In Silico Screening of Nonsteroidal Anti-Inflammatory Drugs and Their Combined Action on Prostaglandin H Synthase-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Kinetic Model of Inhibitory Action of NSAIDs on PGHS-1
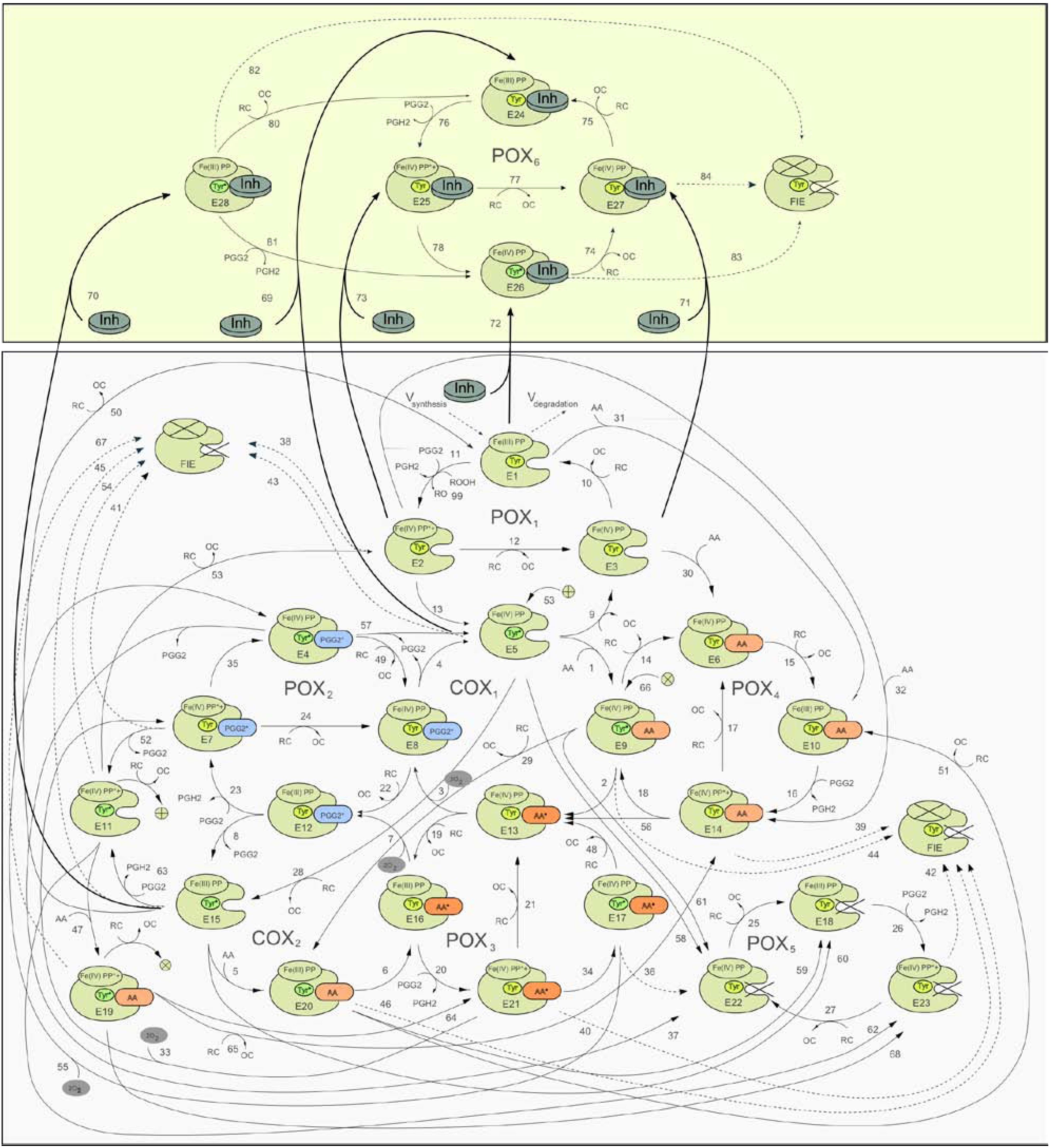
2.1. Detailed kinetic model of PGHS-1
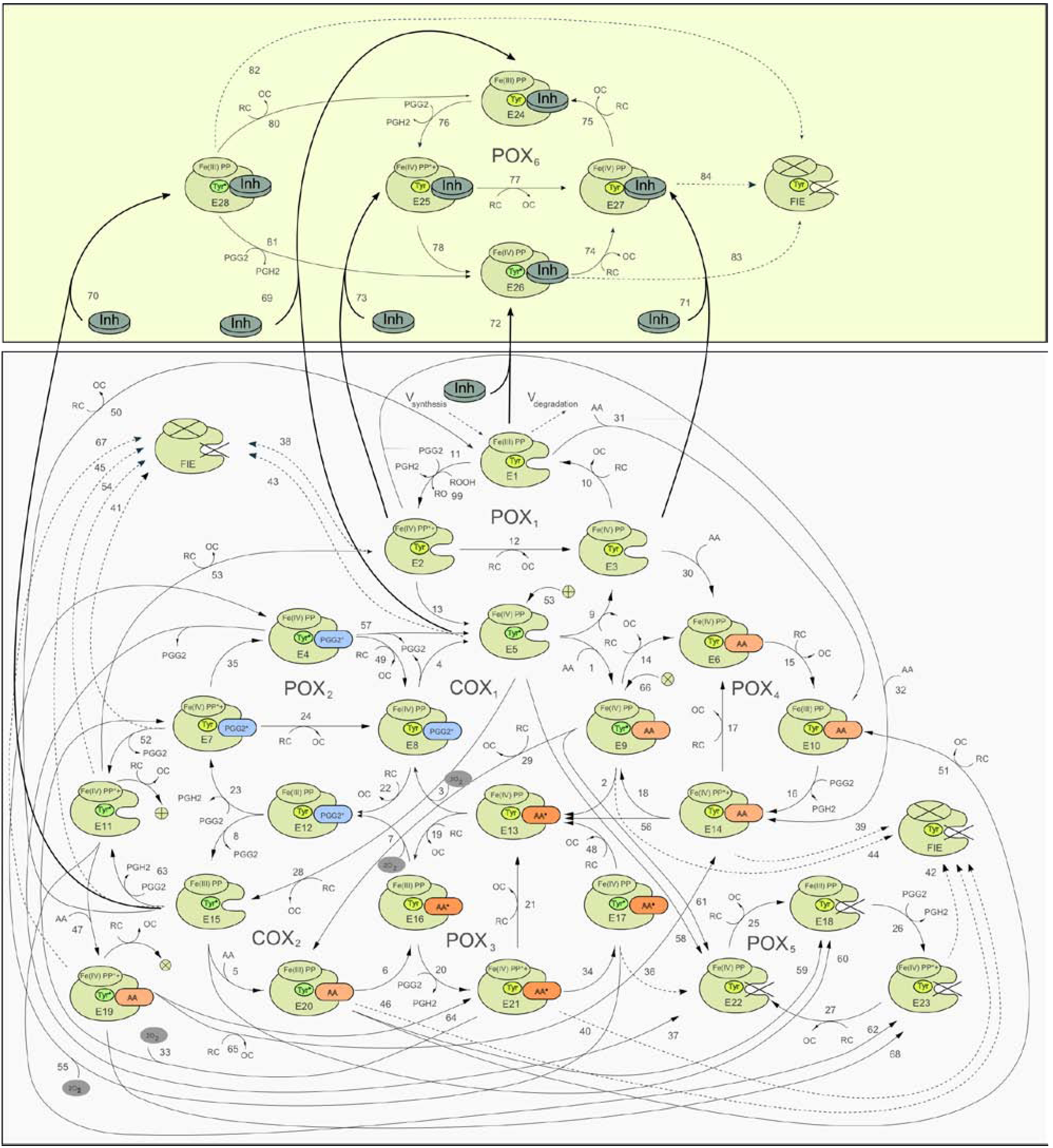
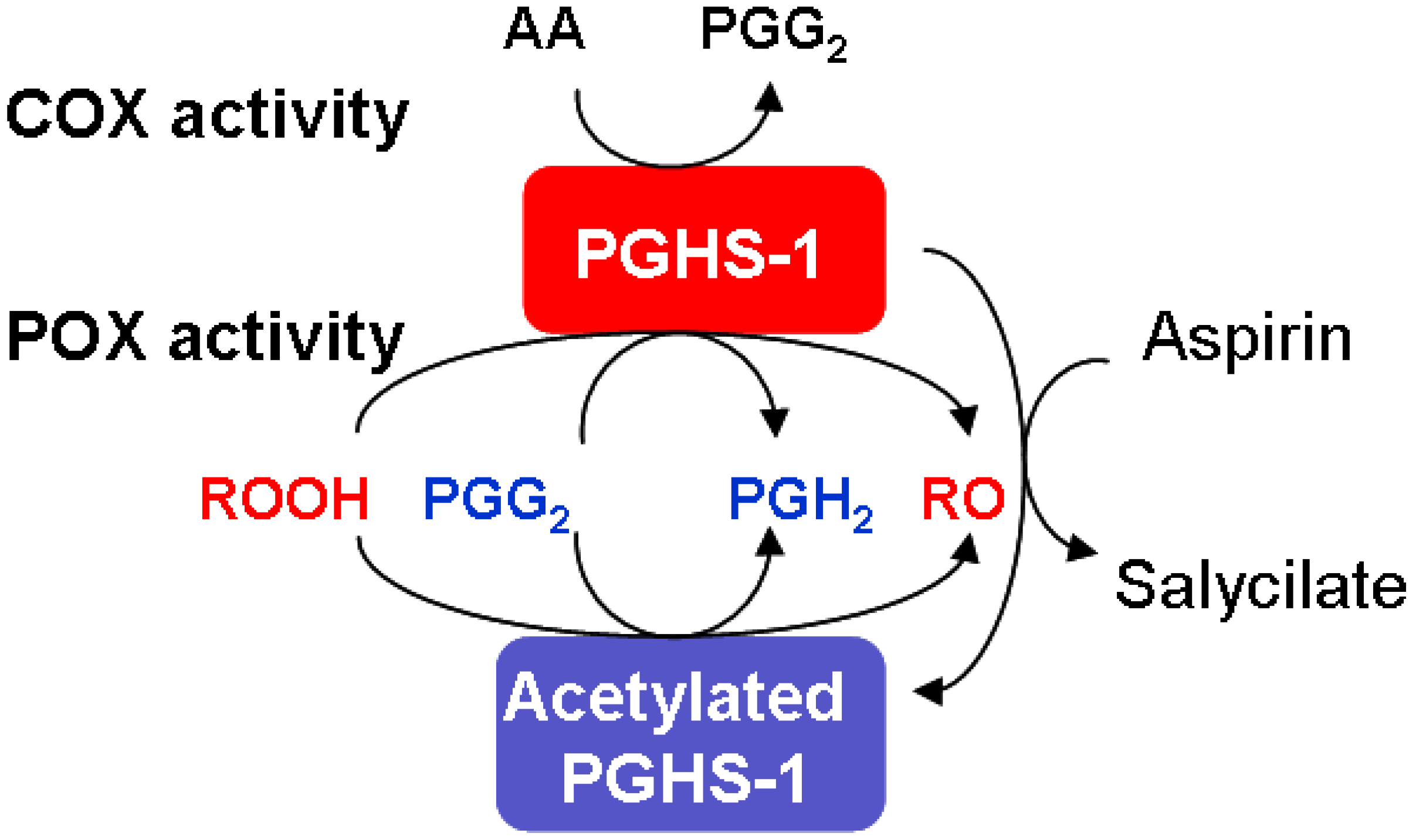
