The Influence of Estrogens on the Biological and Therapeutic Actions of Growth Hormone in the Liver

Abstract
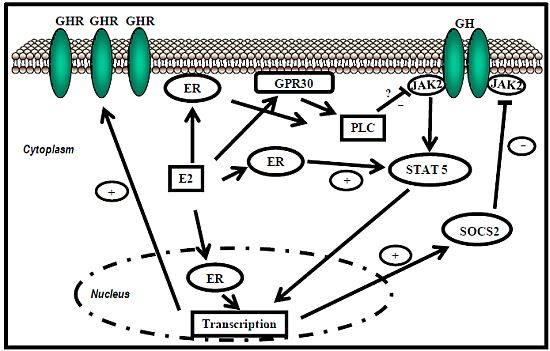
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
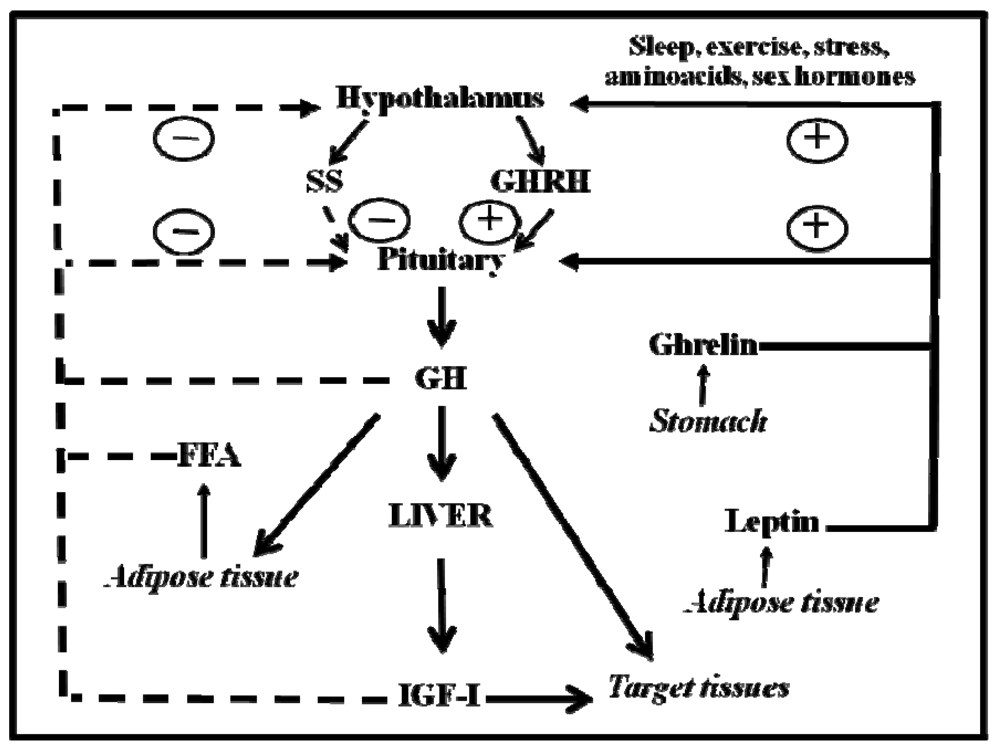
2. Physiological Basis of Pituitary GH Secretion
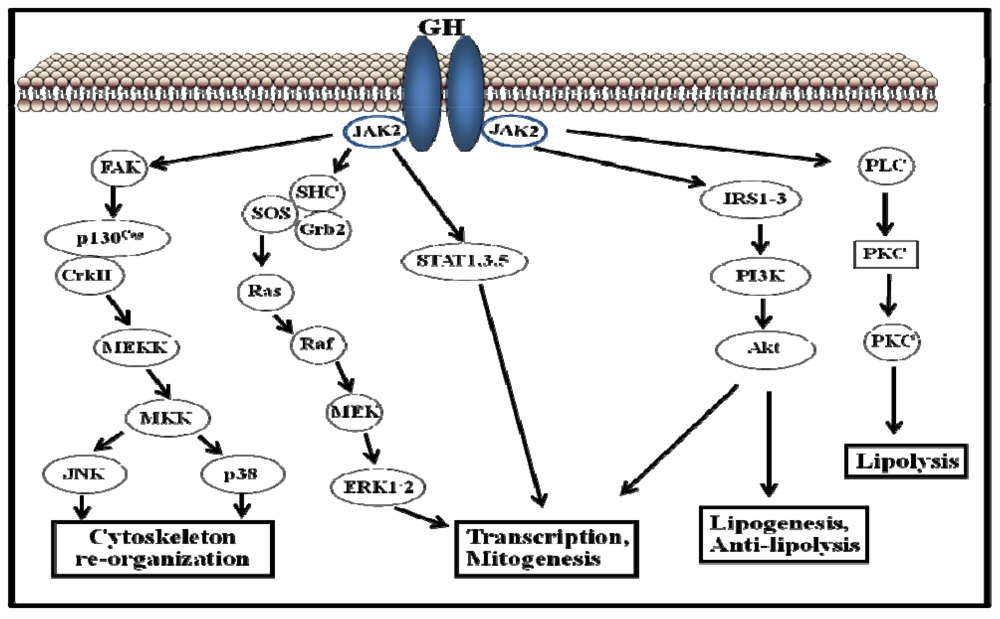
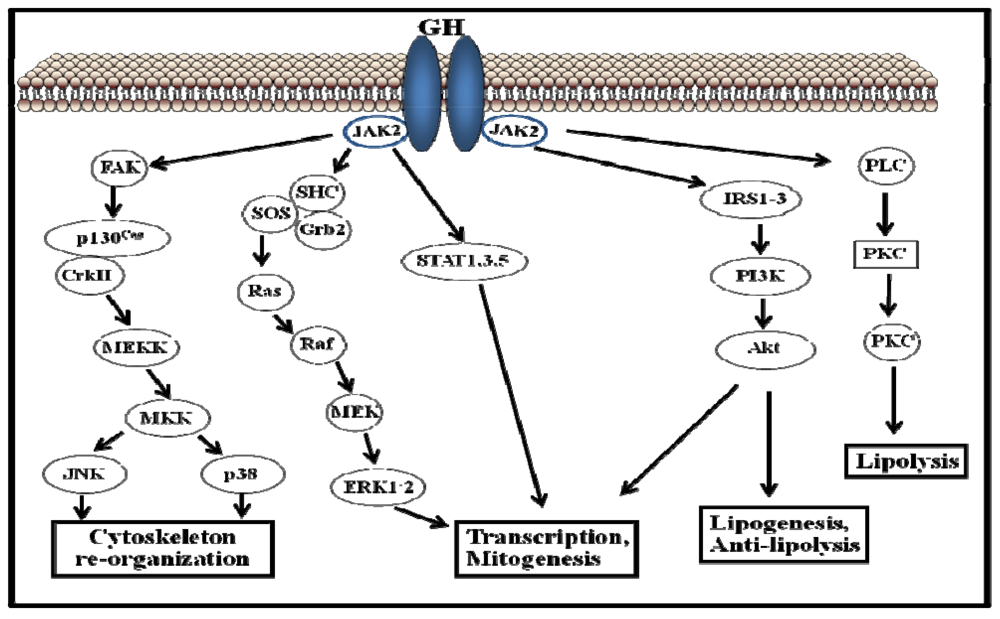
3. The Cellular Regulation of GH Signaling
4. STAT5 Plays a Relevant Role in GH-Dependent Regulation of Body Growth and Composition, Liver Metabolism and Gender-Dependent Dimorphism
4.1. Body Growth
4.2. Metabolism
4.3. Insulin Sensitivity
4.4. Gender Dimorphism in the Liver
5. The Liver Is a Target for Estrogen
5.1. Estrogen Receptor Signaling
 ER-subtype-selective agonists have been developed; PPT and DPN are ERα- and ERβ-selective agonists, respectively. In addition, the ERs bind a wide range of synthetic compounds with strikingly diverse structures, including selective estrogen receptor modulators (SERMs) (e.g., raloxifene). The SERMs are synthetic ER ligands that display tissue-selective pharmacology; as anti-estrogens (antagonists), they oppose the action of estrogens in certain tissues while mimicking the action of endogenous estrogens (agonists) in other tissues [70]. Environmental contaminants (e.g., polycyclic aromatic hydrocarbons, phthalates, pesticides), a class of estrogens termed xenoestrogens, and phytoestrogens also have estrogenic actions. Although their affinity for ERs is mostly 100 to 10,000 times lower than that of E2 [71], it is not questionable whether xeno- and phytoestrogens are biologically relevant in humans and farm animals. The tissue-selective expression of ERs can also determine estrogen physiology. ERα is mainly expressed in reproductive