Differential Cellular and Molecular Effects of Butyrate and Trichostatin A on Vascular Smooth Muscle Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
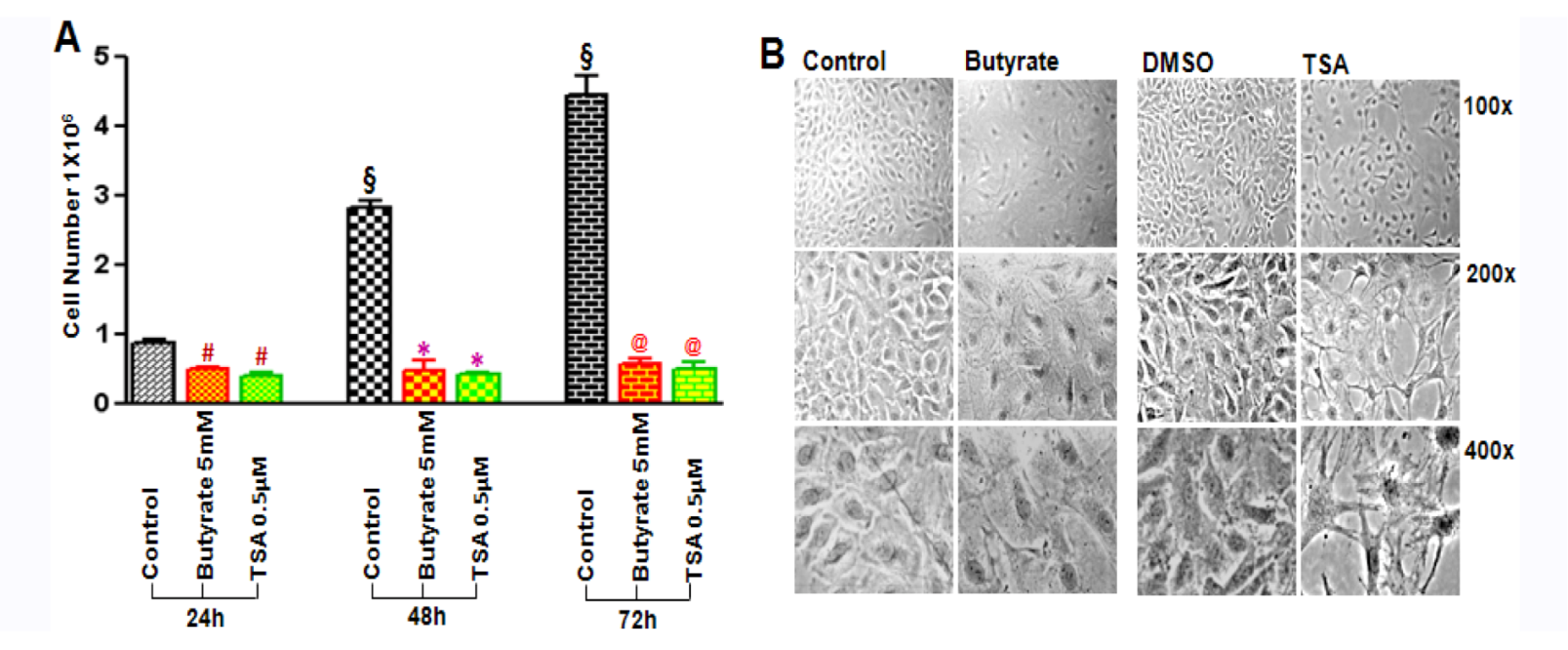
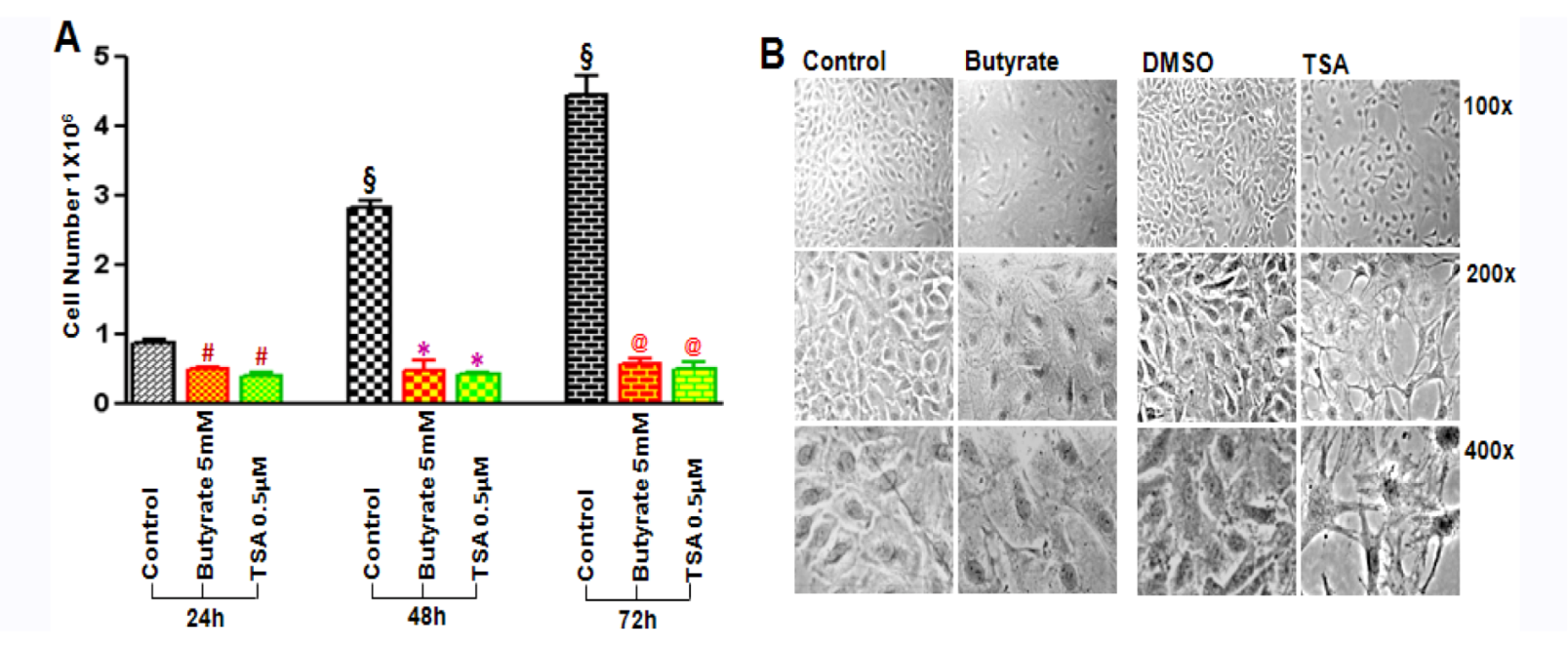
2.1. Cytostatic and Cytotoxic Mode of Inhibition of VSMC Proliferation by Butyrate and TSA
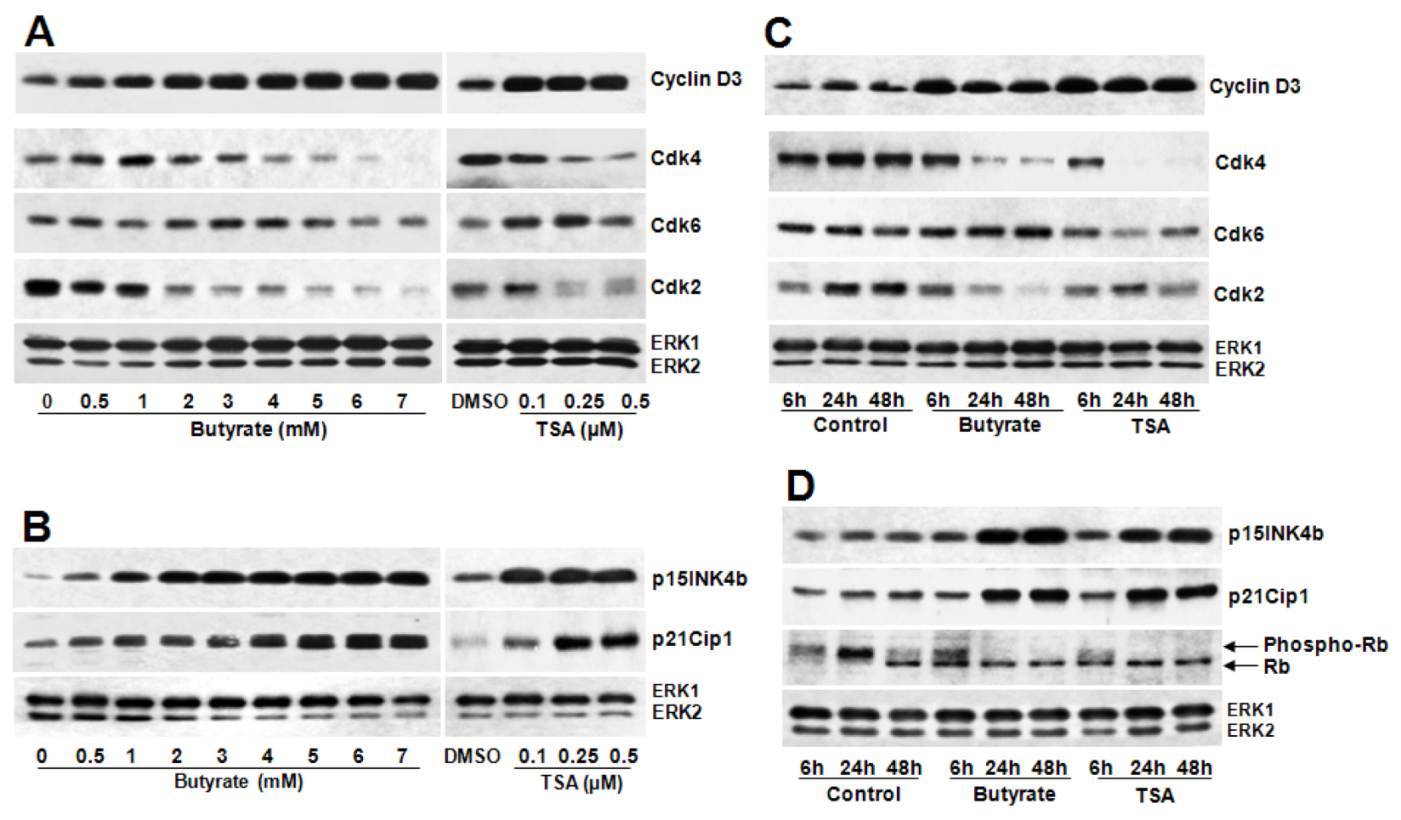
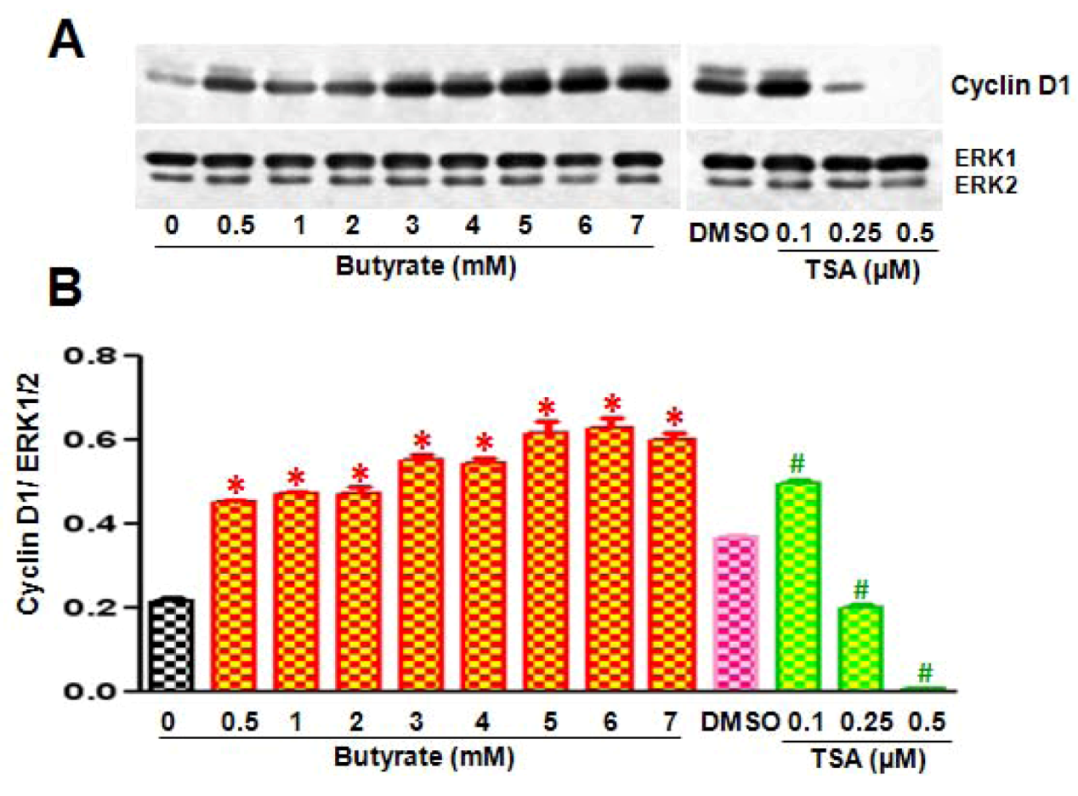
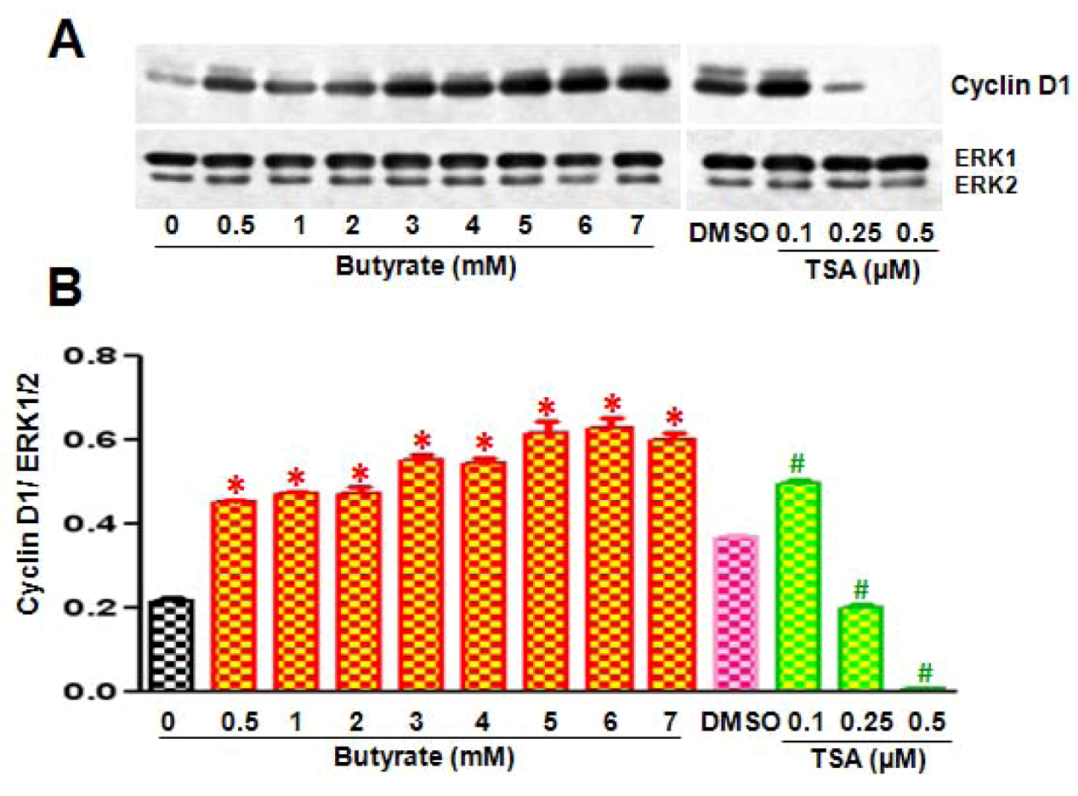
2.2. Disparate Effects of Butyrate and TSA on Cyclin D1 Expression in VSMC
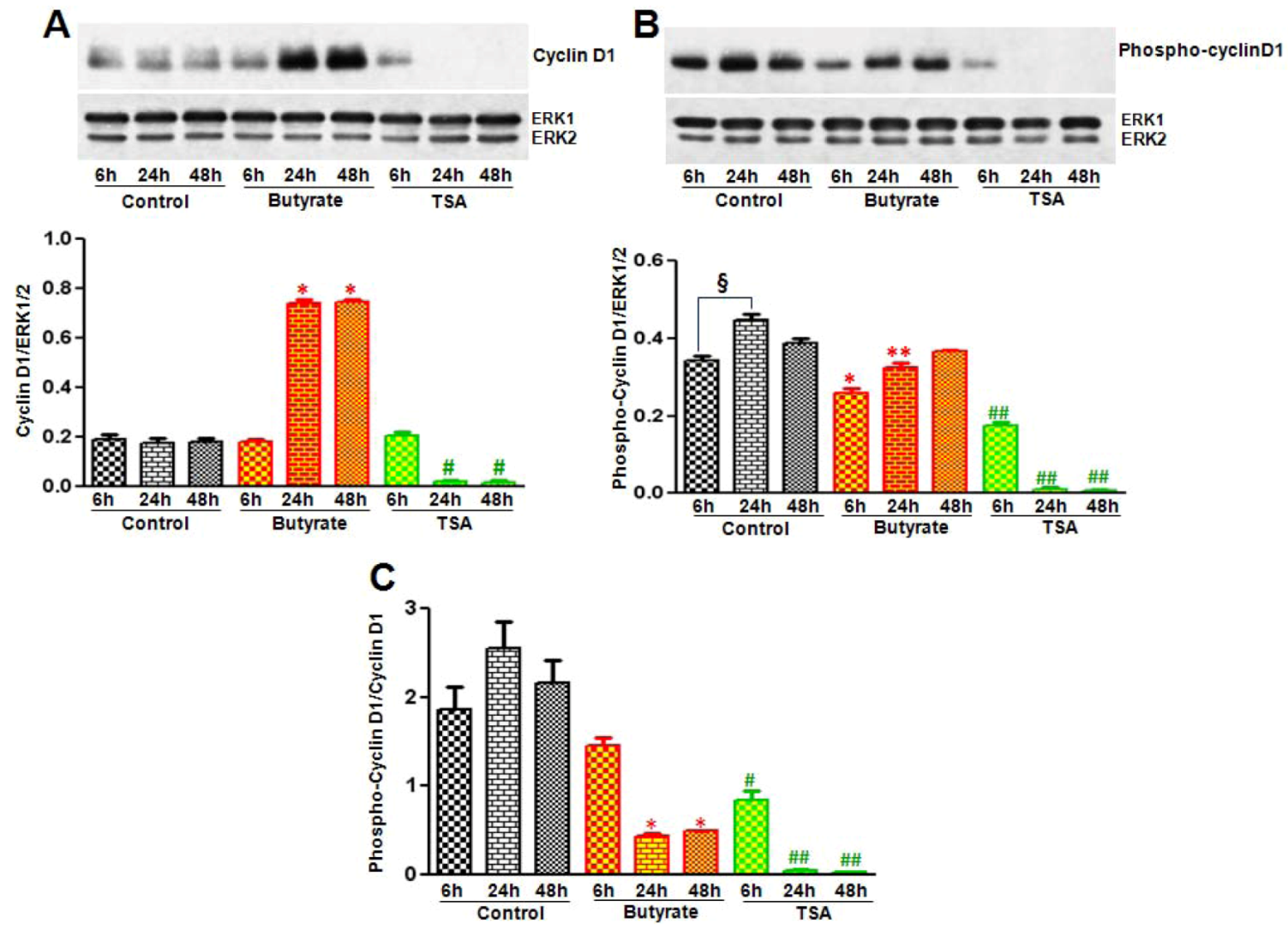
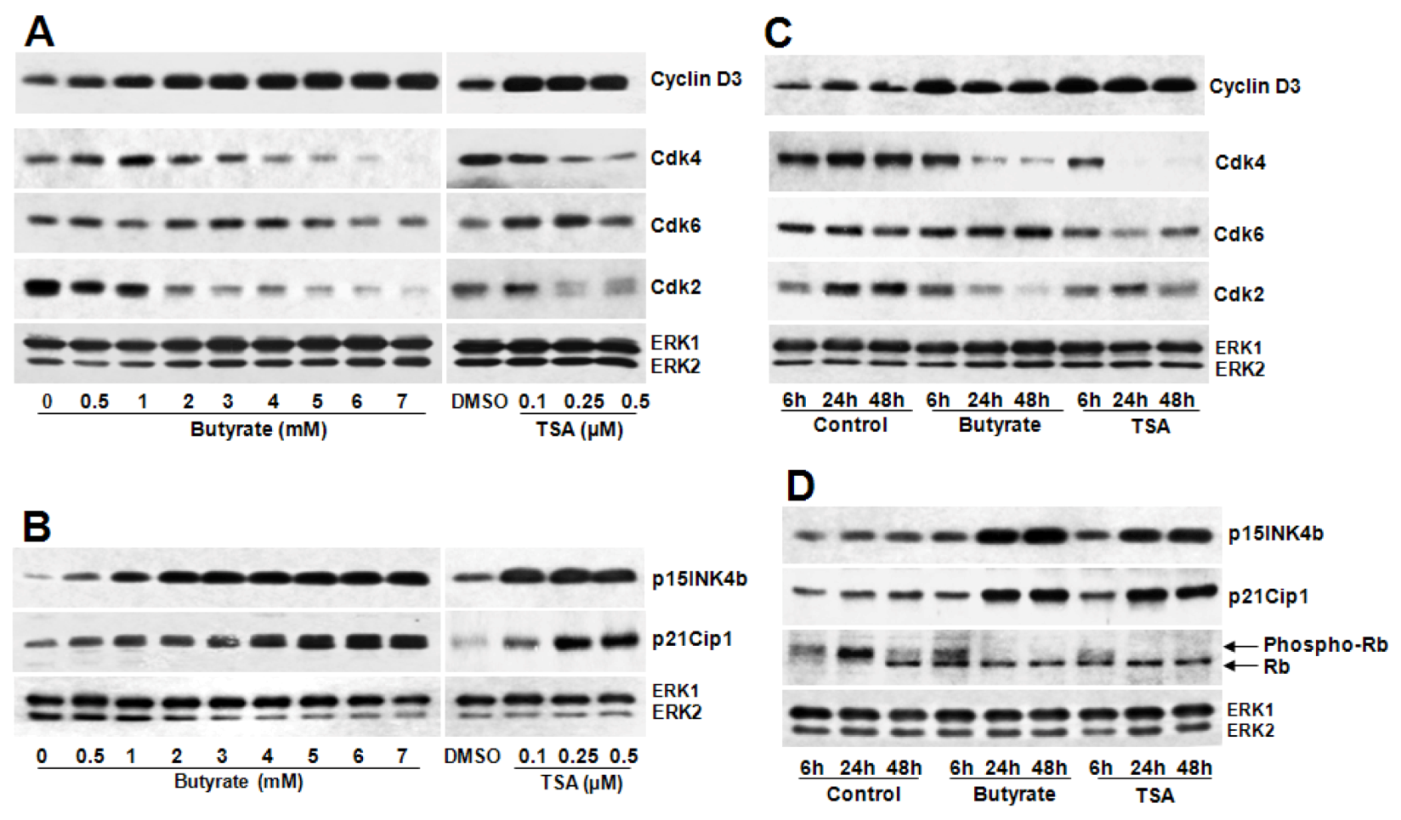
2.2.1. Influence of Butyrate and TSA on Phosphorylation State of Cyclin D1 in VSMC
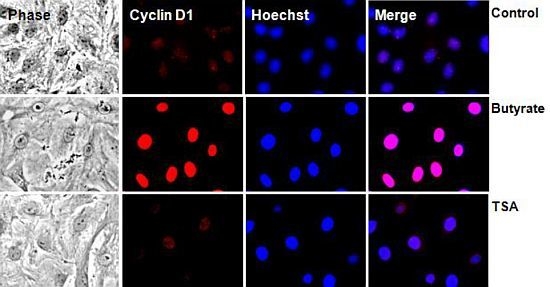
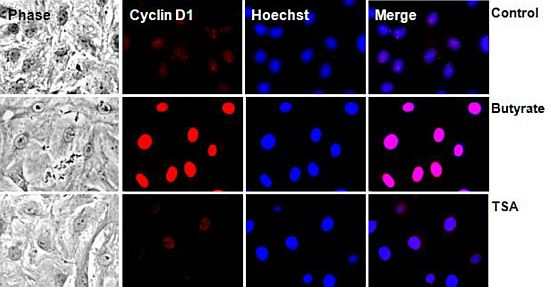
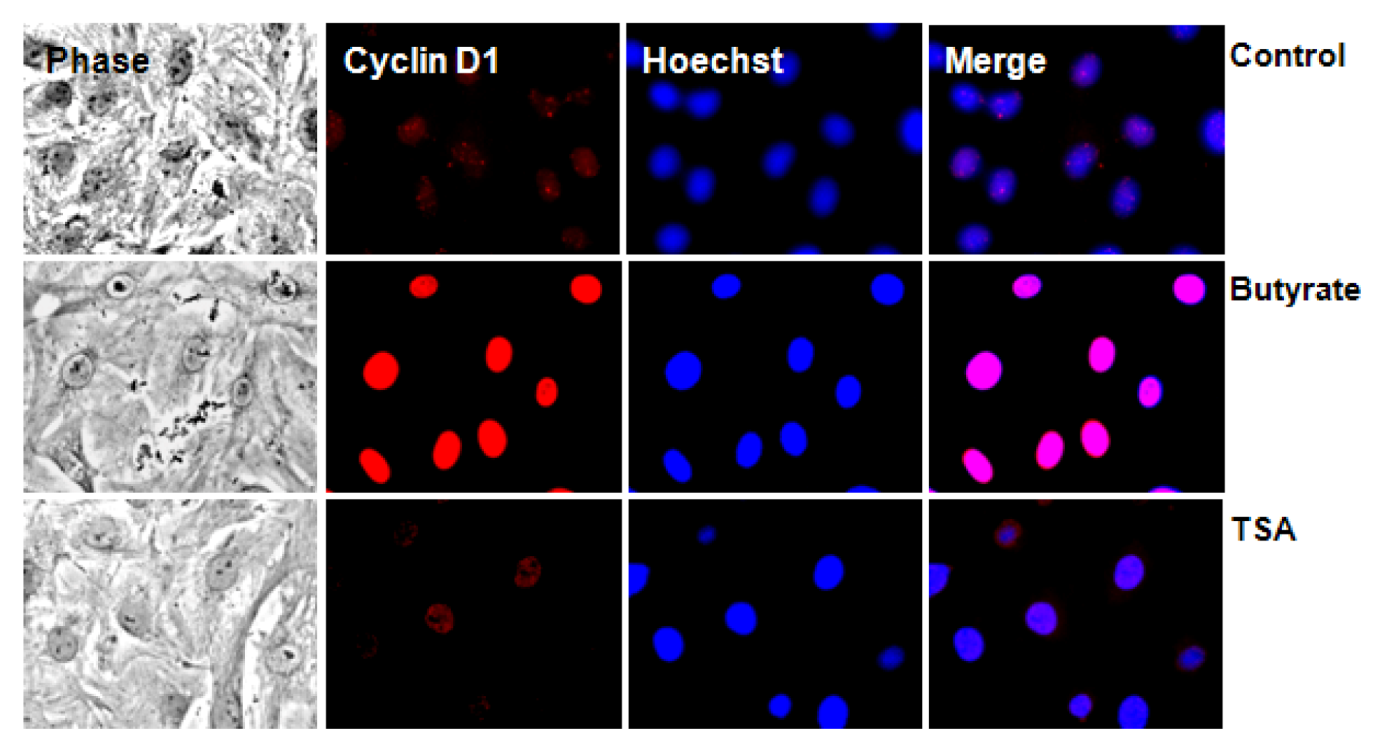
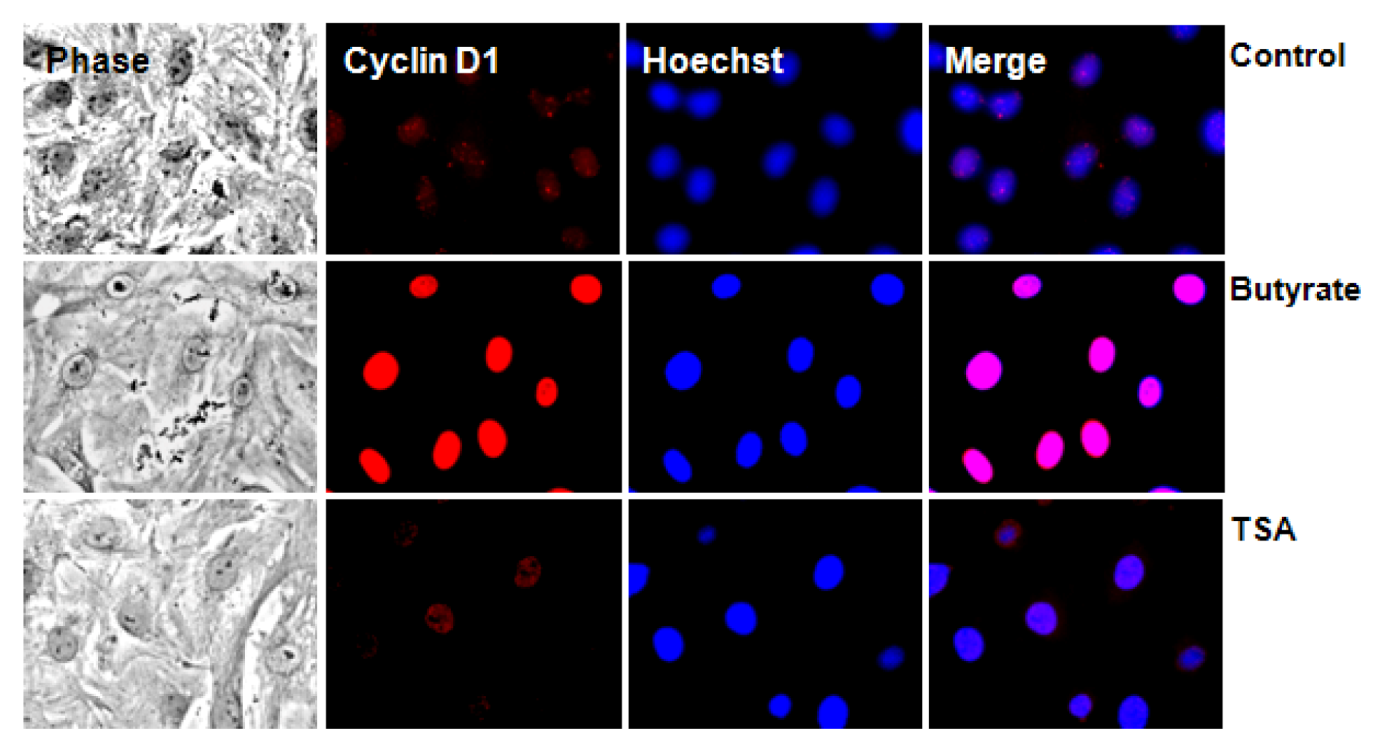
2.2.2. Subcellular Localization of Cyclin D1 Protein in VSMC Treated with HDACi
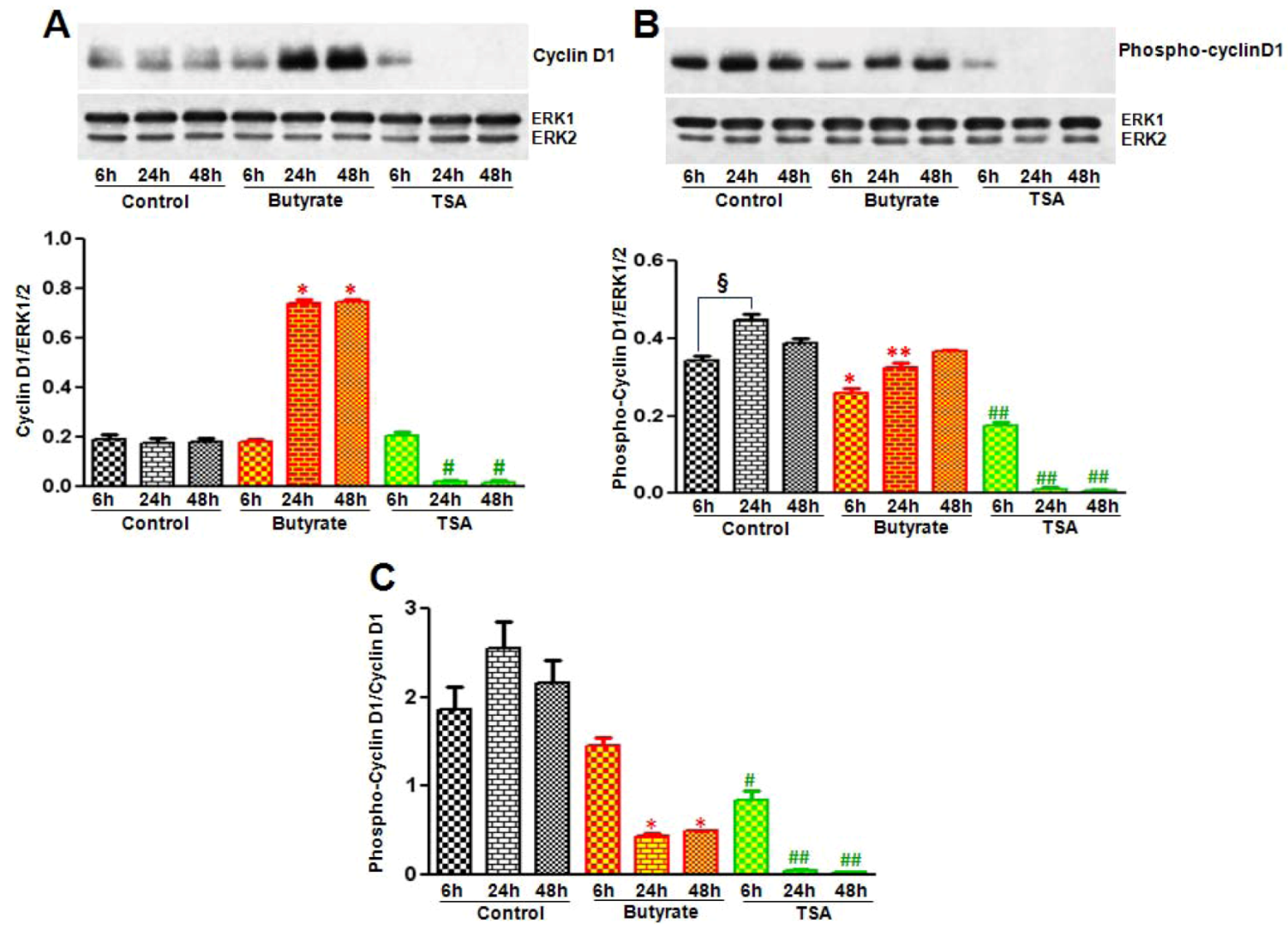
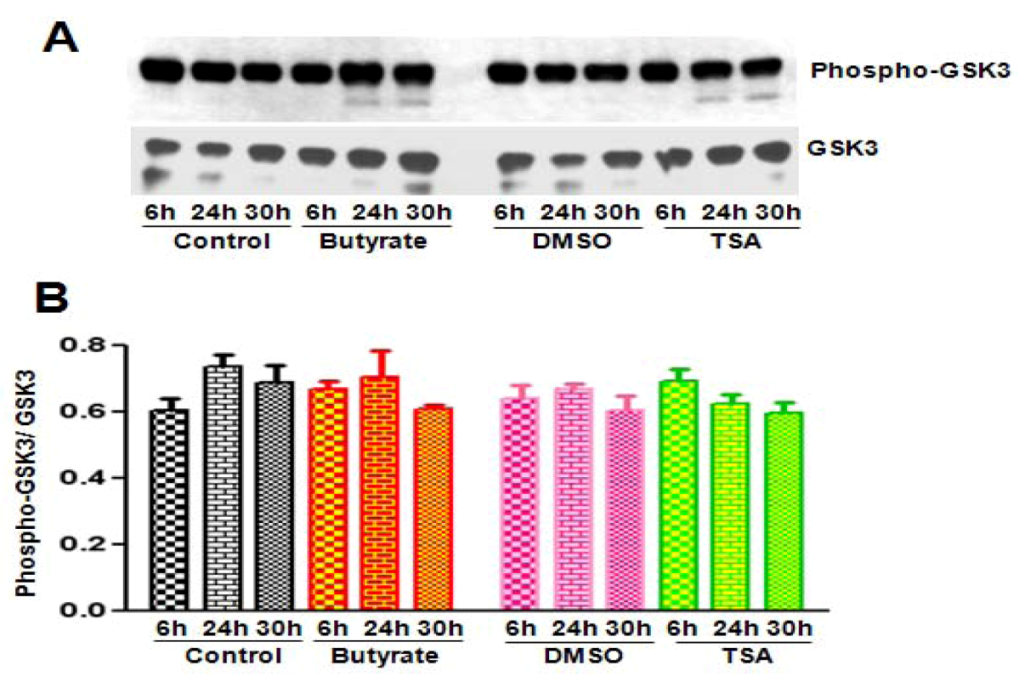
2.2.3. Cyclin D1 Levels Are Not Regulated by GSK3 in VSMC Treated with Butyrate and TSA
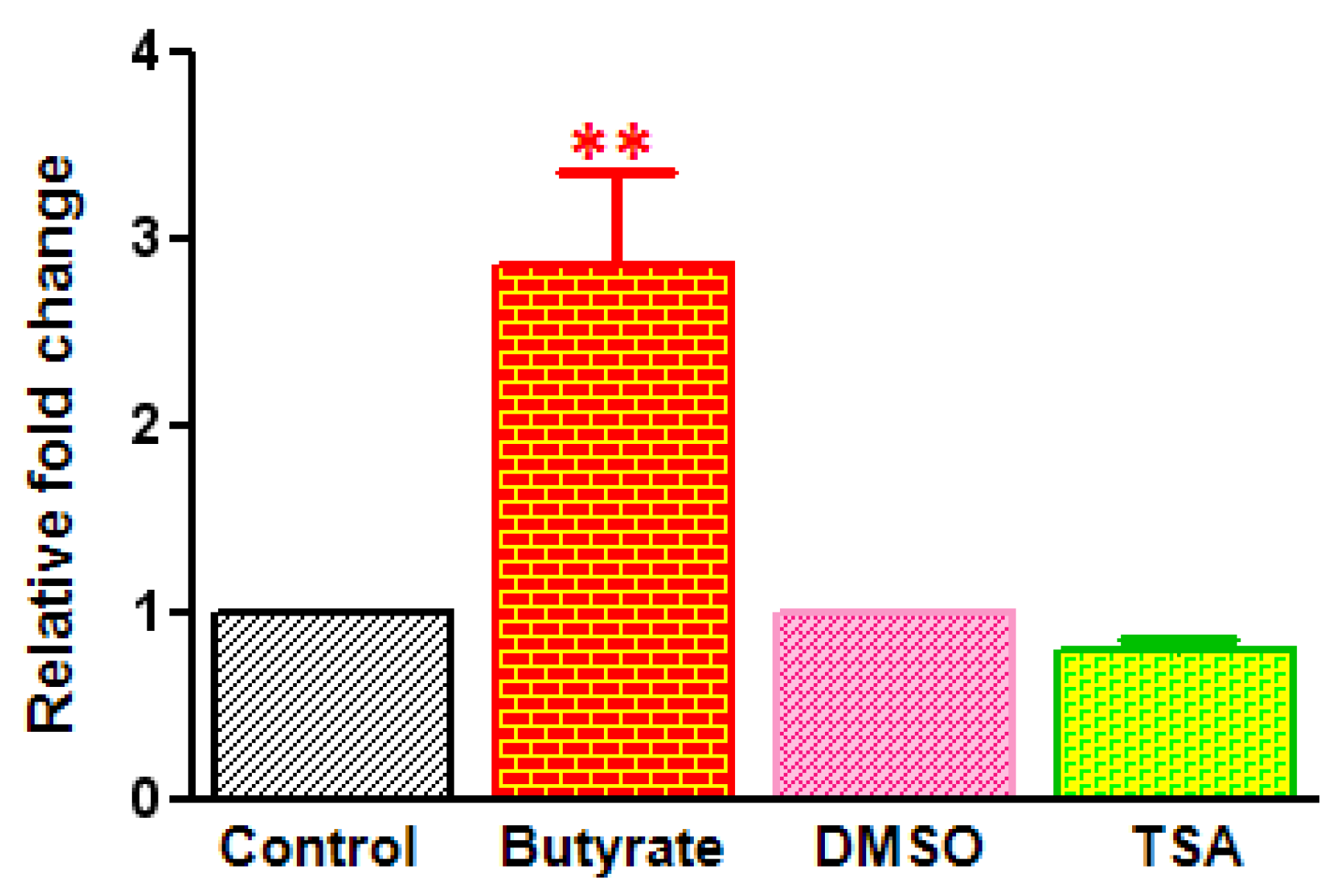
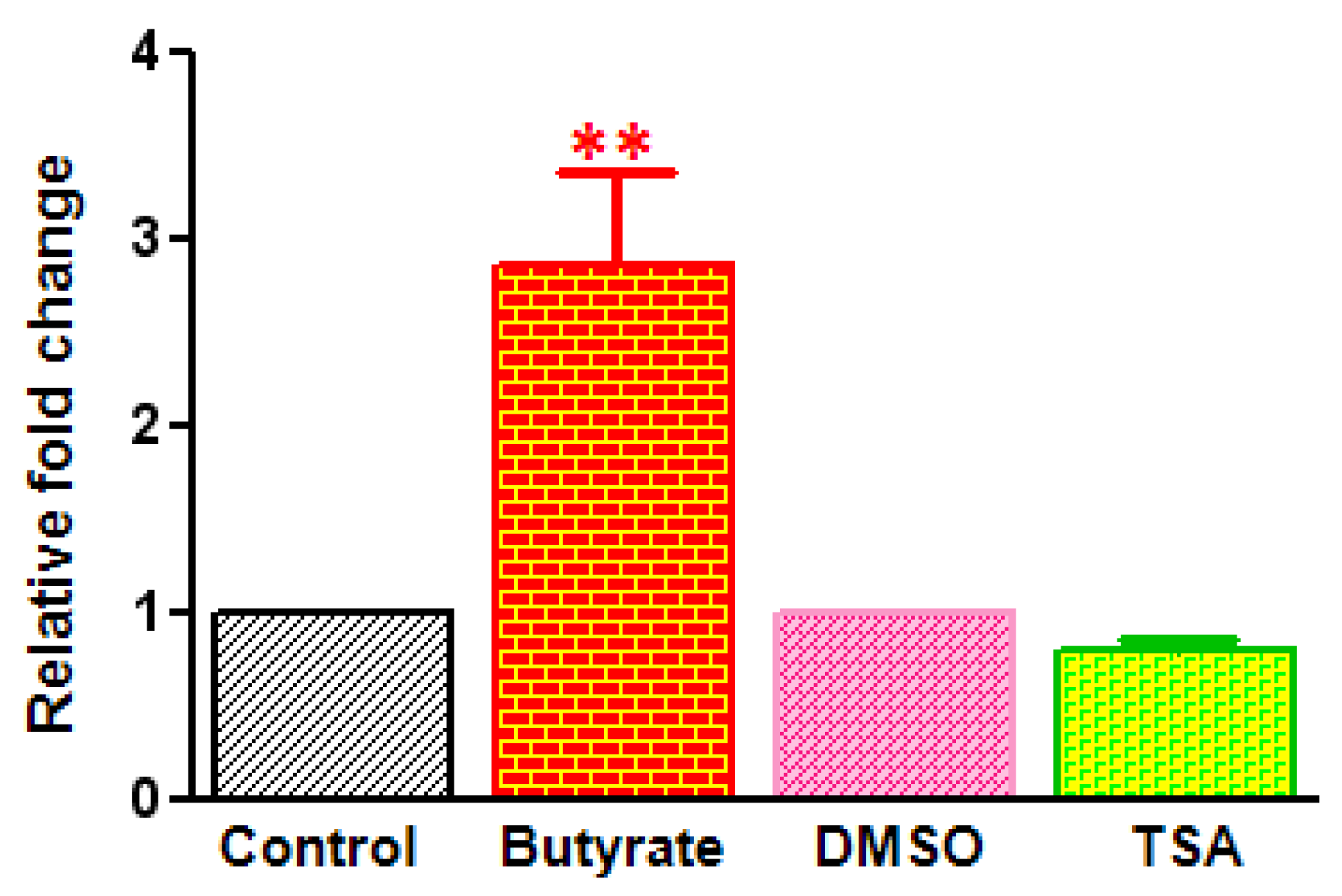
2.3. Differential Effects of HDACi on Cyclin D1 Transcription
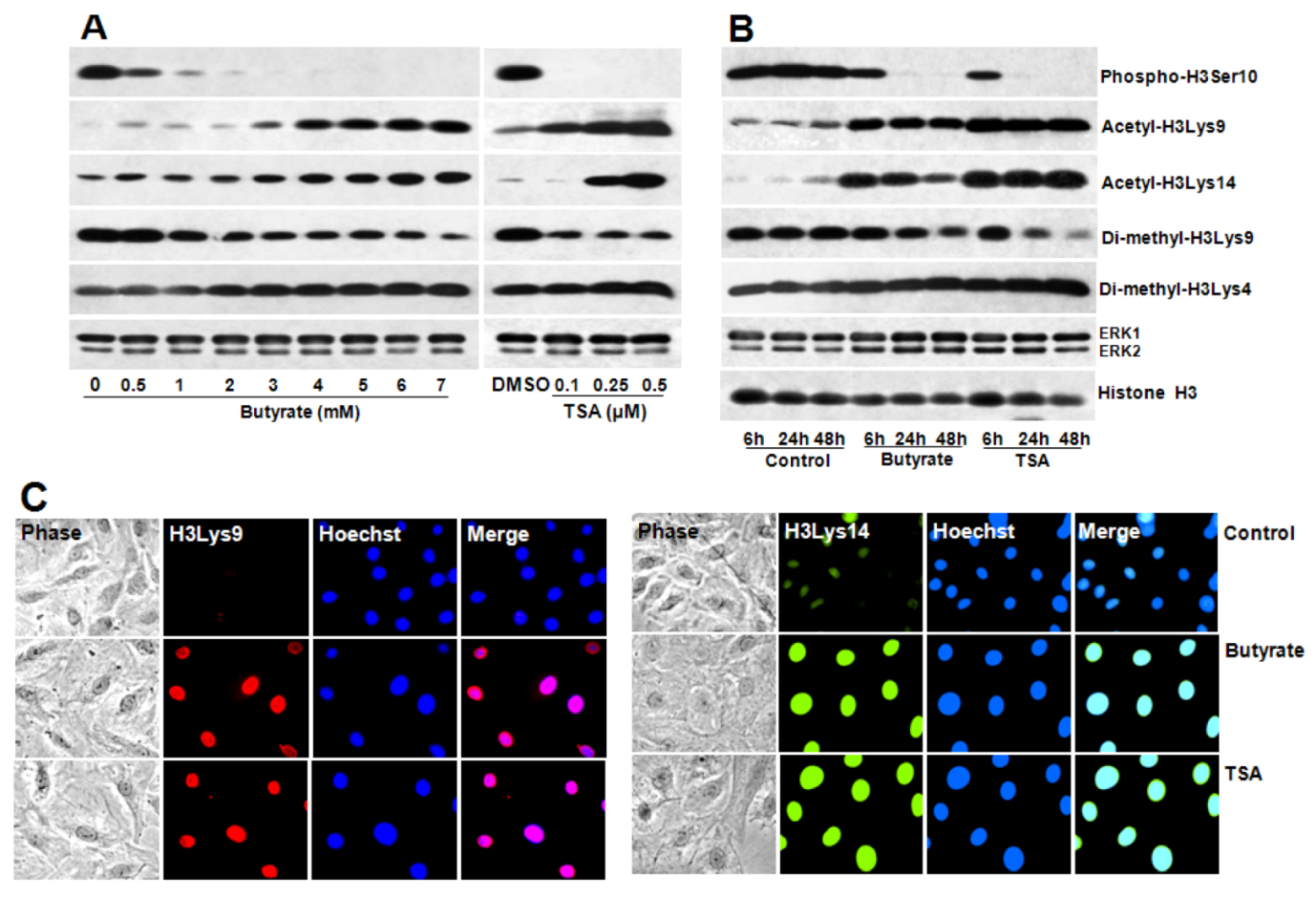
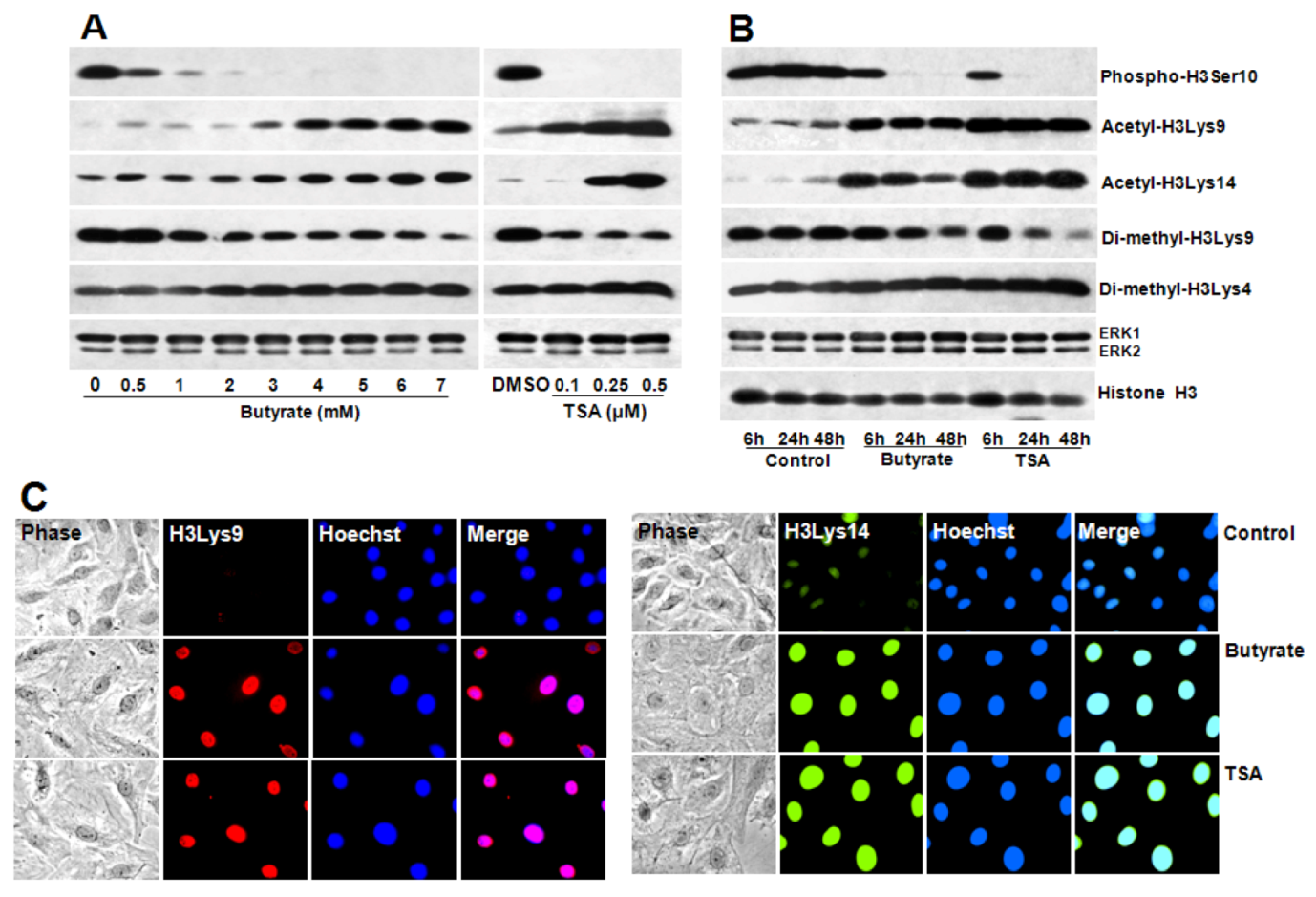
2.4. Portraits of Butyrate and TSA-Induced Site-Specific Posttranslational Modifications of Histone H3 in VSMC
3. Experimental
3.1. Materials
3.2. Cell Culture and Treatments
3.3. Measurement of Cell Proliferation
3.4. Western Analysis
3.5. Immunofluorescence
3.6. Real-Time Quantitative RT-PCR