2.2. Consideration of NSAID Effects in the Model
2.3. The Method of in silico Screening of NSAID Action on PGHS-1
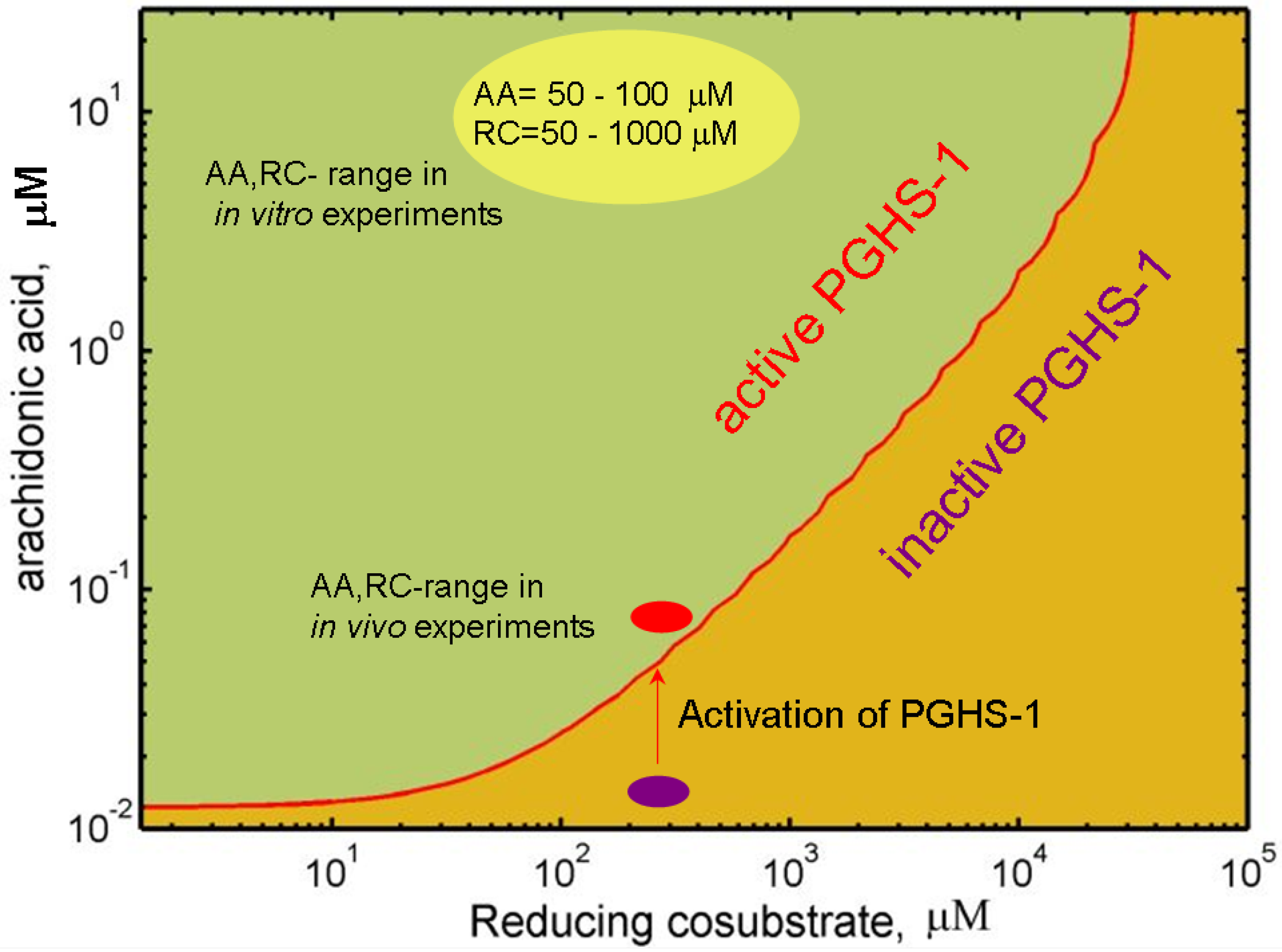
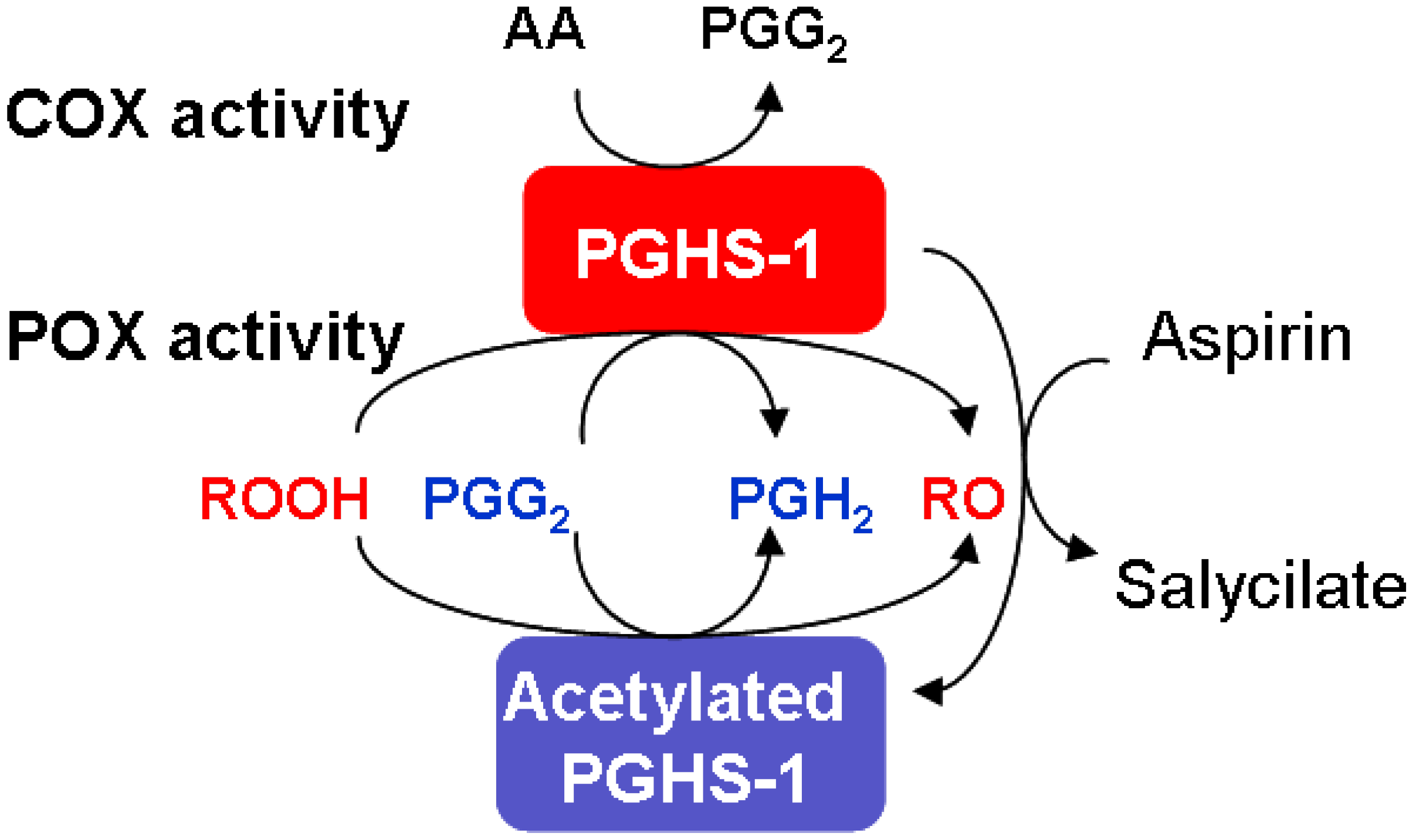
2.4. Mechanism of PGHS-1 regulation by arachidonic acid and reducing cosubstrate
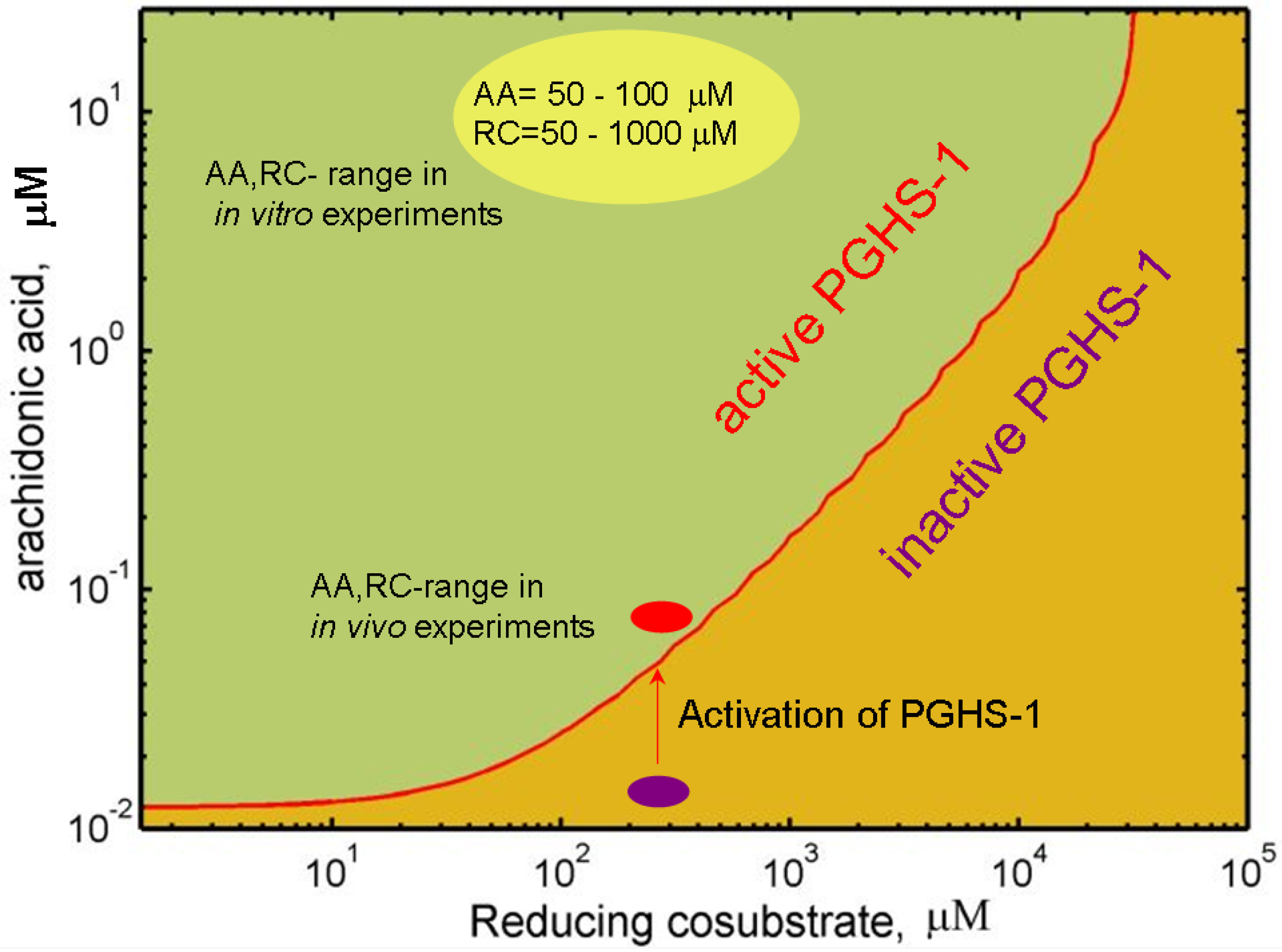
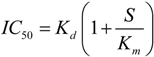