tissues, the kidney, bones, white adipose tissue, and the liver, while ERβ is expressed in the ovary, the prostate, the lungs, the gastrointestinal tract, the bladder, hematopoietic cells, and the central nervous system. Therefore, specific therapeutic actions of estrogens on tissues (e.g., the liver) may be obtained through selective ERα agonists (e.g., PPT) [72].
ER-subtype-selective agonists have been developed; PPT and DPN are ERα- and ERβ-selective agonists, respectively. In addition, the ERs bind a wide range of synthetic compounds with strikingly diverse structures, including selective estrogen receptor modulators (SERMs) (e.g., raloxifene). The SERMs are synthetic ER ligands that display tissue-selective pharmacology; as anti-estrogens (antagonists), they oppose the action of estrogens in certain tissues while mimicking the action of endogenous estrogens (agonists) in other tissues [70]. Environmental contaminants (e.g., polycyclic aromatic hydrocarbons, phthalates, pesticides), a class of estrogens termed xenoestrogens, and phytoestrogens also have estrogenic actions. Although their affinity for ERs is mostly 100 to 10,000 times lower than that of E2 [71], it is not questionable whether xeno- and phytoestrogens are biologically relevant in humans and farm animals. The tissue-selective expression of ERs can also determine estrogen physiology. ERα is mainly expressed in reproductive tissues, the kidney, bones, white adipose tissue, and the liver, while ERβ is expressed in the ovary, the prostate, the lungs, the gastrointestinal tract, the bladder, hematopoietic cells, and the central nervous system. Therefore, specific therapeutic actions of estrogens on tissues (e.g., the liver) may be obtained through selective ERα agonists (e.g., PPT) [72].
