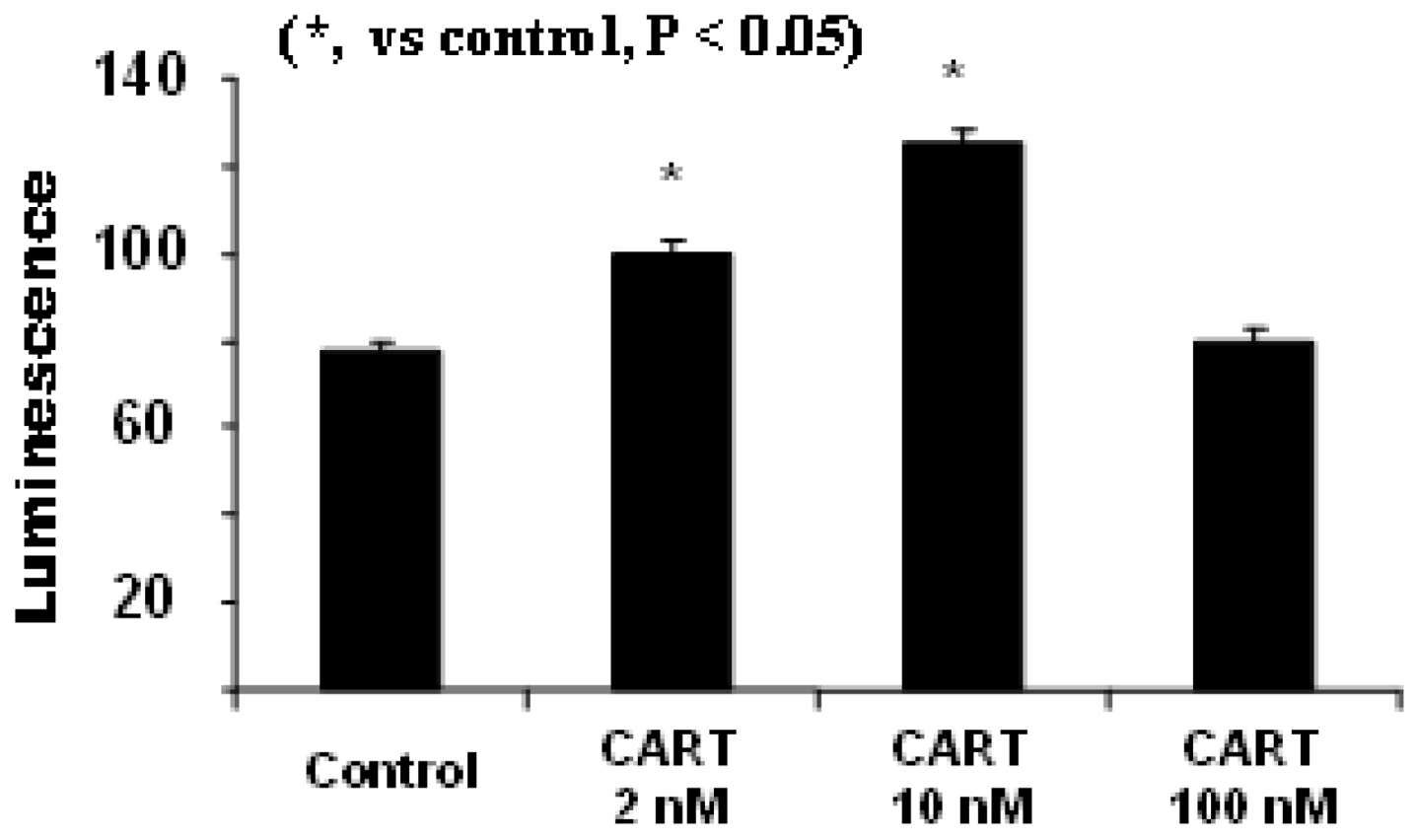
4.1. CART Stimulates the Activities of SDH and Complex II, Increases Cellular ATP Levels
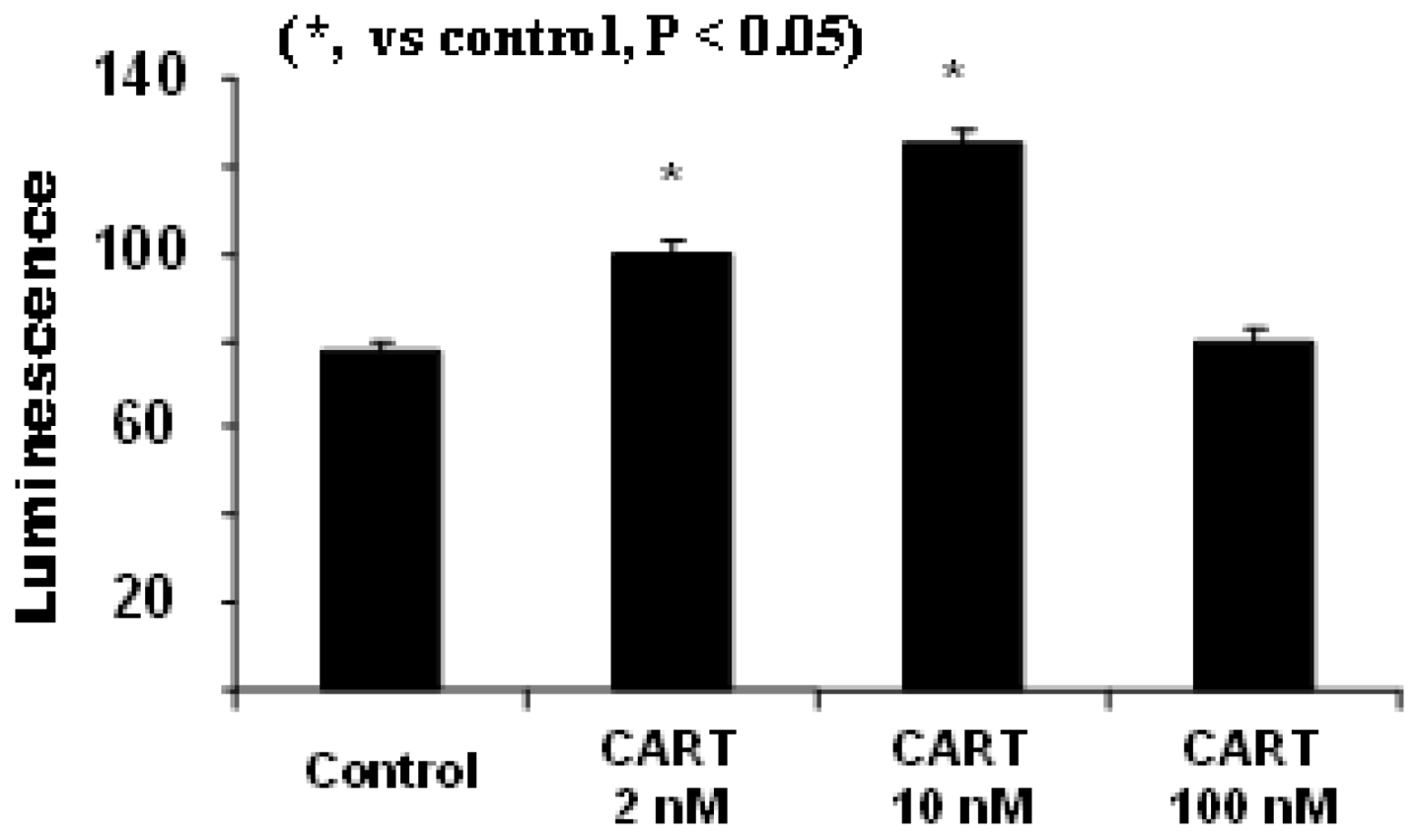
4.2. CART’s Antioxidant Properties
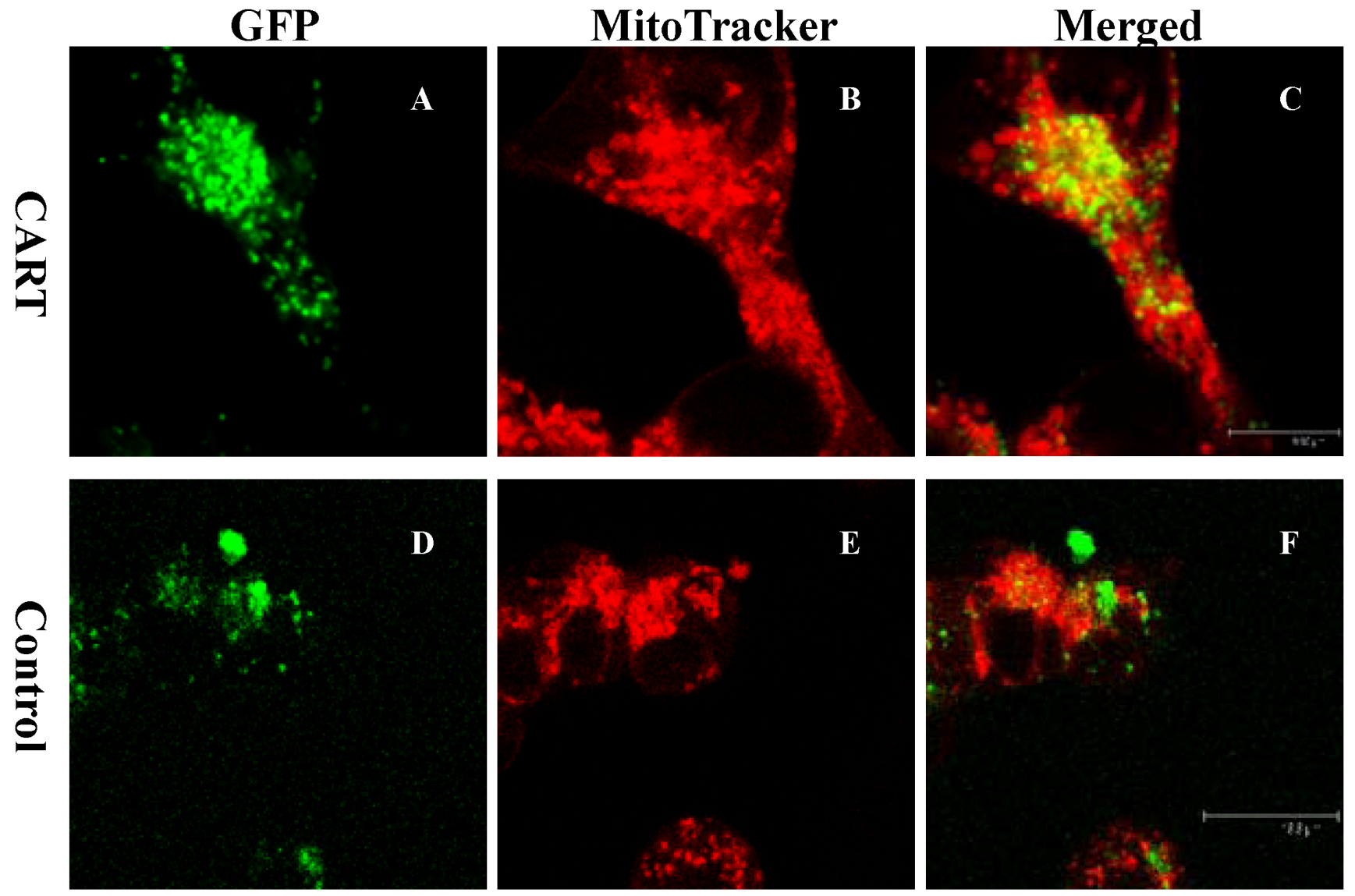
4.3. CART Is Physiologically Associated with the Dopamine System
4.4. The Role of CART in PD Model
4.5. CART as a Potential Therapeutic Target for PD
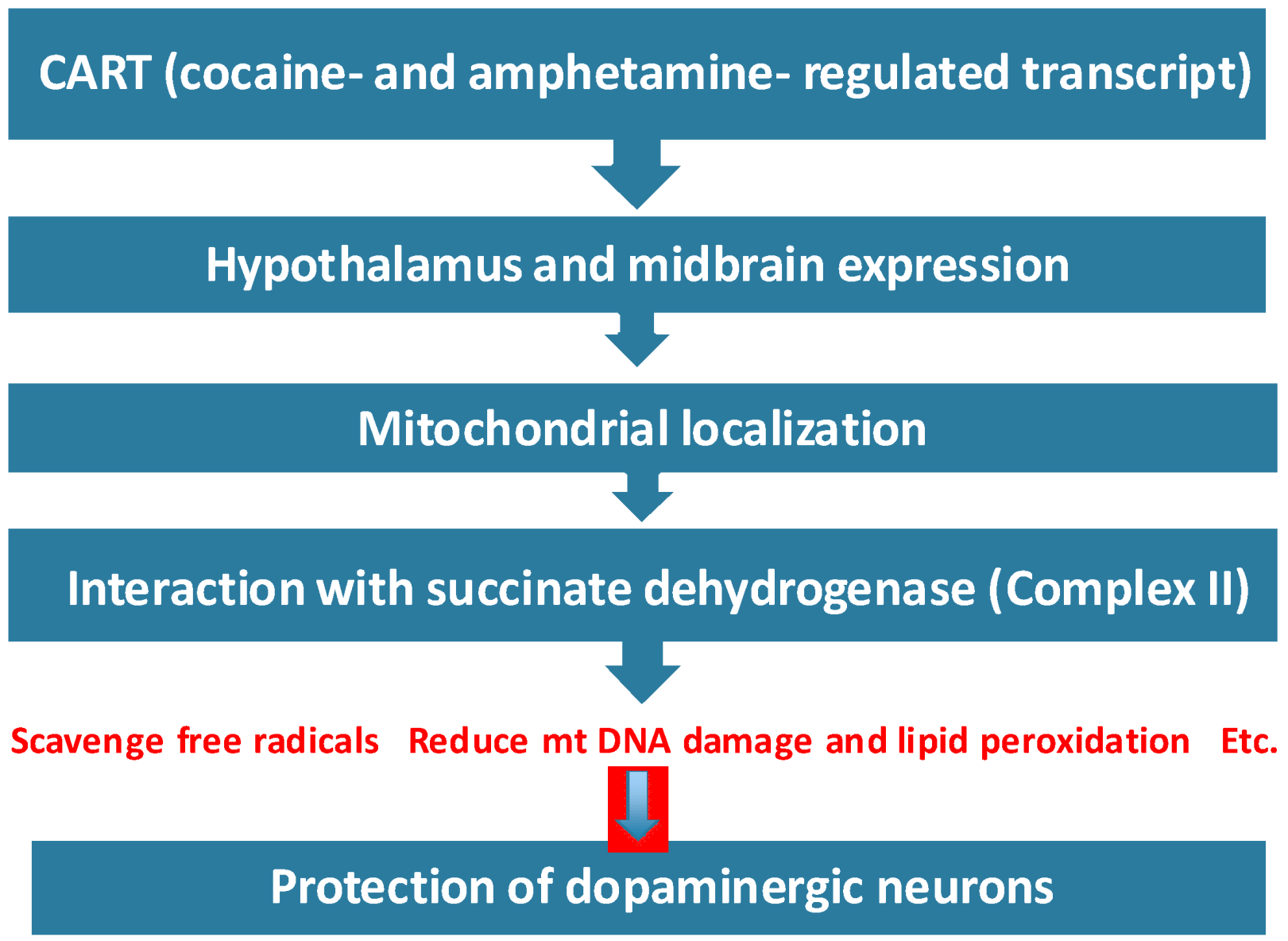
5. Conclusions
Acknowledgments
References
- Tanner, C.M.; Goldman, S.M. Epidemiology of Parkinson's Disease. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Barzilai, A.; Melamed, E. Molecular Mechanisms of Selective Dopaminergic Neuronal Death in Parkinson's Disease. Trends Mol. Med. 2003, 9, 126–132. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular Pathophysiology of Parkinson's Disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef]
- Thomas, B.; Beal, M.F. Parkinson's Disease. Hum. Mol. Genet. 2007, 16 Spec No. 2, R183–R194. [Google Scholar] [CrossRef]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial Dysfunction in the Limelight of Parkinson's Disease Pathogenesis. Biochim. Biophys. Acta 2009, 1792, 651–663. [Google Scholar]