3.7. Data Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Dzau, V.J.; Braun-Dullaeus, R.C.; Sedding, D.G. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat. Med. 2002, 8, 1249–1256. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Pons, D.; de Vries, F.R.; van den Elsen, P.J.; Heijmans, B.T.; Quax, P.H.; Jukema, J.W. Epigenetic histone acetylation modifiers in vascular remodeling: New targets for therapy in cardiovascular disease. Eur. Heart J. 2009, 30, 266–277. [Google Scholar]
- Ranganna, K.; Yatsu, F.M.; Mathew, O.P. Insights into the pathogenesis and intervention of atherosclerosis. Vasc. Dis. Prev. 2006, 3, 375–390. [Google Scholar]
- Ross, R. Cell biology of atherosclerosis. Annu. Rev. Physiol. 1995, 57, 791–804. [Google Scholar] [CrossRef]
- Holmes, D.R., Jr. State of the art in coronary intervention. Am. J. Cardiol. 2003, 91, 50A–53A. [Google Scholar] [CrossRef]
- Ranganna, K.; Yousefipour, Z.; Yatsu, F.M.; Milton, S.G.; Hayes, B.E. Gene expression profile of butyrate-inhibited vascular smooth muscle cell proliferation. Mol. Cell. Biochem. 2003, 254, 21–36. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Goddard, M.; Shanahan, C.; Shapiro, L.; Bennett, M. Differential gene expression in vascular smooth muscle cells in primary atherosclerosis and in-stent stenosis in humans. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2030–2036. [Google Scholar] [CrossRef]
- Ekström, T.J. Epigenetic control of gene expression. Biochim. Biophys. Acta 2009, 1790, 845–846. [Google Scholar] [CrossRef]
- Turunen, M.P.; Aavik, E.; Yla-Herttuala, S. Epigenetics and atherosclerosis. Biochim. Biophys. Acta 2009, 1790, 886–891. [Google Scholar] [CrossRef]
- Ranganna, K.; Yatsu, F.M.; Mathew, O.P. Emerging epigenetic therapy for vascular proliferative diseases. Available online: http://www.intechopen.com/articles/show/title/emerging-epigenetic-therapy-for-vascular-proliferative-diseases/ (accessed on 22 August 2012).
- Kristeleit, R.; Stimson, L.; Workman, P.; Aherne, W. Histone modification enzymes: Novel targets for cancer drugs. Expert Opin. Emerg. Drugs 2004, 9, 135–154. [Google Scholar] [CrossRef]