5.2. E2 Modulates the GH Promoting of Skeletal Growth
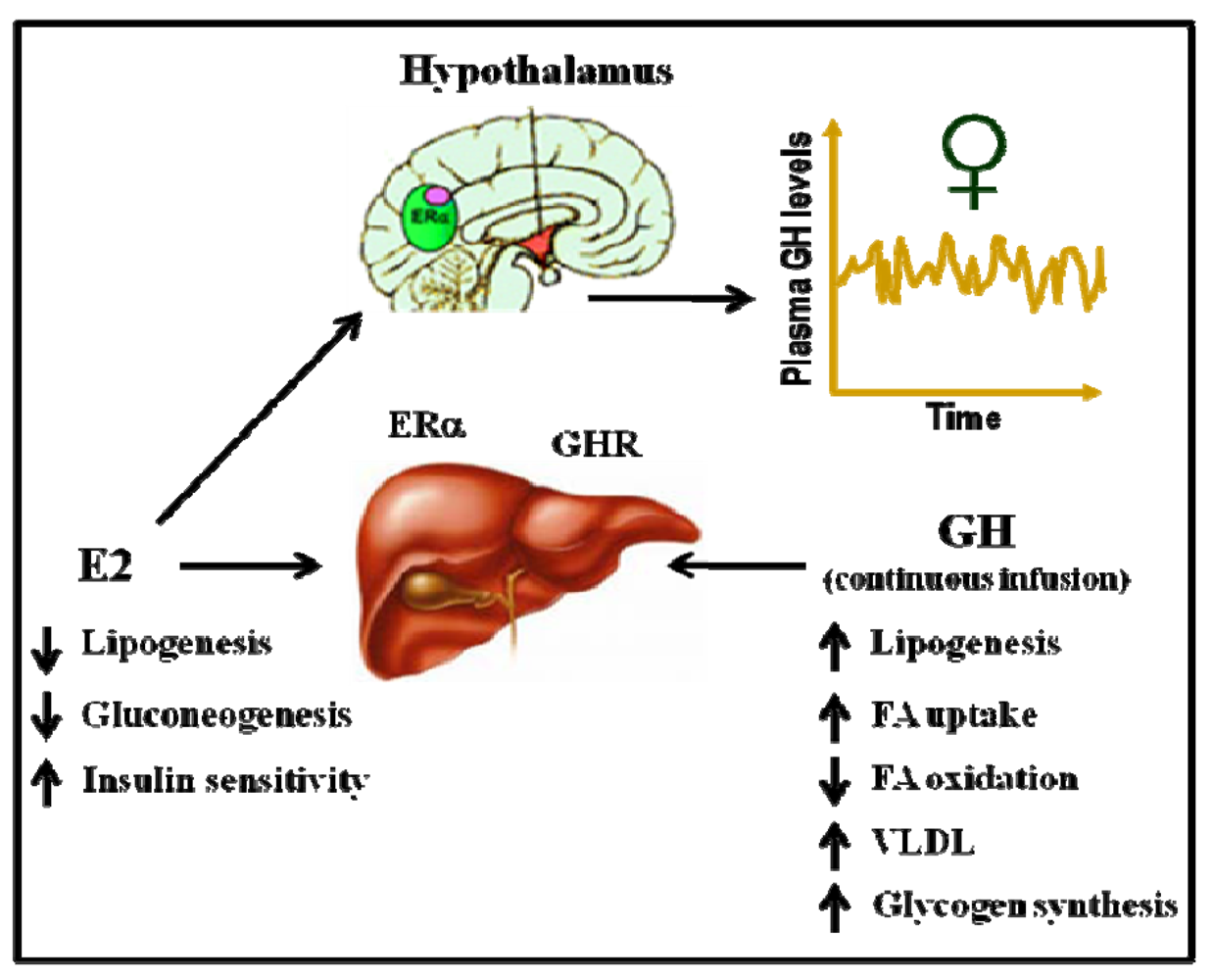
5.3. Gender Dimorphism in the Liver Is Regulated by the Pattern of GH Secretion and Sex Steroids
5.4. E2 Regulates Lipid Metabolism and Insulin Sensitivity: Potential Crosstalk with GH
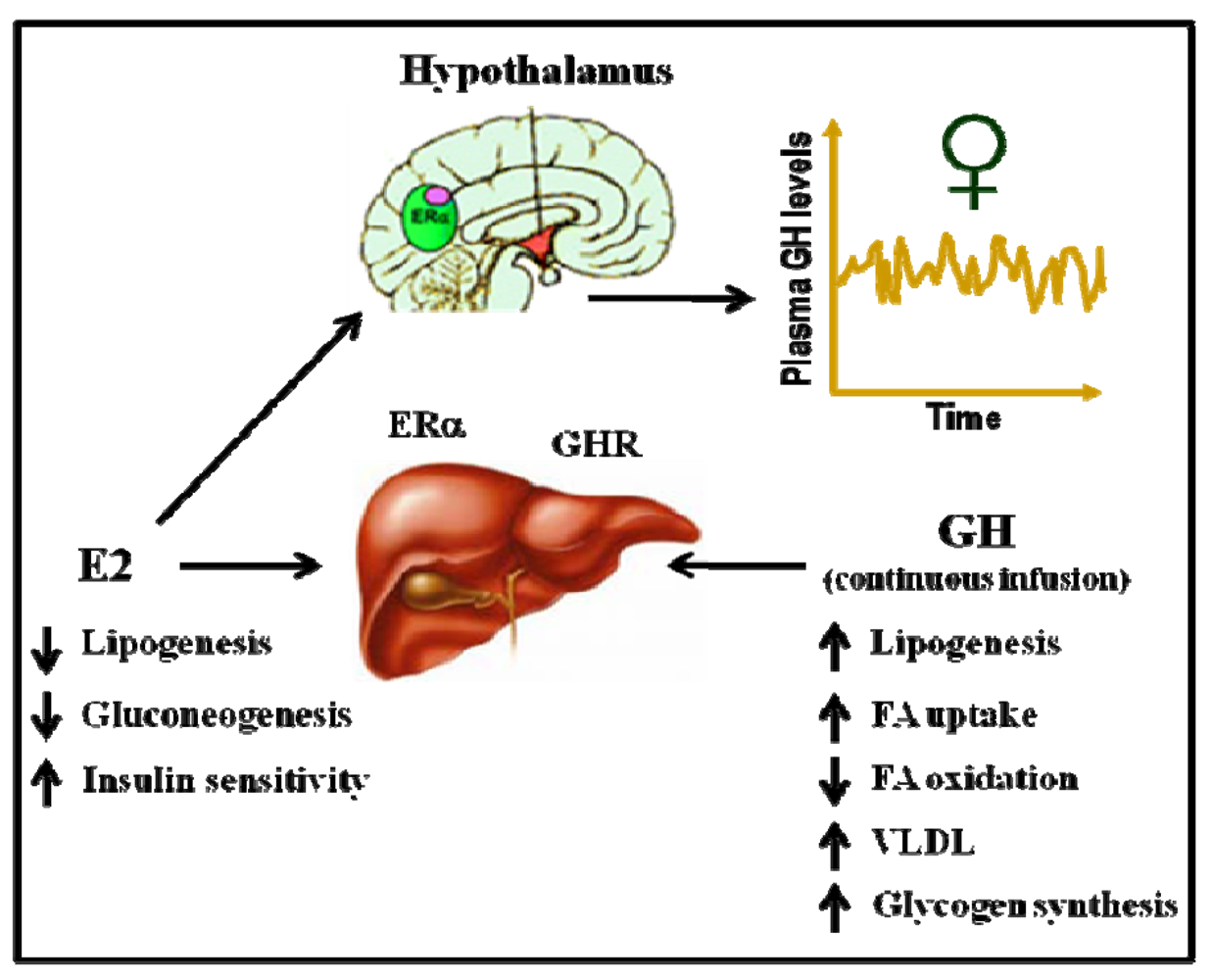
6. The Modulation of GH Actions by Estrogens Is Clinically Relevant
7. Conclusions
Conflict of Interest
Acknowledgements
References
- Butler, A.A.; Le Roith, D. Control of growth by the somatropic axis: Growth hormone and the insulin-like growth factors have related and independent roles. Annu. Rev. Physiol. 2001, 63, 141–164. [Google Scholar] [CrossRef]
- Mode, A.; Gustafsson, J.A. Sex and the liver—A journey through five decades. Drug Metab. Rev. 2006, 38, 197–207. [Google Scholar] [CrossRef]
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor 1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef]
- Lichanska, A.M.; Waters, M.J. How growth hormone controls growth, obesity and sexual dimorphism. Trends Genet. 2008, 24, 41–47. [Google Scholar] [CrossRef]
- Vijayakumar, A.; Novosyadlyy, R.; Wu, Y.; Yakar, S.; LeRoith, D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm. IGF Res. 2010, 20, 1–7. [Google Scholar] [CrossRef]
- Baik, M.; Yu, J.H.; Hennighausen, L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann. NY Acad. Sci. 2011, 1229, 29–37. [Google Scholar] [CrossRef]
- List, E.O.; Sackmann-Sala, L.; Berryman, D.E.; Funk, K.; Kelder, B.; Gosney, E.S.; Okada, S.; Ding, J.; Cruz-Topete, D.; Kopchick, J.J. Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR−/−) mouse. Endocr. Rev. 2011, 32, 356–386. [Google Scholar] [CrossRef]
- Waxman, D.J.; Holloway, M.G. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol. Pharmacol. 2009, 76, 215–228. [Google Scholar] [CrossRef]
- Lichanska, A.M.; Waters, M.J. New insights into growth hormone receptor function and clinical implications. Horm. Res. 2008, 69, 138–145. [Google Scholar] [CrossRef]
- Simpson, E.R.; Misso, M.; Hewitt, K.N.; Hill, R.A.; Boon, W.C.; Jones, M.E.; Kovacic, A.; Zhou, J.; Clyne, C.D. Estrogen—The good, the bad, and the unexpected. Endocr. Rev. 2005, 26, 322–330. [Google Scholar] [CrossRef]
- Faulds, M.H.; Zhao, C.; Dahlman-Wright, K.; Gustafsson, J.A. The diversity of sex steroid action: Regulation of metabolism by estrogen signaling. J. Endocrinol. 2012, 212, 3–12. [Google Scholar] [CrossRef]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef]