3. In silico Screening of NSAID Effects at the Different Microenvironments of PGHS-1
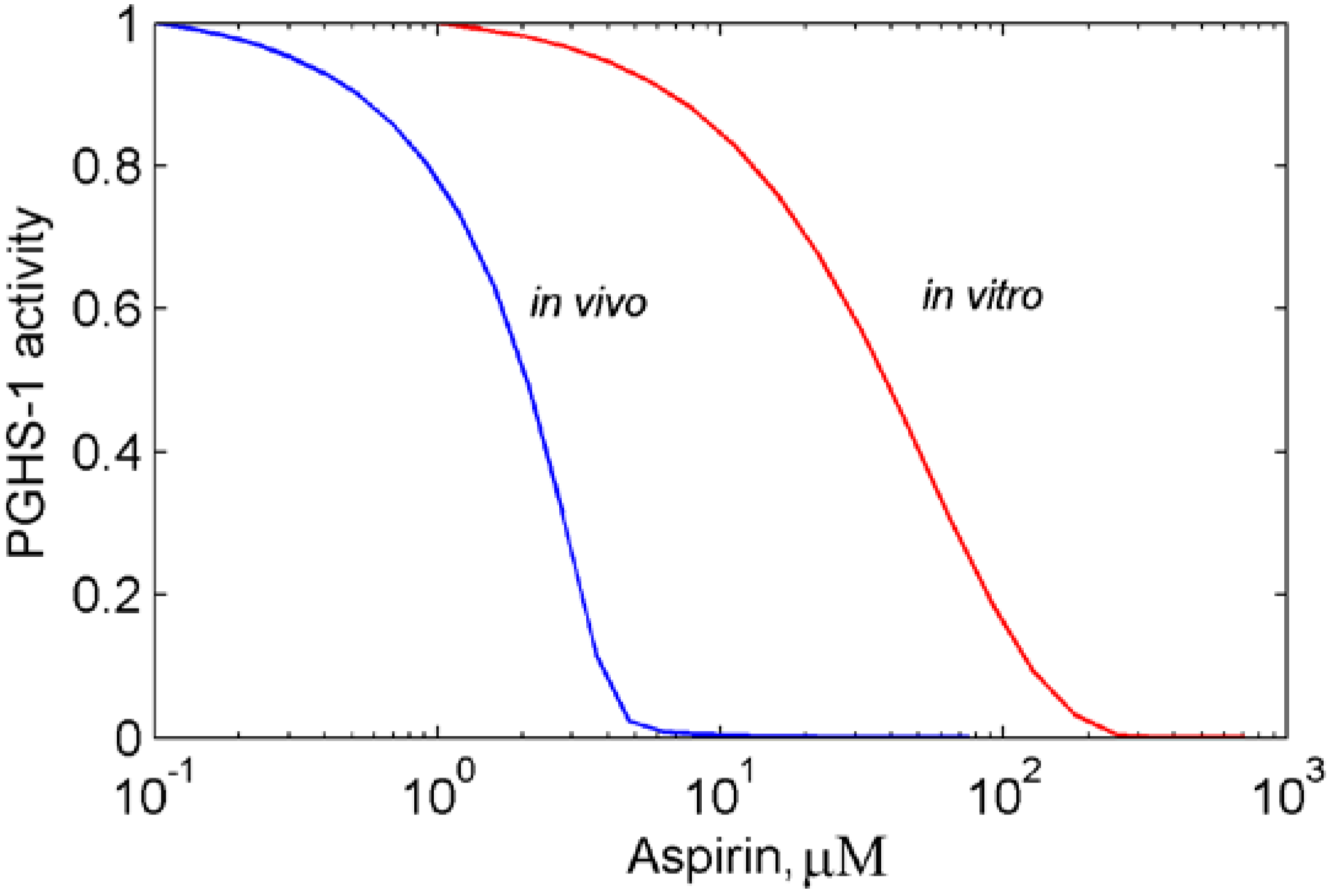
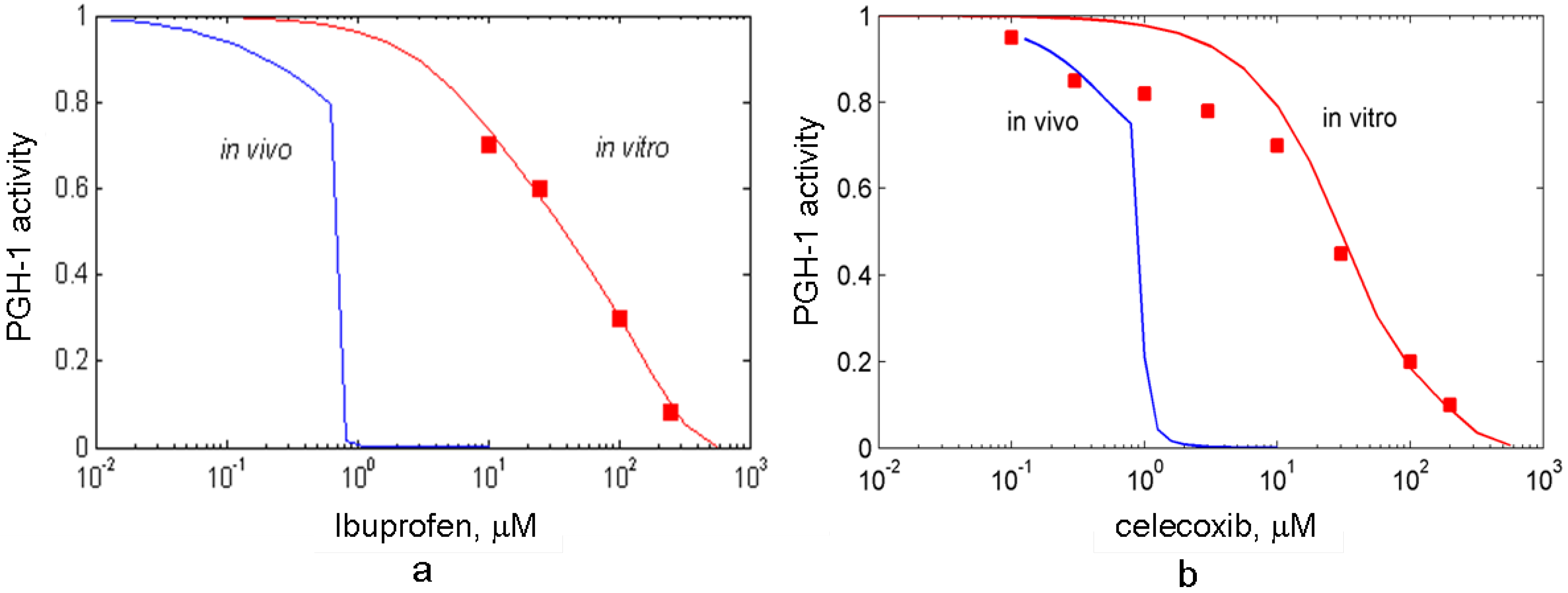
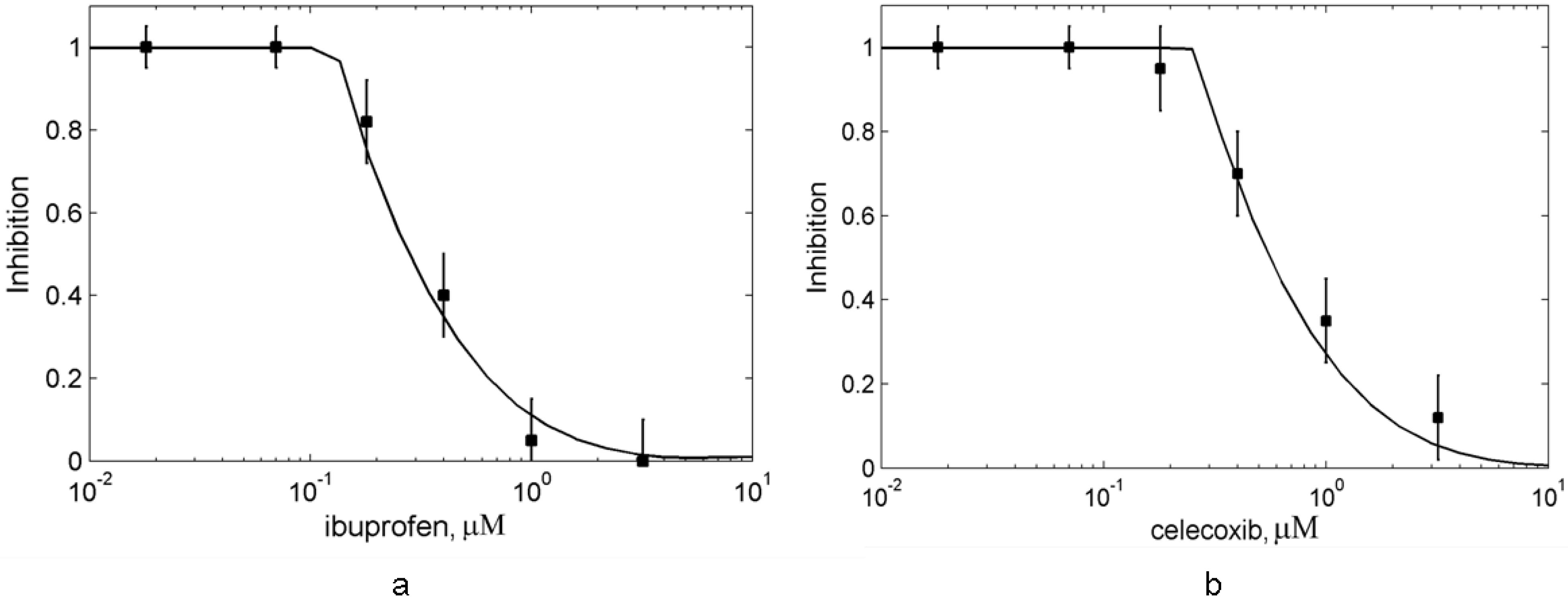
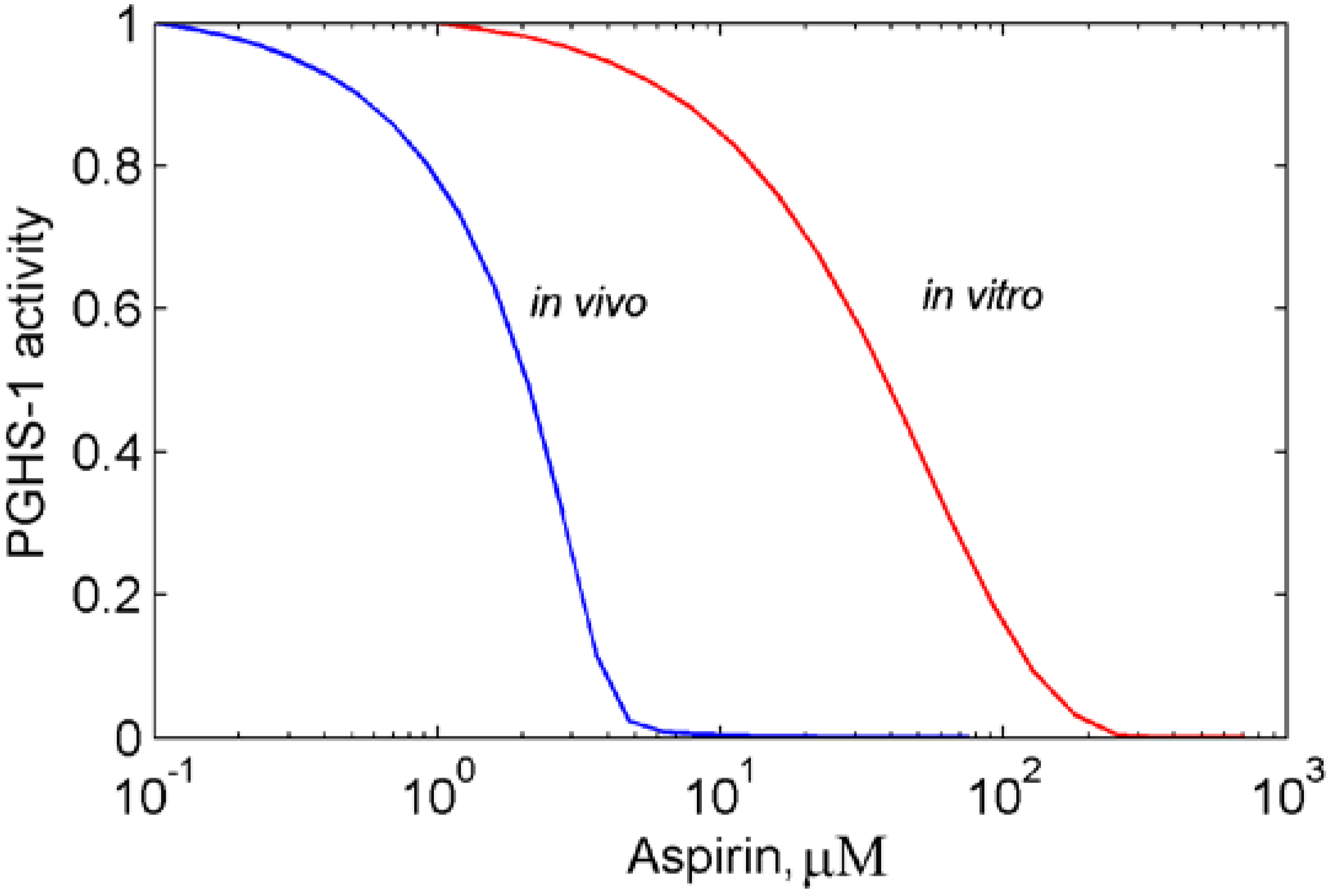
3.1. In silico experiment on aspirin action on PGHS-1 under in vitro/in vivo conditions
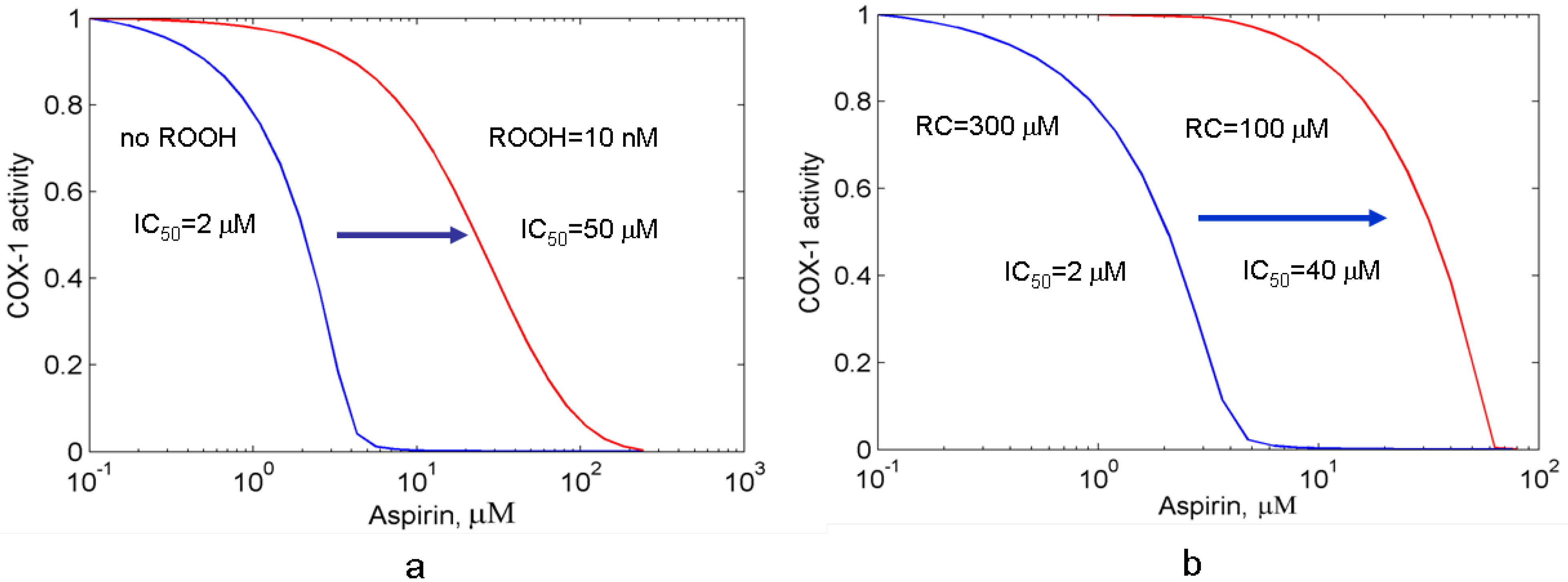