- Ranganna, K.; Yatsu, F.M.; Hayes, B.E. Butyrate, a small pleiotropic molecule with multiple cellular and molecular actions: Its role as an anti-atherogenic agent. Recent Res. Dev. Mol. Cell. Biochem. 2005, 2, 123–151. [Google Scholar]
- Berni Canani, R.; di costanzo, M.; Leone, L. The epigenetic effects of butyrate: Potential therapeutic implications for clinical practice. Clin. Epigenetics 2012, 4, 4. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012, 7, 357–367. [Google Scholar] [CrossRef]
- Bhalla, K.N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar] [CrossRef]
- Mathew, O.P.; Ranganna, K.; Yatsu, F.M. Butyrate, an HDAC inhibitor, stimulates interplay between different posttranslational modifications of histone H3 and differently alters G1-specific cell cycle proteins in vascular smooth muscle cells. Biomed. Pharmacother. 2010, 64, 733–740. [Google Scholar] [CrossRef]
- Cao, D.; Wang, Z.; Zhang, C.L.; Oh, J.; Xing, W.; Li, S.; Richardson, J.A.; Wang, D.Z.; Olson, E.N. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol. Cell. Biol. 2005, 25, 364–376. [Google Scholar]
- Sahar, S.; Reddy, M.A.; Wong, C.; Meng, L.; Wang, M.; Natarajan, R. Cooperation of SRC-1 and p300 with NF-kB and CREB in angiotensin II-induced IL-6 expression in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1528–1534. [Google Scholar] [CrossRef]
- Vinh, A.; Gaspari, T.A.; Liu, H.B.; Dousha, L.F.; Widdop, R.E.; Dear, A.E. A novel histone deacetylase inhibitor reduces abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. J. Vasc. Res. 2008, 45, 143–152. [Google Scholar] [CrossRef]
- Waltregny, D.; Glénisson, W.; Tran, S.L.; North, B.J.; Verdin, E.; Colige, A.; Castronovo, V. Histone deacetylase HDAC8 associates with smooth muscle α-actin and is essential for smooth muscle cell contractility. FASEB J. 2005, 19, 966–968. [Google Scholar]