- Schapira, A.H. Targeting Mitochondria for Neuroprotection in Parkinson's Disease. Antioxid. Redox Signal. 2012, 16, 965–973. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial Diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Wood-Kaczmar, A.; Gandhi, S.; Wood, N.W. Understanding the Molecular Causes of Parkinson's Disease. Trends Mol. Med. 2006, 12, 521–528. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial Dysfunction in Parkinson's Disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Mao, P.; Ardeshiri, A.; Jacks, R.; Yang, S.; Hurn, P.D.; Alkayed, N.J. Mitochondrial Mechanism of Neuroprotection by CART. Eur. J. Neurosci. 2007, 26, 624–632. [Google Scholar] [CrossRef]
- Mao, P.; Meshul, C.K.; Thuillier, P.; Goldberg, N.R.; Reddy, P.H. CART Peptide is a Potential Endogenous Antioxidant and Preferentially Localized in Mitochondria. PLoS One 2012, 7, e29343. [Google Scholar]
- Warner, T.T.; Schapira, A.H. Genetic and Environmental Factors in the Cause of Parkinson's Disease. Ann. Neurol. 2003, 53 Suppl 3, S16–S23; discussion S23–S25. [Google Scholar]
- Schapira, A.H. Etiology and Pathogenesis of Parkinson Disease. Neurol. Clin. 2009, 27, 83–603, v. [Google Scholar] [CrossRef]
- Schapira, A.H. Etiology of Parkinson's Disease. Neurology 2006, 66, S10–S23. [Google Scholar] [CrossRef]
- Lansbury, P.T., Jr; Brice, A. Genetics of Parkinson's Disease and Biochemical Studies of Implicated Gene Products. Curr. Opin. Cell Biol. 2002, 14, 653–660. [Google Scholar] [CrossRef]
- Dodson, M.W.; Guo, M. Pink1, Parkin, DJ-1 and Mitochondrial Dysfunction in Parkinson's Diseas. Curr. Opin. Neurobiol. 2007, 17, 331–337. [Google Scholar] [CrossRef]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic Etiology of Parkinson Disease Associated with Mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 Genes: A Mutation Update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef]