3.2. Effects of peroxide and reducing cosubstrate on PGHS-1 inhibition by aspirin
3.2.1. The suppression effect of external peroxide on aspirin-mediated inhibition of PGHS-1
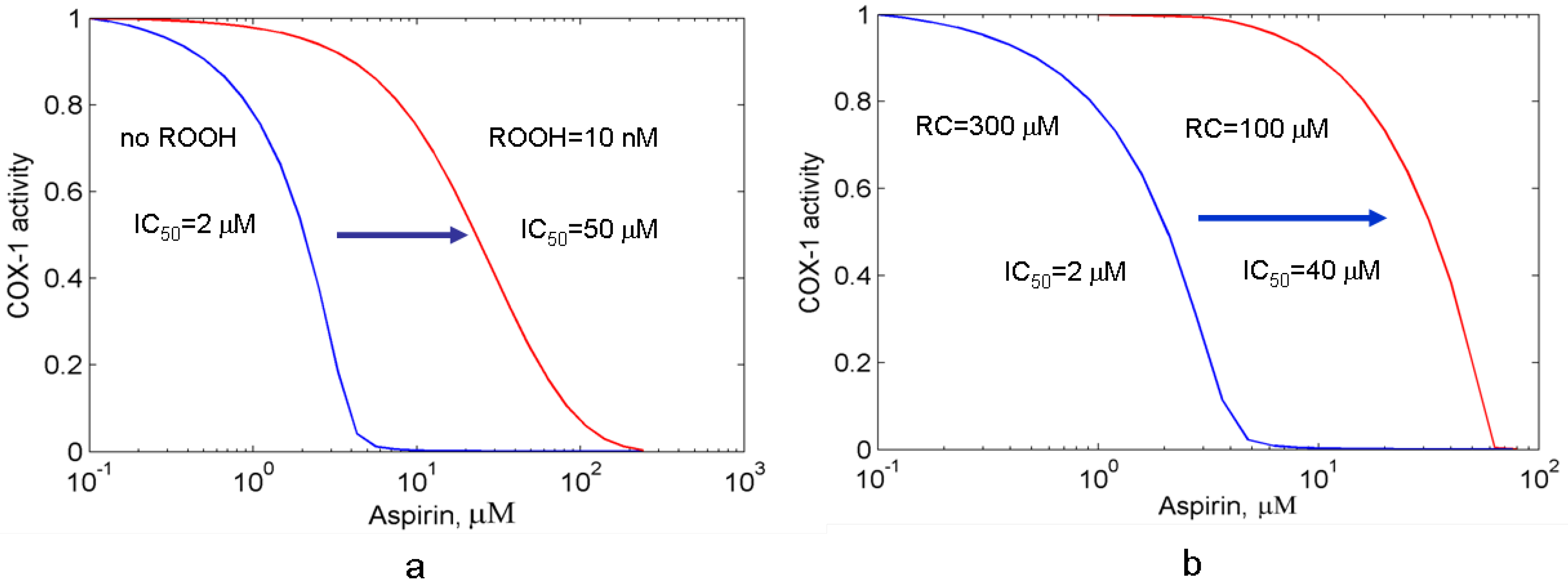
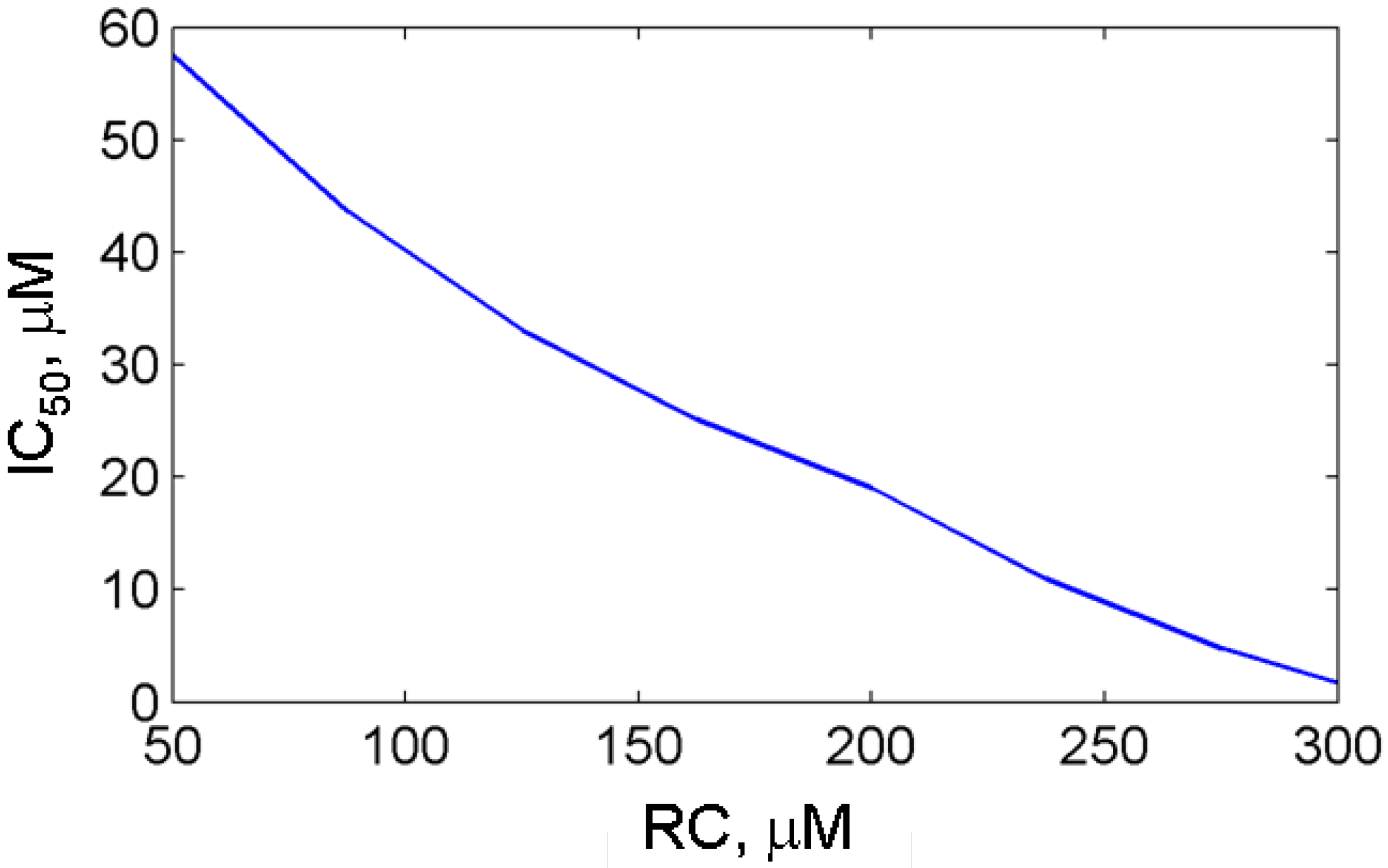
3.2.2. Effect of reducing cosubstrate on the aspirin-mediated inhibition of PGHS-1
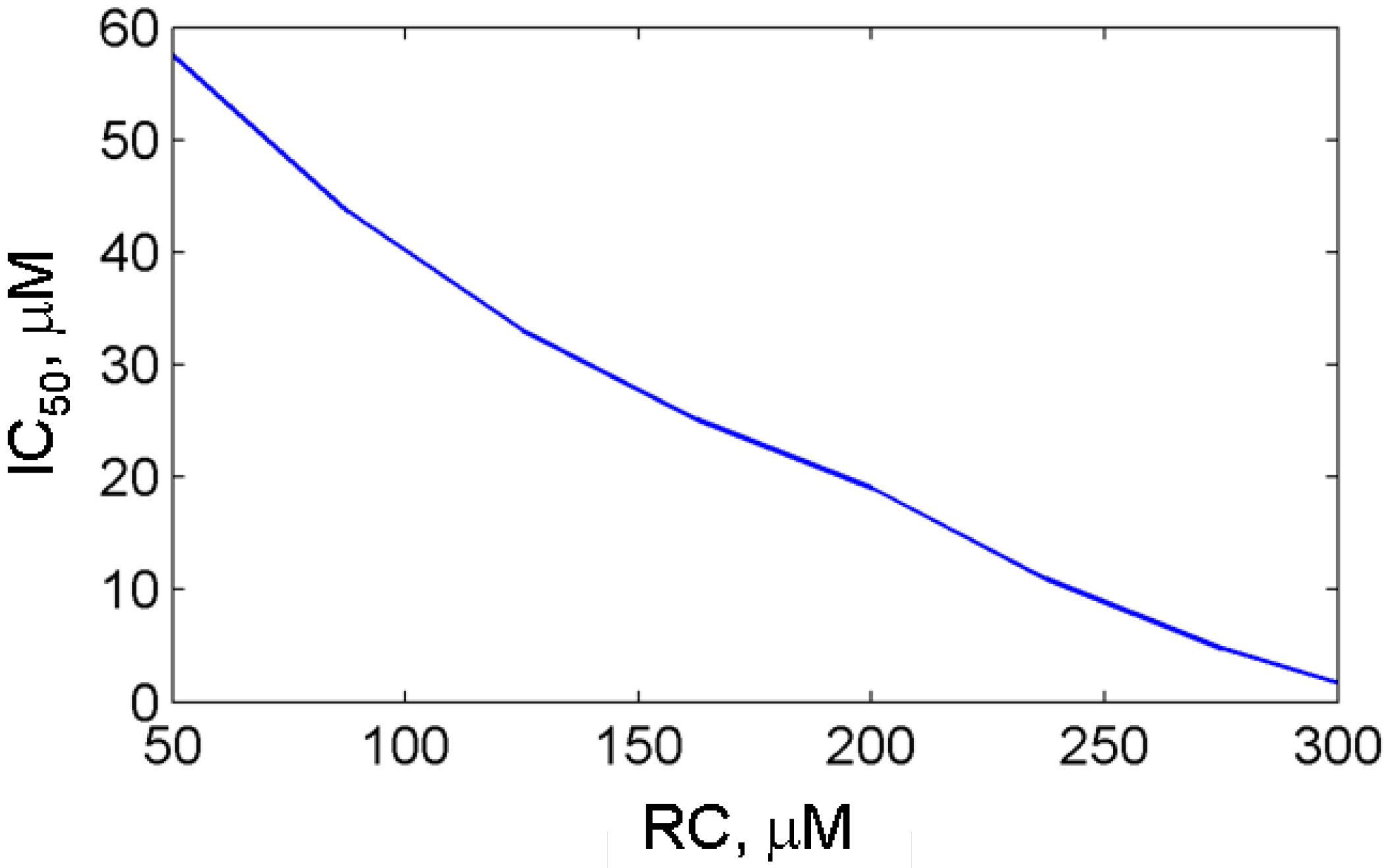