- Xu, X.; Ha, C.H.; Wong, C.; Wang, W.; Hausser, A.; Pfizenmaier, K.; Olson, E.N.; McKinsey, T.A.; Jin, Z.G. Angiotensin II stimulates protein kinase D-dependent histone deacetylase 5 phosphorylation and nuclear export leading to vascular smooth muscle cell hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2355–2362. [Google Scholar] [CrossRef]
- Yan, Z.Q.; Yao, Q.P.; Zhang, M.L.; Qi, Y.X.; Guo, Z.Y.; Shen, B.R.; Jiang, Z.L. Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J. Biomech. 2009, 42, 945–948. [Google Scholar]
- McDonald, O.G.; Wamhoff, B.R.; Hoofnagle, M.H.; Owens, G.K. Control of srf binding to carg boxchromatin regulates smooth muscle gene expression in vivo. J. Clin. Invest. 2006, 116, 36–48. [Google Scholar]
- Okamoto, H.; Fujioka, Y.; Takahashi, A.; Takahashi, T.; Taniguchi, T.; Ishikawa, Y.; Yokoyama, M.; Trichostatin, A. An inhibitor of histone deacetylase, inhibits smooth muscle cell proliferation via induction of p21 (WAF1). J. Atheroscler. Thromb. 2006, 13, 183–191. [Google Scholar] [CrossRef]
- Ranganna, K.; Yatsu, F.M.; Hayes, B.E.; Milton, S.G.; Jayakumar, A. Butyrate inhibits proliferation-induced proliferating cell nuclear antigen expression (PCNA) in rat vascular smooth muscle cells. Mol. Cell. Biochem. 2000, 205, 149–161. [Google Scholar] [CrossRef]
- Yamashita, Y.; Shimada, M.; Harimoto, N.; Rikimaru, T.; Shirabe, K.; Tanaka, S.; Sugimachi, K. Histone deacetylase inhibitor trichostatin A induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. Int. J. Cancer 2003, 103, 572–576. [Google Scholar] [CrossRef]