- Martin, L.J. Biology of Mitochondria in Neurodegenerative Diseases. Prog. Mol. Biol. Transl. Sci. 2012, 107, 355–415. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the Alpha-Synuclein Gene Identified in Families with Parkinson's Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Bower, K.; Fink, A.L. Synergistic Effects of Pesticides and Metals on the Fibrillation of Alpha-Synuclein: Implications for Parkinson's Disease. Neurotoxicology 2002, 23, 527–536. [Google Scholar] [CrossRef]
- Maraganore, D.M.; de Andrade, M.; Elbaz, A.; Farrer, M.J.; Ioannidis, J.P.; Kruger, R.; Rocca, W.A.; Schneider, N.K.; Lesnick, T.G.; Lincoln, S.J.; et al. Collaborative Analysis of Alpha-Synuclein Gene Promoter Variability and Parkinson Disease. JAMA 2006, 296, 661–670. [Google Scholar] [CrossRef]
- Galvin, J.E.; Pollack, J.; Morris, J.C. Clinical Phenotype of Parkinson Disease Dementia. Neurology 2006, 67, 1605–1611. [Google Scholar] [CrossRef]
- Galvin, J.E. Cognitive Change in Parkinson Disease. Alzheimer Dis. Assoc. Disord. 2006, 20, 302–310. [Google Scholar] [CrossRef]
- Janda, E.; Isidoro, C.; Carresi, C.; Mollace, V. Defective Autophagy in Parkinson's Disease: Role of Oxidative Stress. Mol. Neurobiol. 2012.