- Kerrigan, J.R.; Rogol, A.D. The impact of gonadal steroid hormone action on growth hormone secretion during childhood and adolescence. Endocr. Rev. 1992, 13, 281–298. [Google Scholar]
- Waters, M.J. Seeking SOCS and sex steroids. Trends Endocrinol. Metab. 2003, 14, 149–151. [Google Scholar] [CrossRef]
- Leung, K.C.; Johannsson, G.; Leong, G.M.; Ho, K.K. Estrogen regulation of growth hormone action. Endocr. Rev. 2004, 25, 693–721. [Google Scholar] [CrossRef]
- Santana-Farre, R.; Flores-Morale, A.; Fernández-Pérez, L. Growth Hormone, thyroid hormones and estradiol interplay in vivo to regulate gene expression of suppressors of cytokine signalling (SOCS). In 13th International Congress of Endocrinology, Proceedings of the 13th International Congress of Endocrinology, Rio de Janeiro, Brazil, 8–12 November 2008; Godoy-Matos, A., Rio de Janeiro, J.W., Eds.; Medimond, S.r.l.: Bologna, Italy, 2008; pp. 8–12. [Google Scholar]
- Barclay, J.L.; Nelson, C.N.; Ishikawa, M.; Murray, L.A.; Kerr, L.M.; McPhee, T.R.; Powell, E.E.; Waters, M.J. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology 2011, 152, 181–192. [Google Scholar]
- Luo, B.; Zou, T.; Lu, N.; Chai, F.; Ye, X.; Wang, Y.; Qi, Y. Role of suppressor of cytokine signaling 3 in lipid metabolism: Analysis based on a phage-display human liver cDNA library. Biochem. Biophys. Res. Commun. 2011, 416, 39–44. [Google Scholar] [CrossRef]
- Zadjali, F.; Santana-Farre, R.; Vesterlund, M.; Carow, B.; Mirecki-Garrido, M.; Hernandez-Hernandez, I.; Flodstrom-Tullberg, M.; Parini, P.; Rottenberg, M.; Norstedt, G.; et al. SOCS2 deletion protects against hepatic steatosis but worsens insulin resistance in high-fat-diet-fed mice. FASEB J. 2012. [Google Scholar]
- Fisher, B.; Gunduz, N.; Saffer, E.A.; Zheng, S. Relation of estrogen and its receptor to rat liver growth and regeneration. Cancer Res. 1984, 44, 2410–2415. [Google Scholar]
- Vidal, O.; Lindberg, M.K.; Hollberg, K.; Baylink, D.J.; Andersson, G.; Lubahn, D.B.; Mohan, S.; Gustafsson, J.A.; Ohlsson, C. Estrogen receptor specificity in the regulation of skeletal growth and maturation in male mice. Proc. Natl. Acad. Sci. USA 2000, 97, 5474–5479. [Google Scholar]
- Yamamoto, Y.; Moore, R.; Hess, H.A.; Guo, G.L.; Gonzalez, F.J.; Korach, K.S.; Maronpot, R.R.; Negishi, M. Estrogen receptor alpha mediates 17alpha-ethynylestradiol causing hepatotoxicity. J. Biol. Chem. 2006, 281, 16625–16631. [Google Scholar]
- Bigsby, R.M.; Caperell-Grant, A. The role for estrogen receptor-alpha and prolactin receptor in sex-dependent DEN-induced liver tumorigenesis. Carcinogenesis 2011, 32, 1162–1166. [Google Scholar] [CrossRef]