3.3. In silico screening of reversible inhibitor actions in different environmental conditions of PGHS-1
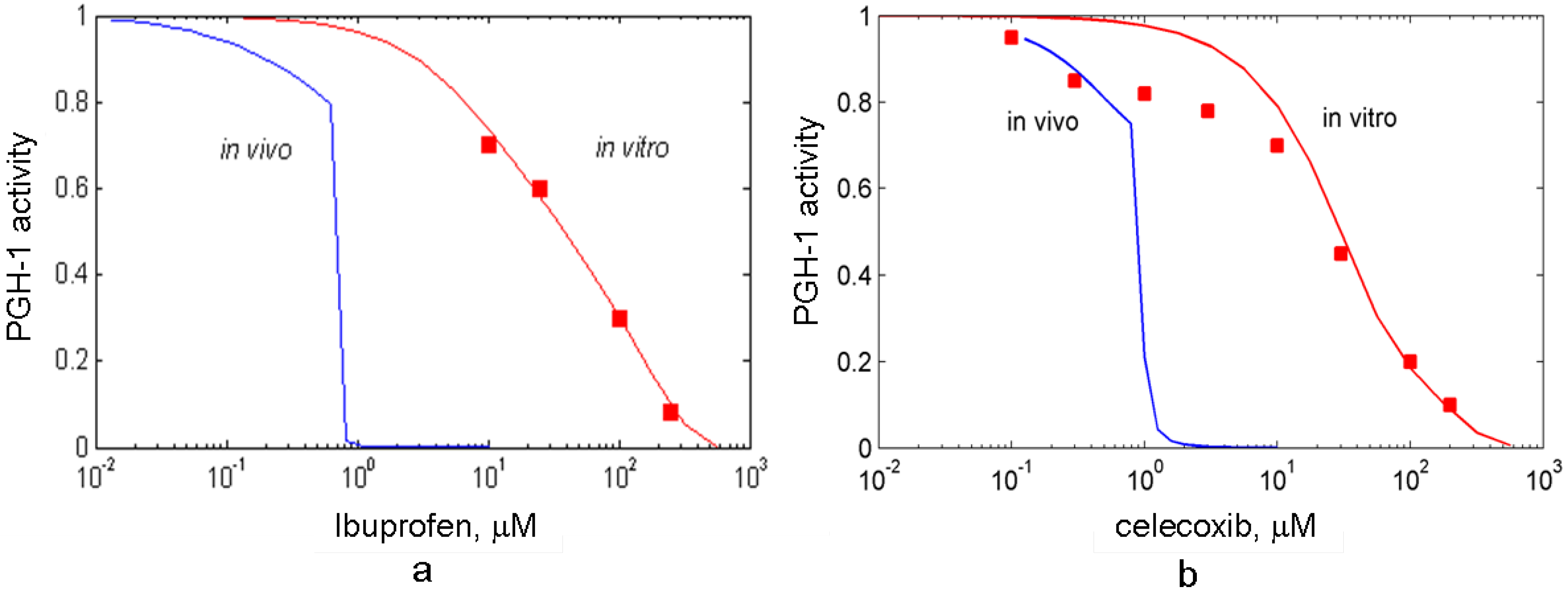
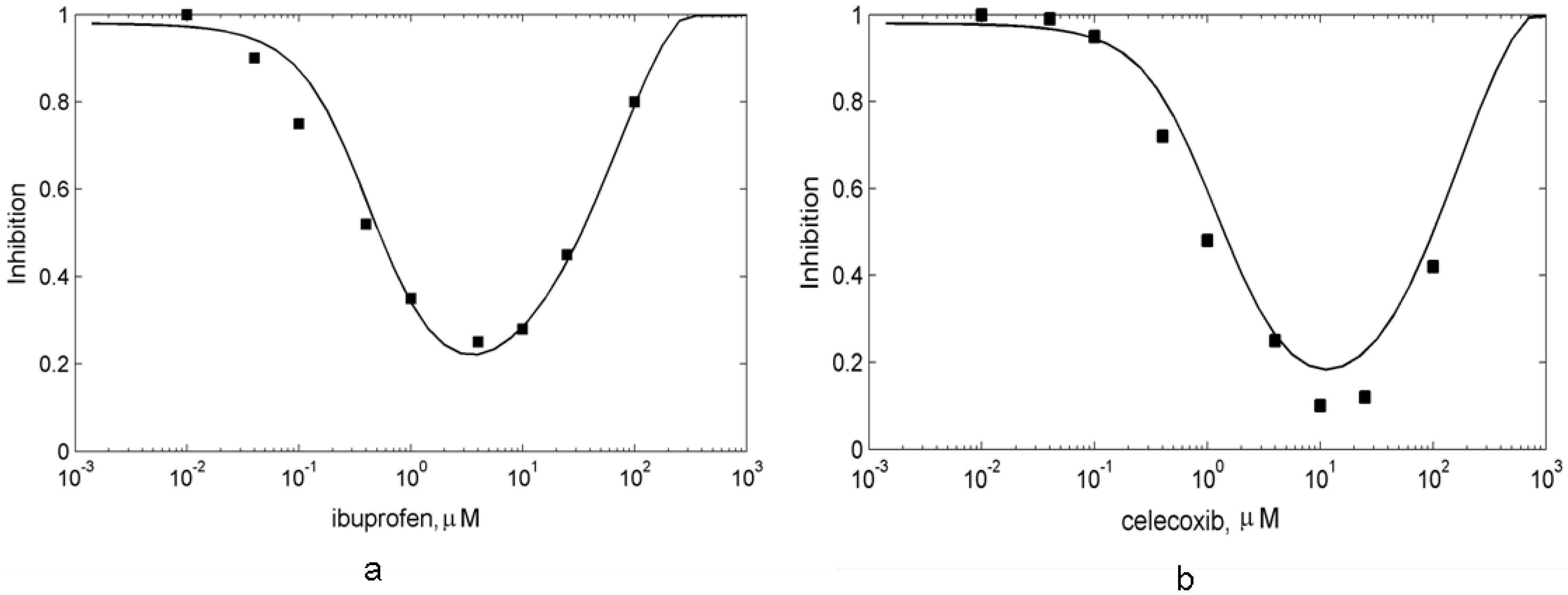
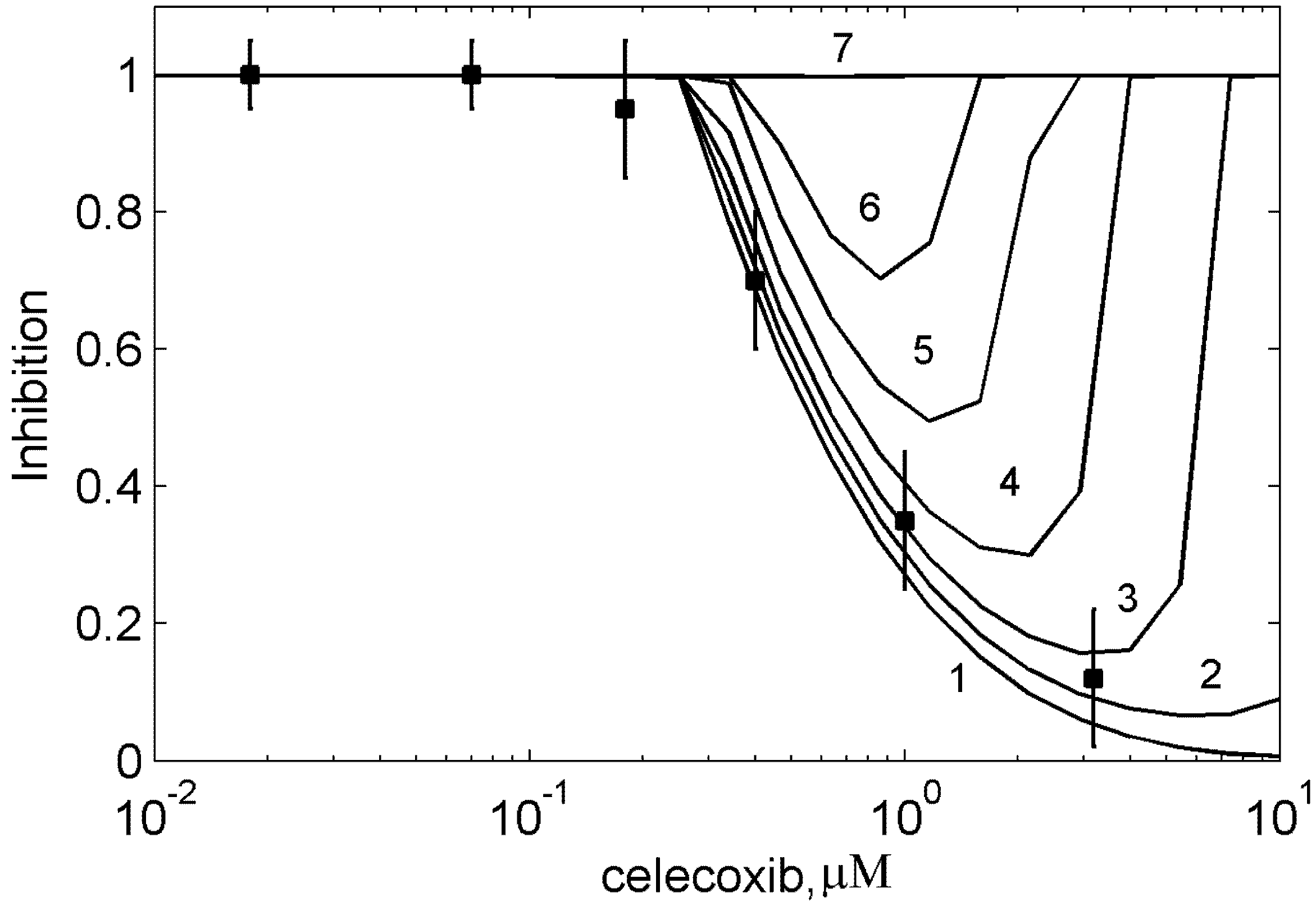
3.4. In silico screening of combined effects of NSAIDs on PGHS-1 inhibition in vitro/in vivo conditions
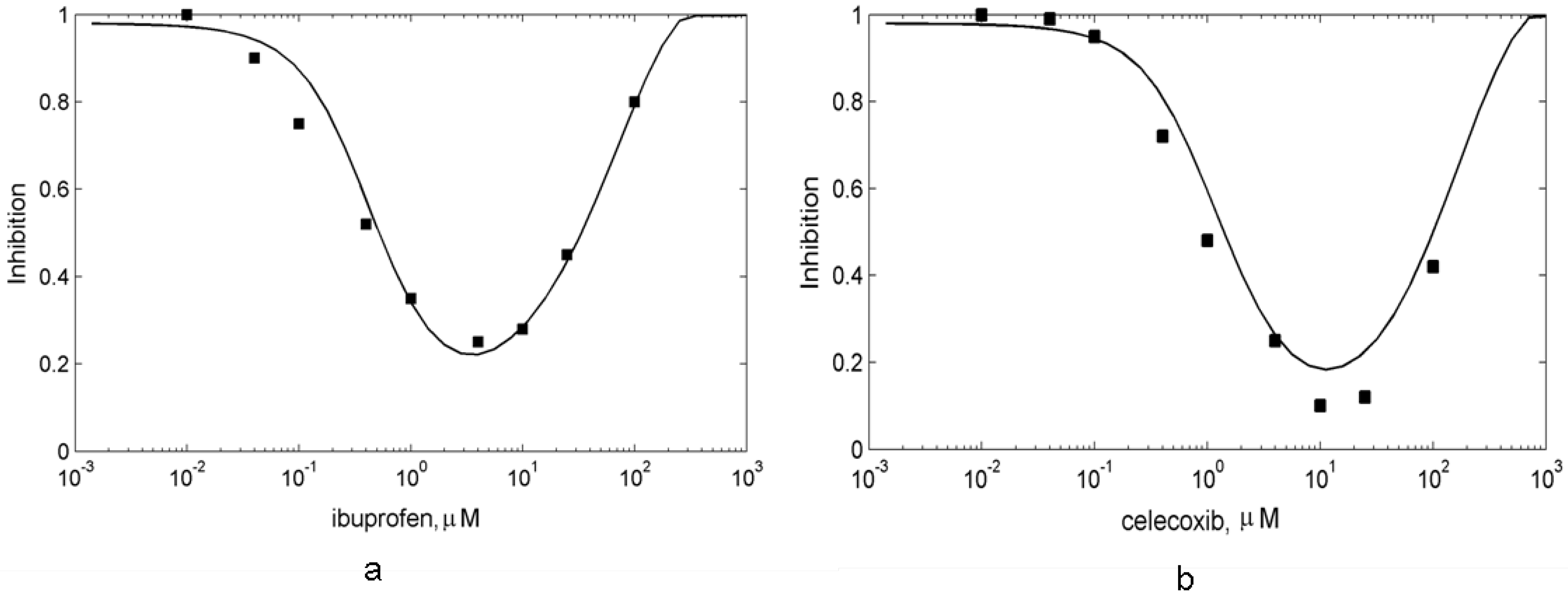
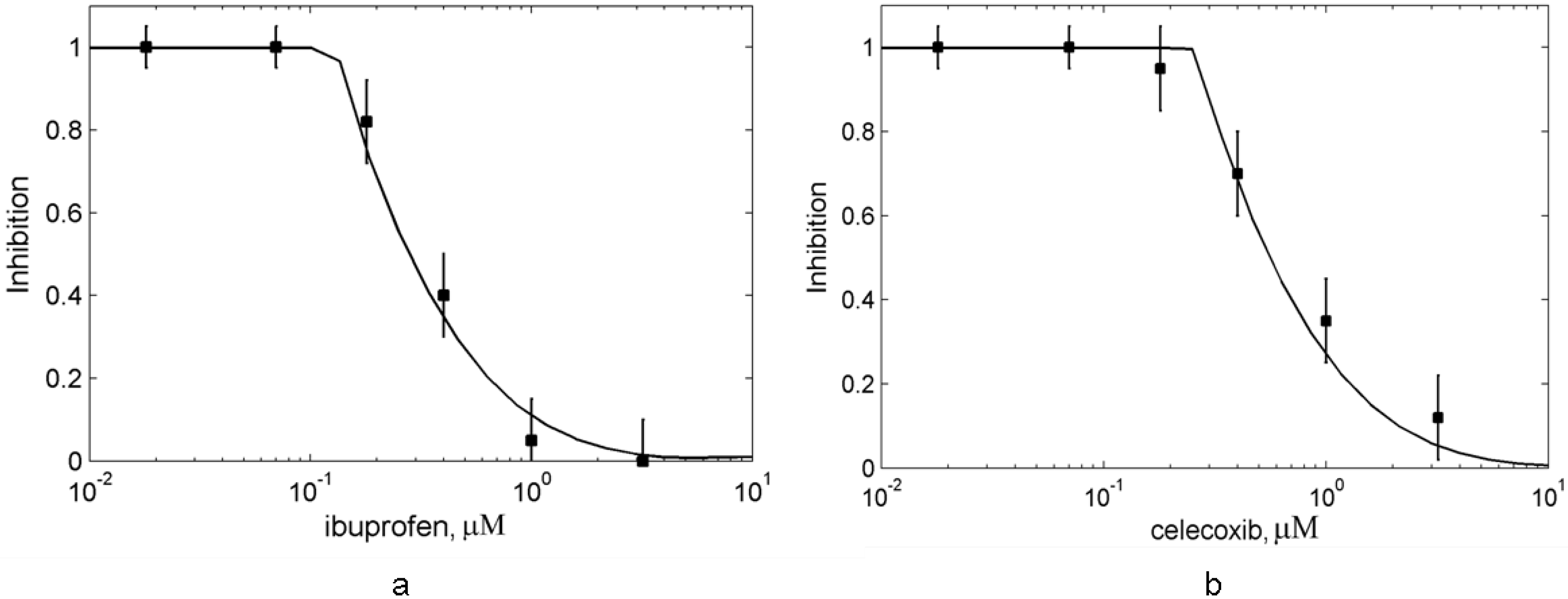
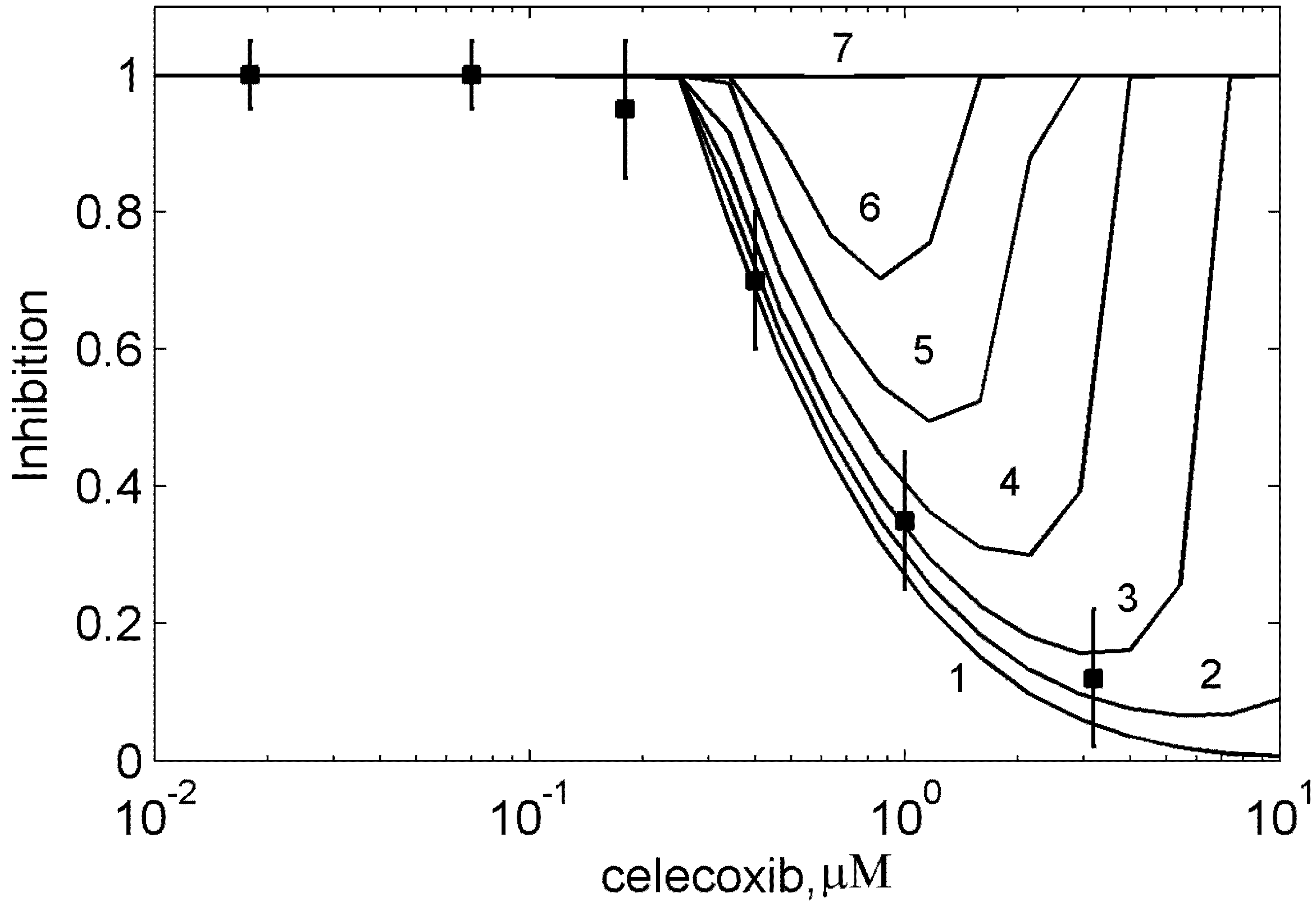
4. Conclusions
- In silico screening of inhibition effects of different types of NSAIDs on PGHS-1 in various environmental conditions of the enzyme;
- Dissection of the key factors, which determine the variability of PGHS-1 response to NSAID action in in vitro assays, platelets, endothelia and other cells;
- In silico comparison of the different NSAIDs and the establishment of bioequivalence of the various drugs in the different experimental conditions;
- Translation of the results on NSAIDs action on PGHS-1, obtained in in vitro experimental conditions to in vivo settings;
- In silico study of the drug interference, underlying the combined action of two NSAIDs on PGHS-1 to predict the potential risks and benefits of NSAID combination therapy.