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic Systemic Pesticide Exposure Reproduces Features of Parkinson's Disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Langston, J.W.; Sastry, S.; Chan, P.; Forno, L.S.; Bolin, L.M.; Di Monte, D.A. Novel Alpha-Synuclein-Immunoreactive Proteins in Brain Samples from the Contursi Kindred, Parkinson's, and Alzheimer's Disease. Exp. Neurol. 1998, 154, 684–690. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson's Disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Giasson, B.I.; Lee, V.M. Are Ubiquitination Pathways Central to Parkinson's Disease? Cell 2003, 114, 1–8. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson's Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial Import and Enzymatic Activity of PINK1 Mutants Associated to Recessive Parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant Stress Evoked by Pacemaking in Dopaminergic Neurons is Attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef]
- Papkovskaia, T.D.; Chau, K.Y.; Inesta-Vaquera, F.; Papkovsky, D.B.; Healy, D.G.; Nishio, K.; Staddon, J.; Duchen, M.R.; Hardy, J.; Schapira, A.H.; et al. G2019S Leucine-Rich Repeat Kinase 2 Causes Uncoupling Protein-Mediated Mitochondrial Depolarization. Hum. Mol. Genet. 2012, 21, 4201–4213. [Google Scholar] [CrossRef]
- Abeliovich, A. Parkinson's Disease: Mitochondrial Damage Control. Nature 2010, 463, 744–745. [Google Scholar] [CrossRef]
- Schapira, A.H.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and Disease Specificity of NADH CoQ1 Reductase (Complex I) Deficiency in Parkinson's Disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative Stress in Parkinson's Disease. Ann. Neurol. 2003, S26–S36; discussion S36–S38. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Moran, M.; Moreno-Lastres, D.; Marin-Buera, L.; Arenas, J.; Martin, M.A.; Ugalde, C. Mitochondrial Respiratory Chain Dysfunction: Implications in Neurodegeneration. Free Radic. Biol. Med. 2012, 53, 595–609. [Google Scholar] [CrossRef]
- Jenner, P. Presymptomatic Detection of Parkinson's Disease. J. Neural Transm. Suppl. 1993, 40, 23–36. [Google Scholar]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative Stress in Parkinson's Disease: A Mechanism of Pathogenic and Therapeutic Significance. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson's Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson's Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Gunzler, S.A.; Shakil, S.; Carlson, N.E.; Nutt, J.G.; Meshul, C.K. Low Doses of Apomorphine Transiently Reduce Locomotor Activity in MPTP-Treated Mice. Neurosci. Lett. 2007, 428, 64–67. [Google Scholar] [CrossRef]
- Meshul, C.K.; Emre, N.; Nakamura, C.M.; Allen, C.; Donohue, M.K.; Buckman, J.F. Time-Dependent Changes in Striatal Glutamate Synapses Following a 6-Hydroxydopamine Lesion. Neuroscience 1999, 88, 1–16. [Google Scholar] [CrossRef]
- Robinson, S.; Freeman, P.; Moore, C.; Touchon, J.C.; Krentz, L.; Meshul, C.K. Acute and Subchronic MPTP Administration Differentially Affects Striatal Glutamate Synaptic Function. Exp. Neurol. 2003, 180, 74–87. [Google Scholar] [CrossRef]
- Fox, S.H.; Brotchie, J.M. The MPTP-Lesioned Non-Human Primate Models of Parkinson's Disease. Past, Present, and Future. Prog. Brain Res. 2010, 184, 133–157. [Google Scholar] [CrossRef]
- Iderberg, H.; Francardo, V.; Pioli, E.Y. Animal Models of L-DOPA-Induced Dyskinesia: An Update on the Current Options. Neuroscience 2012, 211, 13–27. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and Interplay of Parkin, PINK1, and DJ1 in Neurodegenerative and Neuroinflammatory Disorders. Free Radic. Biol. Med. 2012, 53, 983–992. [Google Scholar] [CrossRef]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The Herbicide Paraquat Causes Up-Regulation and Aggregation of Alpha-Synuclein in Mice: Paraquat and Alpha-Synuclein. J. Biol. Chem. 2002, 277, 1641–1644. [Google Scholar]