- Jeon, H.G.; Yoon, C.Y.; Yu, J.H.; Park, M.J.; Lee, J.E.; Jeong, S.J.; Hong, S.K.; Byun, S.S.; Lee, S.E. Induction of caspase mediated apoptosis and down-regulation of nuclear factor-κB and Akt signaling are involved in the synergistic antitumor effect of gemcitabine and the histone deacetylase inhibitor trichostatin A in human bladder cancer cells. J. Urol. 2011, 186, 2084–2093. [Google Scholar]
- Francisco, R.; Pérez-Perarnau, A.; Cortés, C.; Gil, J.; Tauler, A.; Ambrosio, S. Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett. 2012, 318, 42–52. [Google Scholar] [CrossRef]
- Wetzel, M.; Premkumar, D.R.; Arnold, B.; Pollack, I.F. Effect of trichostatin A, a histone deacetylase inhibitor, on glioma proliferation in vitro by inducing cell cycle arrest and apoptosis. J. Neurosurg. 2005, 103, 549–556. [Google Scholar]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. GenesDev. 1999, 13, 1501–1512. [Google Scholar]
- Lundberg, A.S.; Weinberg, R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 1998, 18, 753–7561. [Google Scholar]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef]
- Lallemand, F.; Courilleau, D.; Sabbah, M.; Redeuilh, G.; Mester, J. Direct inhibition of the expression of cyclin D1 gene by sodium butyrate. Biochem. Biophys. Res. Commun. 1996, 229, 163–169. [Google Scholar] [CrossRef]
- Siavoshian, S.; Segain, J.P.; Kornprobst, M.; Bonnet, C.; Cherbut, C.; Galmiche, J.P.; Blottière, H.M. Butyrate and trichostatin A effects on the proliferation/differentiation of human intestinal epithelial cells: Induction of cyclin D3 and p21 expression. Gut 2000, 46, 507–514. [Google Scholar] [CrossRef]
- Davis, T.; Kennedy, C.; Chiew, Y.E.; Clarke, C.L.; DeFazio, A. Histone deacetylase inhibitors decrease proliferation and modulate cell cycle gene expression in normal mammary epithelial cells. Clin. CancerRes. 2000, 6, 4334–4342. [Google Scholar]
- Vaziri, C.; Stice, L.; Faller, D.V. Butyrate-induced G1 arrest results from p21-independent disruption of retinoblastoma protein-mediated signals. CellGrowthDiffer. 1998, 9, 465–474. [Google Scholar]