- Della Torre, S.; Rando, G.; Meda, C.; Stell, A.; Chambon, P.; Krust, A.; Ibarra, C.; Magni, P.; Ciana, P.; Maggi, A. Amino acid-dependent activation of liver estrogen receptor alpha integrates metabolic and reproductive functions via IGF-1. Cell Metab. 2011, 13, 205–214. [Google Scholar] [CrossRef]
- Foryst-Ludwig, A.; Kintscher, U. Metabolic impact of estrogen signalling through ERalpha and ERbeta. J. Steroid Biochem. Mol. Biol. 2010, 122, 74–81. [Google Scholar] [CrossRef]
- Meinhardt, U.J.; Ho, K.K. Regulation of growth hormone action by gonadal steroids. Endocrinol. Metab. Clin. North Am. 2007, 36, 57–73. [Google Scholar] [CrossRef]
- Waters, M.J.; Shang, C.A.; Behncken, S.N.; Tam, S.P.; Li, H.; Shen, B.; Lobie, P.E. Growth hormone as a cytokine. Clin. Exp. Pharmacol. Physiol. 1999, 26, 760–764. [Google Scholar] [CrossRef]
- Kaplan, S.A.; Cohen, P. The somatomedin hypothesis 2007: 50 Years later. J. Clin. Endocrinol. Metab. 2007, 92, 4529–4535. [Google Scholar] [CrossRef]
- Carro, E.; Senaris, R.; Considine, R.V.; Casanueva, F.F.; Dieguez, C. Regulation of in vivo growth hormone secretion by leptin. Endocrinology 1997, 138, 2203–2206. [Google Scholar] [CrossRef]
- Howard, A.D.; Feighner, S.D.; Cully, D.F.; Arena, J.P.; Liberator, P.A.; Rosenblum, C.I.; Hamelin, M.; Hreniuk, D.L.; Palyha, O.C.; Anderson, J.; et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 1996, 273, 974–977. [Google Scholar]
- Holst, B.; Schwartz, T.W. Ghrelin receptor mutations—Too little height and too much hunger. J. Clin. Invest. 2006, 116, 637–641. [Google Scholar] [CrossRef]
- Svensson, J.; Lonn, L.; Jansson, J.O.; Murphy, G.; Wyss, D.; Krupa, D.; Cerchio, K.; Polvino, W.; Gertz, B.; Boseaus, I.; et al. Two-month treatment of obese subjects with the oral growth hormone (GH) secretagogue MK-677 increases GH secretion, fat-free mass, and energy expenditure. J. Clin. Endocrinol. Metab. 1998, 83, 362–369. [Google Scholar] [CrossRef]
- Lanning, N.J.; Carter-Su, C. Recent advances in growth hormone signaling. Rev. Endocr. Metab. Disord. 2006, 7, 225–235. [Google Scholar]
- Vidal, O.M.; Merino, R.; Rico-Bautista, E.; Fernandez-Perez, L.; Chia, D.J.; Woelfle, J.; Ono, M.; Lenhard, B.; Norstedt, G.; Rotwein, P.; et al. In vivo transcript profiling and phylogenetic analysis identifies suppressor of cytokine signaling 2 as a direct signal transducer and activator of transcription 5b target in liver. Mol. Endocrinol. 2007, 21, 293–311. [Google Scholar]