- Dawson, T.M.; Dawson, V.L. Molecular Pathways of Neurodegeneration in Parkinson's Disease. Science 2003, 302, 819–822. [Google Scholar] [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. Rejuvenation' Protects Neurons in Mouse Models of Parkinson's Disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Chinta, S.J.; Andersen, J.K. Reversible Inhibition of Mitochondrial Complex I Activity Following Chronic Dopaminergic Glutathione Depletion in Vitro: Implications for Parkinson's Disease. Free Radic. Biol. Med. 2006, 41, 1442–1448. [Google Scholar] [CrossRef]
- Chinta, S.J.; Kumar, M.J.; Hsu, M.; Rajagopalan, S.; Kaur, D.; Rane, A.; Nicholls, D.G.; Choi, J.; Andersen, J.K. Inducible Alterations of Glutathione Levels in Adult Dopaminergic Midbrain Neurons Result in Nigrostriatal Degeneration. J. Neurosci. 2007, 27, 13997–14006. [Google Scholar] [CrossRef]
- Sarkar, C.; Basu, B.; Chakroborty, D.; Dasgupta, P.S.; Basu, S. The Immunoregulatory Role of Dopamine: An Update. Brain Behav. Immun. 2010, 24, 525–528. [Google Scholar] [CrossRef]
- Rasheed, N.; Alghasham, A. Central Dopaminergic System and its Implications in Stress-Mediated Neurological Disorders and Gastric Ulcers: Short Review. Adv. Pharmacol. Sci. 2012, 2012, 182671. [Google Scholar]
- Hastings, T.G. The Role of Dopamine Oxidation in Mitochondrial Dysfunction: Implications for Parkinson's Disease. J. Bioenerg. Biomembr. 2009, 41, 469–472. [Google Scholar] [CrossRef]
- Mao, P.; Reddy, P.H. Aging and Amyloid Beta-Induced Oxidative DNA Damage and Mitochondrial Dysfunction in Alzheimer's Disease: Implications for Early Intervention and Therapeutics. Biochim. Biophys. Acta 2011.
- Duncan, J.; Johnson, S.; Ou, X.M. Monoamine Oxidases in Major Depressive Disorder and Alcoholism. Drug Discov. Ther. 2012, 6, 112–122. [Google Scholar]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA Damage in the Parkinsonian Brain: An Apparent Selective Increase in 8-Hydroxyguanine Levels in Substantia Nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar]
- Alam, Z.I.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Jenner, P.; Halliwell, B. A Generalised Increase in Protein Carbonyls in the Brain in Parkinson's but Not Incidental Lewy Body Disease. J. Neurochem. 1997, 69, 1326–1329. [Google Scholar]
- Jenner, P. Oxidative Stress and Parkinson's Disease. Handb. Clin. Neurol. 2007, 83, 507–520. [Google Scholar] [CrossRef]
- Seet, R.C.; Lee, C.Y.; Lim, E.C.; Tan, J.J.; Quek, A.M.; Chong, W.L.; Looi, W.F.; Huang, S.H.; Wang, H.; Chan, Y.H.; et al. Oxidative Damage in Parkinson Disease: Measurement using Accurate Biomarkers. Free Radic. Biol. Med. 2010, 48, 560–566. [Google Scholar] [CrossRef]
- Douglass, J.; McKinzie, A.A.; Couceyro, P. PCR Differential Display Identifies a Rat Brain mRNA that is Transcriptionally Regulated by Cocaine and Amphetamine. J. Neurosci. 1995, 15, 2471–2481. [Google Scholar]
- Thim, L.; Kristensen, P.; Nielsen, P.F.; Wulff, B.S.; Clausen, J.T. Tissue-Specific Processing of Cocaine- and Amphetamine-Regulated Transcript Peptides in the Rat. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 2722–2727. [Google Scholar]
- Larsen, P.J.; Seier, V.; Fink-Jensen, A.; Holst, J.J.; Warberg, J.; Vrang, N. Cocaine- and Amphetamine-Regulated Transcript is Present in Hypothalamic Neuroendocrine Neurones and is Released to the Hypothalamic-Pituitary Portal Circuit. J. Neuroendocrinol. 2003, 15, 219–226. [Google Scholar] [CrossRef]