- Li, C.; Liu, W.; Meng, F.; Huang, W.; Zhou, J.; Sun, H.; Feng, Y. Effect and comparison of sodium butyrate and trichostatin A on the proliferation/differentiation of K562. J. Huazhong Univ. Sci. Technol. Med. Sci. 2003, 23, 249–253. [Google Scholar] [CrossRef]
- Alao, J.P.; Stavropoulou, A.V.; Lam, E.W.-F.; Coombes, R.C.; Vigushin, D.M. Histone deacetylase inhibitor, Trichostatin A induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast cancer cells. BMC Mol. Cancer 2006, 5, 8. [Google Scholar]
- Jeon, H.G.; Yoon, C.Y.; Yu, J.H.; Park, M.J.; Lee, J.E.; Jeong, S.J.; Hong, S.K.; Byun, S.S.; Lee, S.E. Induction of caspase mediated apoptosis and down-regulation of nuclear factor-κB and Akt signaling are involved in the synergistic antitumor effect of gemcitabine and the histone deacetylase inhibitor trichostatin A in human bladder cancer cells. J. Urol. 2011, 186, 2084–2093. [Google Scholar]
- Bajbouj, K.; Mawrin, C.; Hartig, R.; Schulze-Luehrmann, J.; Wilisch-Neumann, A.; Roessner, A.; Schneider-Stock, R. P53-dependent antiproliferative and pro-apoptotic effects of trichostatin A (TSA) in glioblastoma cells. J. Neurooncol. 2012, 107, 503–516. [Google Scholar] [CrossRef]
- Feng, P.; Ge, L.; Akyhani, N.; Liau, G. Sodium butyrate is a potent modulator of smooth muscle cell proliferation and gene expression. Cell Prolif. 1996, 29, 231–241. [Google Scholar]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar]
- Takahashi-Yanaga, F.; Sasaguri, T. GSK-3β regulates cyclin D1 expression: A new target for chemotherapy. Cell. Signal. 2008, 20, 581–589. [Google Scholar] [CrossRef]
- Alt, J.R.; Cleveland, J.L.; Hannink, M.; Diehl, J.A. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000, 14, 3102–3114. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from cell Biology and animal models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar]