- Rowland, J.E.; Lichanska, A.M.; Kerr, L.M.; White, M.; d’Aniello, E.M.; Maher, S.L.; Brown, R.; Teasdale, R.D.; Noakes, P.G.; Waters, M.J. In vivo analysis of growth hormone receptor signaling domains and their associated transcripts. Mol. Cell Biol. 2005, 25, 66–77. [Google Scholar] [CrossRef]
- Flores-Morales, A.; Greenhalgh, C.J.; Norstedt, G.; Rico-Bautista, E. Negative regulation of growth hormone receptor signaling. Mol. Endocrinol. 2006, 20, 241–253. [Google Scholar]
- Fernandez, L.; Flores-Morales, A.; Lahuna, O.; Sliva, D.; Norstedt, G.; Haldosen, L.A.; Mode, A.; Gustafsson, J.A. Desensitization of the growth hormone-induced Janus kinase 2 (Jak 2)/signal transducer and activator of transcription 5 (Stat5)-signaling pathway requires protein synthesis and phospholipase C. Endocrinology 1998, 139, 1815–1824. [Google Scholar]
- Waxman, D.J.; Ram, P.A.; Park, S.H.; Choi, H.K. Intermittent plasma growth hormone triggers tyrosine phosphorylation and nuclear translocation of a liver-expressed, Stat 5-related DNA binding protein. Proposed role as an intracellular regulator of male-specific liver gene transcription. J. Biol. Chem. 1995, 270, 13262–13270. [Google Scholar]
- Waxman, D.J.; O’Connor, C. Growth hormone regulation of sex-dependent liver gene expression. Mol. Endocrinol. 2006, 20, 2613–2629. [Google Scholar] [CrossRef]
- Kelly, P.A.; Djiane, J.; Postel-Vinay, M.C.; Edery, M. The prolactin/growth hormone receptor family. Endocr. Rev. 1991, 12, 235–251. [Google Scholar] [CrossRef]
- Stofega, M.R.; Wang, H.; Ullrich, A.; Carter-Su, C. Growth hormone regulation of SIRP and SHP-2 tyrosyl phosphorylation and association. J. Biol. Chem. 1998, 273, 7112–7117. [Google Scholar]
- Rico-Bautista, E.; Flores-Morales, A.; Fernandez-Perez, L. Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev. 2006, 17, 431–439. [Google Scholar] [CrossRef]
- Colao, A.; Merola, B.; Ferone, D.; Lombardi, G. Acromegaly. J. Clin. Endocrinol. Metab. 1997, 82, 2777–2781. [Google Scholar] [CrossRef]
- Horvat, S.; Medrano, J.F. Lack of Socs2 expression causes the high-growth phenotype in mice. Genomics 2001, 72, 209–212. [Google Scholar] [CrossRef]
- Miller, M.E.; Michaylira, C.Z.; Simmons, J.G.; Ney, D.M.; Dahly, E.M.; Heath, J.K.; Lund, P.K. Suppressor of cytokine signaling-2: A growth hormone-inducible inhibitor of intestinal epithelial cell proliferation. Gastroenterology 2004, 127, 570–581. [Google Scholar]
- Iglesias, D. Personal communication, CPR, University of Copenhagen, Copenhagen, Denmark, 2012.