- Koylu, E.O.; Couceyro, P.R.; Lambert, P.D.; Ling, N.C.; DeSouza, E.B.; Kuhar, M.J. Immunohistochemical Localization of Novel CART Peptides in Rat Hypothalamus, Pituitary and Adrenal Gland. J. Neuroendocrinol. 1997, 9, 823–833. [Google Scholar]
- Koylu, E.O.; Balkan, B.; Kuhar, M.J.; Pogun, S. Cocaine and Amphetamine Regulated Transcript (CART) and the Stress Response. Peptides 2006, 27, 1956–1969. [Google Scholar] [CrossRef]
- Mao, P. Potential Antidepressant Role of Neurotransmitter CART: Implications for Mental Disorders. Depress Res. Treat. 2011, 2011, 762139. [Google Scholar]
- Kuhar, M.J.; Adams, S.; Dominguez, G.; Jaworski, J.; Balkan, B. CART Peptides. Neuropeptides 2002, 36, 1–8. [Google Scholar] [CrossRef]
- Hunter, R.G.; Kuhar, M.J. CART Peptides as Targets for CNS Drug Development. Curr. Drug Targets CNS Neurol. Disord. 2003, 2, 201–205. [Google Scholar] [CrossRef]
- Rogge, G.; Jones, D.; Hubert, G.W.; Lin, Y.; Kuhar, M.J. CART Peptides: Regulators of Body Weight, Reward and Other Functions. Nat. Rev. Neurosci. 2008, 9, 747–758. [Google Scholar]
- del Giudice, E.M.; Santoro, N.; Cirillo, G.; D'Urso, L.; Di Toro, R.; Perrone, L. Mutational Screening of the CART Gene in Obese Children: Identifying a Mutation (Leu34Phe) Associated with Reduced Resting Energy Expenditure and Cosegregating with Obesity Phenotype in a Large Family. Diabetes 2001, 50, 2157–2160. [Google Scholar] [CrossRef]
- Yanik, T.; Dominguez, G.; Kuhar, M.J.; Del Giudice, E.M.; Loh, Y.P. The Leu34Phe ProCART Mutation Leads to Cocaine- and Amphetamine-Regulated Transcript (CART) Deficiency: A Possible Cause for Obesity in Humans. Endocrinology 2006, 147, 39–43. [Google Scholar]
- Mao, P.; Jacks, R. Transcriptional Activity by Cocaine-Amphetamine-Regulated Transcript. Mol. Psychiatry 2007, 12, 223–224. [Google Scholar] [CrossRef]
- Wu, B.; Hu, S.; Yang, M.; Pan, H.; Zhu, S. CART Peptide Promotes the Survival of Hippocampal Neurons by Upregulating Brain-Derived Neurotrophic Factor. Biochem. Biophys. Res. Commun. 2006, 347, 656–661. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, W.; Klaus, J.; Young, J.; Koerner, I.; Sheldahl, L.C.; Hurn, P.D.; Martinez-Murillo, F.; Alkayed, N.J. Role of Cocaine- and Amphetamine-Regulated Transcript in Estradiol-Mediated Neuroprotection. Proc. Natl. Acad. Sci. USA 2006, 103, 14489–14494. [Google Scholar]
- Zhang, M.; Han, L.; Xu, Y. Roles of Cocaine- and Amphetamine-Regulated Transcript in the Central Nervous System. Clin. Exp. Pharmacol. Physiol. 2012, 39, 586–592. [Google Scholar] [CrossRef]
- Mao, P. Recent progress and concerns in dementia: distinguishing Alzheimer's disease and dementia with Lewy bodies via biochemical markers in the cerebrospinal fluid. Advances in Biological Chemistry 2012, 2, 176–190. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Newell, K.L. "Untangling" the Relationship between Alzheimer Disease and Dementia with Lewy Bodies. Neurology 2012, 79, 1938–1939. [Google Scholar] [CrossRef]
- Cecchini, G. Function and Structure of Complex II of the Respiratory Chain. Annu. Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef]
- Rustin, P.; Rotig, A. Inborn Errors of Complex II--Unusual Human Mitochondrial Diseases. Biochim. Biophys. Acta 2002, 1553, 117–122. [Google Scholar] [CrossRef]
- Bertoni-Freddari, C.; Fattoretti, P.; Paoloni, R.; Caselli, U.; Galeazzi, L.; Meier-Ruge, W. Synaptic Structural Dynamics and Aging. Gerontology 1996, 42, 170–180. [Google Scholar] [CrossRef]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Kerlan, V.; Plouin, P.F.; Rotig, A.; Jeunemaitre, X. Functional Consequences of a SDHB Gene Mutation in an Apparently Sporadic Pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 4771–4774. [Google Scholar] [CrossRef]