- Yang, K.; Guo, Y.; Stacey, W.C.; Harwalkar, J.; Fretthold, J.; Hitomi, M.; Stacey, D.W. Glycogen synthase kinase 3 has a limited role in cell cycle regulation of cyclin D1 levels. BMC Cell Biol. 2006, 7, 33. [Google Scholar] [CrossRef]
- Zou, Y.; Ewton, D.Z.; Deng, X.; Mercer, S.E.; Friedman, E. Mirk/dyrk1B kinase destabilizes cyclin D1 by phosphorylation at threonine 288. J. Biol. Chem. 2004, 279, 27790–27798. [Google Scholar]
- Ito, T. Role of histone modifications in chromatin dynamics. J. Biochem. 2007, 141, 609–614. [Google Scholar] [CrossRef]
- McManus, K.J.; Hendzel, M.J. The relationship between histone H3 phosphorylation and acetylation throughout the mammalian cell cycle. Biochem. Cell Biol. 2006, 84, 640–657. [Google Scholar] [CrossRef]
- Edmondson, D.G.; Davie, J.K.; Zhou, J.; Mirnikjoo, B.; Tatchell, K.; Dent, S.Y. Site-specific loss of acetylation upon phosphorylation of histone H3. J. Biol. Chem. 2002, 277, 29496–29502. [Google Scholar]
- Eberlin, A.; Grauffel, C.; Oulad-Abdelghani, M.; Robert, F.; Torres-Padilla, M.E.; Lambrot, R.; Spehner, D.; Ponce-Perez, L.; Würtz, J.M.; Stote, R.H.; et al. Histone H3 tails containing dimethylated lysine and adjacent phosphorylated serine modifications adopt a specific conformation during mitosis and meiosis. Mol. Cell Biol. 2008, 28, 1739–1754. [Google Scholar]
- Ducasse, M.; Brown, M.A. Epigenetic aberrations and cancer. Mol. Cancer 2006, 5, 60. [Google Scholar] [CrossRef]
- Ramos, K.; Cox, L.R. Primary cultures of rat aortic endothelial and smooth muscle cells: I. An in vitro model to study xenobiotic-induced vascular cytotoxicity. In Vitro Cell. Dev. Biol. 1987, 23, 288–296. [Google Scholar] [CrossRef]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar]