- Flores-Morales, A.; Stahlberg, N.; Tollet-Egnell, P.; Lundeberg, J.; Malek, R.L.; Quackenbush, J.; Lee, N.H.; Norstedt, G. Microarray analysis of the in vivo effects of hypophysectomy and growth hormone treatment on gene expression in the rat. Endocrinology 2001, 142, 3163–3176. [Google Scholar] [CrossRef]
- Tollet-Egnell, P.; Parini, P.; Stahlberg, N.; Lonnstedt, I.; Lee, N.H.; Rudling, M.; Flores-Morales, A.; Norstedt, G. Growth hormone-mediated alteration of fuel metabolism in the aged rat as determined from transcript profiles. Physiol. Genomics 2004, 16, 261–267. [Google Scholar]
- Stahlberg, N.; Merino, R.; Hernandez, L.H.; Fernandez-Perez, L.; Sandelin, A.; Engstrom, P.; Tollet-Egnell, P.; Lenhard, B.; Flores-Morales, A. Exploring hepatic hormone actions using a compilation of gene expression profiles. BMC Physiol. 2005, 5, 8. [Google Scholar] [CrossRef]
- Udy, G.B.; Towers, R.P.; Snell, R.G.; Wilkins, R.J.; Park, S.H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar]
- Rotwein, P. Mapping the growth hormone—Stat5b—IGF-I transcriptional circuit. Trends Endocrinol. Metab. 2012, 23, 186–193. [Google Scholar] [CrossRef]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef]
- Klover, P.; Hennighausen, L. Postnatal body growth is dependent on the transcription factors signal transducers and activators of transcription 5a/b in muscle: A role for autocrine/paracrine insulin-like growth factor I. Endocrinology 2007, 148, 1489–1497. [Google Scholar]
- Engblom, D.; Kornfeld, J.W.; Schwake, L.; Tronche, F.; Reimann, A.; Beug, H.; Hennighausen, L.; Moriggl, R.; Schutz, G. Direct glucocorticoid receptor-Stat5 interaction in hepatocytes controls body size and maturation-related gene expression. Genes Dev. 2007, 21, 1157–1162. [Google Scholar] [CrossRef]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Corpas, E.; Harman, S.M.; Blackman, M.R. Human growth hormone and human aging. Endocr. Rev. 1993, 14, 20–39. [Google Scholar]
- Cui, Y.; Hosui, A.; Sun, R.; Shen, K.; Gavrilova, O.; Chen, W.; Cam, M.C.; Gao, B.; Robinson, G.W.; Hennighausen, L. Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology 2007, 46, 504–513. [Google Scholar]
- Olsson, B.; Bohlooly, Y.M.; Brusehed, O.; Isaksson, O.G.; Ahren, B.; Olofsson, S.O.; Oscarsson, J.; Tornell, J. Bovine growth hormone-transgenic mice have major alterations in hepatic expression of metabolic genes. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E504–E511. [Google Scholar]
- Sos, B.C.; Harris, C.; Nordstrom, S.M.; Tran, J.L.; Balazs, M.; Caplazi, P.; Febbraio, M.; Applegate, M.A.; Wagner, K.U.; Weiss, E.J. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte-specific deletion of JAK2. J. Clin. Invest. 2011, 121, 1412–1423. [Google Scholar]
- Wang, Z.; Masternak, M.M.; Al-Regaiey, K.A.; Bartke, A. Adipocytokines and the regulation of lipid metabolism in growth hormone transgenic and calorie-restricted mice. Endocrinology 2007, 148, 2845–2853. [Google Scholar] [CrossRef]
- Vidarsdottir, S.; Walenkamp, M.J.; Pereira, A.M.; Karperien, M.; van Doorn, J.; van Duyvenvoorde, H.A.; White, S.; Breuning, M.H.; Roelfsema, F.; Kruithof, M.F.; et al. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. J. Clin. Endocrinol. Metab. 2006, 91, 3482–3485. [Google Scholar]
- Cho, Y.; Ariga, M.; Uchijima, Y.; Kimura, K.; Rho, J.Y.; Furuhata, Y.; Hakuno, F.; Yamanouchi, K.; Nishihara, M.; Takahashi, S. The novel roles of liver for compensation of insulin resistance in human growth hormone transgenic rats. Endocrinology 2006, 147, 5374–5384. [Google Scholar]
- Dominici, F.P.; Turyn, D. Growth hormone-induced alterations in the insulin-signaling system. Exp. Biol. Med. (Maywood) 2002, 227, 149–157. [Google Scholar]
- Yakar, S.; Setser, J.; Zhao, H.; Stannard, B.; Haluzik, M.; Glatt, V.; Bouxsein, M.L.; Kopchick, J.J.; LeRoith, D. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J. Clin. Invest. 2004, 113, 96–105. [Google Scholar]
- Wauthier, V.; Sugathan, A.; Meyer, R.D.; Dombkowski, A.A.; Waxman, D.J. Intrinsic sex differences in the early growth hormone responsiveness of sex-specific genes in mouse liver. Mol. Endocrinol. 2010, 24, 667–678. [Google Scholar] [CrossRef]
- Wolfrum, C.; Asilmaz, E.; Luca, E.; Friedman, J.M.; Stoffel, M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 2004, 432, 1027–1032. [Google Scholar]
- Sampath, H.; Ntambi, J.M. Polyunsaturated fatty acid regulation of genes of lipid metabolism. Annu. Rev. Nutr. 2005, 25, 317–340. [Google Scholar] [CrossRef]
- Tollet-Egnell, P.; Flores-Morales, A.; Stahlberg, N.; Malek, R.L.; Lee, N.; Norstedt, G. Gene expression profile of the aging process in rat liver: Normalizing effects of growth hormone replacement. Mol. Endocrinol. 2001, 15, 308–318. [Google Scholar] [CrossRef]
- Sjoberg, A.; Oscarsson, J.; Boren, J.; Eden, S.; Olofsson, S.O. Mode of growth hormone administration influences triacylglycerol synthesis and assembly of apolipoprotein B-containing lipoproteins in cultured rat hepatocytes. J. Lipid Res. 1996, 37, 275–289. [Google Scholar]
- Katzenellenbogen, B.S.; Sun, J.; Harrington, W.R.; Kraichely, D.M.; Ganessunker, D.; Katzenellenbogen, J.A. Structure-function relationships in estrogen receptors and the characterization of novel selective estrogen receptor modulators with unique pharmacological profiles. Ann. NY Acad. Sci. 2001, 949, 6–15. [Google Scholar]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar]
- Lundholm, L.; Bryzgalova, G.; Gao, H.; Portwood, N.; Falt, S.; Berndt, K.D.; Dicker, A.; Galuska, D.; Zierath, J.R.; Gustafsson, J.A.; et al. The estrogen receptor {alpha}-selective agonist propyl pyrazole triol improves glucose tolerance in ob/ob mice; potential molecular mechanisms. J. Endocrinol. 2008, 199, 275–286. [Google Scholar] [CrossRef]
- Faulds, M.H.; Pettersson, K.; Gustafsson, J.A.; Haldosen, L.A. Cross-talk between ERs and signal transducer and activator of transcription 5 is E2 dependent and involves two functionally separate mechanisms. Mol. Endocrinol. 2001, 15, 1929–1940. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Farrar, W.L.; Yang, X. Transcriptional crosstalk between nuclear receptors and cytokine signal transduction pathways in immunity. Cell Mol. Immunol. 2004, 1, 416–424. [Google Scholar]
- Barros, R.P.; Gustafsson, J.A. Estrogen receptors and the metabolic network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef]
- Venken, K.; Schuit, F.; van Lommel, L.; Tsukamoto, K.; Kopchick, J.J.; Coschigano, K.; Ohlsson, C.; Moverare, S.; Boonen, S.; Bouillon, R.; et al. Growth without growth hormone receptor: Estradiol is a major growth hormone-independent regulator of hepatic IGF-I synthesis. J. Bone Miner. Res. 2005, 20, 2138–2149. [Google Scholar] [CrossRef]
- Fernández-Pérez, L.; Santana-Farré, R.; Mirecki-Garrido, M.; Guerra, B.; Flores-Morales, A. Influence of estradiol on the Growth Hormone-regulated liver transcriptome in hypothyroid rats. University of Las Palmas de G.C., Las Palmas de G.C., Spain. Unpublished work, 2012.