- Maier-Woelfle, M.; Brandle, M.; Komminoth, P.; Saremaslani, P.; Schmid, S.; Locher, T.; Heitz, P.U.; Krull, I.; Galeazzi, R.L.; Schmid, C.; et al. A Novel Succinate Dehydrogenase Subunit B Gene Mutation, H132P, Causes Familial Malignant Sympathetic Extraadrenal Paragangliomas. J. Clin. Endocrinol. Metab. 2004, 89, 362–367. [Google Scholar] [CrossRef]
- Chinta, S.J.; Andersen, J.K. Dopaminergic Neurons. Int. J. Biochem. Cell Biol. 2005, 37, 942–946. [Google Scholar] [CrossRef]
- Fagergren, P.; Hurd, Y. CART mRNA Expression in Rat Monkey and Human Brain: Relevance to Cocaine Abuse. Physiol. Behav. 2007, 92, 218–225. [Google Scholar] [CrossRef]
- Dallvechia-Adams, S.; Smith, Y.; Kuhar, M.J. CART Peptide-Immunoreactive Projection from the Nucleus Accumbens Targets Substantia Nigra Pars Reticulata Neurons in the Rat. J. Comp. Neurol. 2001, 434, 29–39. [Google Scholar] [CrossRef]
- Dallvechia-Adams, S.; Kuhar, M.J.; Smith, Y. Cocaine- and Amphetamine-Regulated Transcript Peptide Projections in the Ventral Midbrain: Colocalization with Gamma-Aminobutyric Acid, Melanin-Concentrating Hormone, Dynorphin, and Synaptic Interactions with Dopamine Neurons. J. Comp. Neurol. 2002, 448, 360–372. [Google Scholar] [CrossRef]
- Upadhya, M.A.; Nakhate, K.T.; Kokare, D.M.; Singh, U.; Singru, P.S.; Subhedar, N.K. CART Peptide in the Nucleus Accumbens Shell Acts Downstream to Dopamine and Mediates the Reward and Reinforcement Actions of Morphine. Neuropharmacology 2012, 62, 1823–1833. [Google Scholar] [CrossRef]
- Moffett, M.C.; Song, J.; Kuhar, M.J. CART Peptide Inhibits Locomotor Activity Induced by Simultaneous Stimulation of D1 and D2 Receptors, but Not by Stimulation of Individual Dopamine Receptors. Synapse 2011, 65, 1–7. [Google Scholar] [CrossRef]
- Hostetler, C.M.; Kowalczyk, A.S.; Griffin, L.L.; Bales, K.L. CART Peptide Following Social Novelty in the Prairie Vole (Microtus Ochrogaster). Brain Res. 2011, 1414, 32–40. [Google Scholar] [CrossRef]
- Brischoux, F.; Griffond, B.; Fellmann, D.; Risold, P.Y. Early and Transient Ontogenetic Expression of the Cocaine- and Amphetamine-Regulated Transcript Peptide in the Rat Mesencephalon: Correlation with Tyrosine Hydroxylase Expression. J. Neurobiol. 2002, 52, 221–229. [Google Scholar] [CrossRef]
- Goldberg, N.R.; Haack, A.K.; Lim, N.S.; Janson, O.K.; Meshul, C.K. Dopaminergic and Behavioral Correlates of Progressive Lesioning of the Nigrostriatal Pathway with 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine. Neuroscience 2011, 180, 256–271. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mao, P.; Meshul, C.K.; Thuillier, P.; Reddy, P.H. Neurotransmitter CART as a New Therapeutic Candidate for Parkinson’s Disease. Pharmaceuticals 2013, 6, 108-123. https://doi.org/10.3390/ph6010108
Mao P, Meshul CK, Thuillier P, Reddy PH. Neurotransmitter CART as a New Therapeutic Candidate for Parkinson’s Disease. Pharmaceuticals. 2013; 6(1):108-123. https://doi.org/10.3390/ph6010108
Chicago/Turabian StyleMao, Peizhong, Charles K. Meshul, Philippe Thuillier, and P. Hemachandra Reddy. 2013. "Neurotransmitter CART as a New Therapeutic Candidate for Parkinson’s Disease" Pharmaceuticals 6, no. 1: 108-123. https://doi.org/10.3390/ph6010108
APA StyleMao, P., Meshul, C. K., Thuillier, P., & Reddy, P. H. (2013). Neurotransmitter CART as a New Therapeutic Candidate for Parkinson’s Disease. Pharmaceuticals, 6(1), 108-123. https://doi.org/10.3390/ph6010108