- Ranganna, K.; Yatsu, F.M. Inhibition of platelet derived growth factor BB-induced expression of glyceraldehyde-3-phosphate dehydrogenase by sodium butyrate in rat vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3420–3427. [Google Scholar] [CrossRef]
- Fu, M.; Wang, C.; Li, Z.; Sakamaki, T.; Pestell, R.G. Cyclin D1: Normal and abnormal functions. Endocrinology 2004, 145, 5439–5447. [Google Scholar]
- Marampon, F.; Casimiro, M.C.; Fu, M.; Powell, M.J.; Popov, V.M.; Lindsay, J.; Zani, B.M.; Ciccarelli, C.; Watanabe, G.; Lee, R.J.; et al. Nerve growth factor regulation of cyclin D1 in PC12 cells through a p21RAS extracellular signal-regulated kinase pathway requires cooperative interactions between Sp1 and nuclear factor-kB. Mol. Biol. Cell 2008, 19, 2566–2578. [Google Scholar] [CrossRef]
- Fu, M.; Wang, C.; Rao, M.; Wu, X.; Bouras, T.; Zhang, X.; Li, Z.; Jiao, X.; Yang, J.; Li, A.; et al. Cyclin D1 represses p300 transactivation through a cyclin-dependent kinase-independent mechanism. J. Biol. Chem. 2005, 280, 29728–29742. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Milton, S.G.; Mathew, O.P.; Yatsu, F.M.; Ranganna, K. Differential Cellular and Molecular Effects of Butyrate and Trichostatin A on Vascular Smooth Muscle Cells. Pharmaceuticals 2012, 5, 925-943. https://doi.org/10.3390/ph5090925
Milton SG, Mathew OP, Yatsu FM, Ranganna K. Differential Cellular and Molecular Effects of Butyrate and Trichostatin A on Vascular Smooth Muscle Cells. Pharmaceuticals. 2012; 5(9):925-943. https://doi.org/10.3390/ph5090925
Chicago/Turabian StyleMilton, Shirlette G., Omana P. Mathew, Frank M. Yatsu, and Kasturi Ranganna. 2012. "Differential Cellular and Molecular Effects of Butyrate and Trichostatin A on Vascular Smooth Muscle Cells" Pharmaceuticals 5, no. 9: 925-943. https://doi.org/10.3390/ph5090925