- Coutant, R.; de Casson, F.B.; Rouleau, S.; Douay, O.; Mathieu, E.; Gatelais, F.; Bouhours-Nouet, N.; Voinot, C.; Audran, M.; Limal, J.M. Divergent effect of endogenous and exogenous sex steroids on the insulin-like growth factor I response to growth hormone in short normal adolescents. J. Clin. Endocrinol. Metab. 2004, 89, 6185–6192. [Google Scholar]
- Burman, P.; Johansson, A.G.; Siegbahn, A.; Vessby, B.; Karlsson, F.A. Growth hormone (GH)-deficient men are more responsive to GH replacement therapy than women. J. Clin. Endocrinol. Metab. 1997, 82, 550–555. [Google Scholar] [CrossRef]
- Gibney, J.; Johannsson, G.; Leung, K.C.; Ho, K.K. Comparison of the metabolic effects of raloxifene and oral estrogen in postmenopausal and growth hormone-deficient women. J. Clin. Endocrinol. Metab. 2005, 90, 3897–3903. [Google Scholar] [CrossRef]
- Huang, D.S.; O’Sullivan, A.J. Short-term oral oestrogen therapy dissociates the growth hormone/insulin-like growth factor-I axis without altering energy metabolism in premenopausal women. Growth Horm. IGF Res. 2009, 19, 162–167. [Google Scholar] [CrossRef]
- LeRoith, D. Gender differences in metabolic disorders. Gend. Med. 2009, 6, 1–3. [Google Scholar] [CrossRef]
- Birzniece, V.; Meinhardt, U.J.; Umpleby, M.A.; Handelsman, D.J.; Ho, K.K. Interaction between testosterone and growth hormone on whole-body protein anabolism occurs in the liver. J. Clin. Endocrinol. Metab. 2011, 96, 1060–1067. [Google Scholar]
- Rogol, A.D. Sex steroids, growth hormone, leptin and the pubertal growth spurt. Endocr. Dev. 2010, 17, 77–85. [Google Scholar] [CrossRef]
- Leung, K.C.; Brce, J.; Doyle, N.; Lee, H.J.; Leong, G.M.; Sjogren, K.; Ho, K.K. Regulation of growth hormone signaling by selective estrogen receptor modulators occurs through suppression of protein tyrosine phosphatases. Endocrinology 2007, 148, 2417–2423. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Mirecki-Garrido, M.; Guerra, B.; Mateos-Díaz, C.; Jiménez-Monzón, R.; Díaz-Chico, N.; Díaz-Chico, J.C.; Fernández-Pérez, L. The Influence of Estrogens on the Biological and Therapeutic Actions of Growth Hormone in the Liver. Pharmaceuticals 2012, 5, 758-778. https://doi.org/10.3390/ph5070758
De Mirecki-Garrido M, Guerra B, Mateos-Díaz C, Jiménez-Monzón R, Díaz-Chico N, Díaz-Chico JC, Fernández-Pérez L. The Influence of Estrogens on the Biological and Therapeutic Actions of Growth Hormone in the Liver. Pharmaceuticals. 2012; 5(7):758-778. https://doi.org/10.3390/ph5070758
Chicago/Turabian StyleDe Mirecki-Garrido, Mercedes, Borja Guerra, Carlos Mateos-Díaz, Roberto Jiménez-Monzón, Nicolás Díaz-Chico, Juan C. Díaz-Chico, and Leandro Fernández-Pérez. 2012. "The Influence of Estrogens on the Biological and Therapeutic Actions of Growth Hormone in the Liver" Pharmaceuticals 5, no. 7: 758-778. https://doi.org/10.3390/ph5070758
APA StyleDe Mirecki-Garrido, M., Guerra, B., Mateos-Díaz, C., Jiménez-Monzón, R., Díaz-Chico, N., Díaz-Chico, J. C., & Fernández-Pérez, L. (2012). The Influence of Estrogens on the Biological and Therapeutic Actions of Growth Hormone in the Liver. Pharmaceuticals, 5(7), 758-778. https://doi.org/10.3390/ph5070758