Acknowledgements
References
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [PubMed]
- Kulmacz, R.J.; van der Donk, W.A.; Tsai, A.L. Comparison of the properties of prostaglandin H synthase-1 and -2. Prog. Lipid Res. 2003, 42, 377–404. [Google Scholar]
- Tsai, A.L.; Kulmacz, R.J. Prostaglandin H synthase: Resolved and unresolved mechanistic issues. Arch. Biochem. Biophys. 2010, 493, 103–124. [Google Scholar]
- Kulmacz, R.J. Cellular regulation of prostaglandin H synthase catalysis. FEBS Lett. 1998, 430, 154–157. [Google Scholar]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar]
- Bala, M.; Chin, C.N.; Logan, A.T.; Amin, T.; Marnett, L.J.; Boutaud, O.; Oates, J.A. Acetylation of prostaglandin H2 synthases by aspirin is inhibited by redox cycling of the peroxidase. Biochem. Pharmacol. 2008, 75, 1472–1481. [Google Scholar]
- Gierse, J.K.; Koboldt, C.M.; Walker, M.C.; Seibert, K.; Isakson, P.C. Kinetic basis for selective inhibition of cyclo-oxygenases. Biochem. J. 1999, 339, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, M.; Percival, M.D. Effect of inhibitor time-dependency on selectivity towards cyclooxygenase isoforms. Biochem. J. 1995, 306, 247–251. [Google Scholar]
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: A full in vitro analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 7563–7568. [Google Scholar]
- Brooks, P.; Emery, P.; Evans, J.F.; Fenner, H.; Hawkey, C.J.; Patrono, C.; Smolen, J.; Breedveld, F.; Day, R.; Dougados, M.; Ehrich, E.W.; Gijon-Baños, J.; Kvien, T.K.; Van Rijswijk, M.H.; Warner, T.; Zeidler, H. Interpreting the clinical significance of the differential inhibition of cyclooxygenase-1 and cyclooxygenase-2. Rheumatology 1999, 38, 779–788. [Google Scholar] [PubMed]
- Ouellet, M.; Riendeau, D.; Percival, M.D. A high level of cyclooxygenase-2 inhibitor selectivity is associated with a reduced interference of platelet cyclooxygenase-1 inactivation by aspirin. Proc. Nat. Acad. Sci. USA 2001, 98, 14583–14588. [Google Scholar]
- Callan, O.H.; So, O.-Y.; Swinney, D.C. The kinetic factors that determine the affinity and selectivity for slow binding inhibition of human prostaglandin H synthase 1 and 2 by indomethacin and flurbiprofen. J. Biol. Chem. 1996, 271, 3548–3554. [Google Scholar]
- Mitchell, J.A.; Akarasereenont, P.; Thiemermann, C.; Flower, R.J.; Vane, J.R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. USA 1994, 90, 11693–11697. [Google Scholar]
- Kargman, A.; Wong, E.; Greig, G.M.; Falgueyret, J.P.; Cromlish, W.; Ethier, D.; Yergey, J.A.; Riendeau, D.; Evans, J.F.; Kennedy, B.; Tagari, P.; Francis, D.A.; O'Neill, G.P. Mechanism of selective inhibition of human prostaglandin G/H synthase-1 and -2 in intact cells. Biochem. Pharmacol. 1996, 52, 1113–1125. [Google Scholar]
- Burch, J.W.; Baenziger, N.L.; Stanford, N.; Majerus, P.W. Sensitivity of fatty acid cyclooxygenase from human aorta to acetylation by aspirin. Proc. Natl. Acad. Sci. USA 1978, 75, 5181–5184. [Google Scholar]
- Rome, L.H.; Lands, W.E.M. Structural requirements for time-dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc. Nat. Acad. Sci. USA 1975, 72, 4863–4865. [Google Scholar]
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Stronger inhibition by nonsteroid anti-inflammatory drugs of cyclooxygenase-1 in endothelial cells than platelets offers an explanation for increased risk of thrombotic events. FASEB J. 2006, 20, 2468–2475. [Google Scholar]
- Grosser, T.; Fries, A.; FitzGerald, G.A. Biological basis for the cardiovascular consequences of COX-2 inhibition: Therapeutic challenges and opportunities. J. Clin. Invest. 2006, 116, 4–15. [Google Scholar]
- Sciulli, M.G.; Renda, G.; Capone, M.L.; Tacconelli, S.; Ricciotti, E.; Manarini, S.; Evangelista, V.; Rebuzzi, A.; Patrignani, P. Heterogeneity in the suppression of platelet cyclooxygenase-1 activity by aspirin in coronary heart disease. Clin. Pharmacol. Ther. 2006, 80, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dor, I.; Kleiman, N.S.; Lev, E. Assessment, mechanisms, and clinical implication of variability in platelet response to aspirin and clopidogrel therapy. Am. J. Cardiol. 2009, 104, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Folts, J.D.; Schafer, A.I.; Loscalzo, J.; Willerson, J.T.; Muller, J.E. A perspective on the potential problems with aspirin as an antithrombotic agent: A comparison of studies in an animal model with clinical trials. J. Am. Coll. Cardiol. 1999, 33, 295–303. [Google Scholar]
- Steinhubl, S.R.; Charnigo, R.; Moliterno, D.J. Resistance to antiplatelet resistance is it justified? J. Am. Coll. Cardiol. 2005, 45, 1757–1758. [Google Scholar] [PubMed]
- Steinhubl, S.R. The use of anti-inflammatory analgesics in the patient with cardiovascular disease: What a pain. J. Am. Coll. Cardiol. 2005, 45, 1302–1303. [Google Scholar]
- Blobaum, A.L.; Marnett, L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar]
- Huntjens, D.R.; Danhof, M.; Della Pasqua, O.E. Pharmacokinetic-pharmacodynamic correlations and biomarkers in the development of COX-2 inhibitors. Rheumatology 2005, 44, 846–859. [Google Scholar]
- Capone, M.L.; Sciulli, M.G.; Tacconelli, S.; Grana, M.; Ricciotti, E.; Renda, G.; Di Gregorio, P.; Merciaro, G.; Patrignani, P. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J. Am. Coll. Cardiol. 2005, 45, 1295–1301. [Google Scholar]
- Curtis, J.P.; Krumholz, H.M. The case for an adverse interaction between aspirin and non-steroidal anti-inflammatory drugs: Is it time to believe the hype? J. Am. Coll. Cardiol. 2004, 43, 991–993. [Google Scholar] [PubMed]
- Patrono, C.; García Rodríguez, L.A.; Landolfi, R.; Baigent, C. Low-dose aspirin for the prevention of atherothrombosis. N. Engl. J. Med. 2005, 353, 2373–2383. [Google Scholar]
- Goltsov, A.; Maryashkin, A.; Swat, M.; Kosinsky, Y.; Humphery-Smith, I.; Demin, O.; Goryanin, I.; Lebedeva, G. Kinetic modelling of NSAID action on COX-1: Focus on in vitro/in vivo aspects and drug combinations. Eur. J. Pharm. Sci. 2009, 36, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Dietz, R.; Nastainczyk, W.; Ruf, H.H. Higher oxidation states of prostaglandin H synthase. Rapid electronic spectroscopy detected two spectral intermediates during the peroxidase reaction with prostaglandin G2. Eur. J. Biochem. 1988, 171, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Kulmacz, R.J.; Tsai, A.L. Comparison of branched-chain and tightly coupled reaction mechanisms for prostaglandin H synthase. Biochem. 1995, 34, 8499–8512. [Google Scholar]
- Lu, G.; Tsai, A.L.; Van Wart, H.E.; Kulmacz, R.J. Comparison of the peroxidase reaction kinetics of prostaglandin H synthase-1 and -2. J. Biol. Chem. 1999, 74, 16162–16177. [Google Scholar]
- Wei, C.; Timothy, R.; Pawelek; Kulmacz, R.J. Hydroperoxide dependence and cooperative cyclooxygenase kinetics in prostaglandin H synthase-1 and -2. J. Biol. Chem. 1999, 274, 20301–20306. [Google Scholar] [PubMed]
- Bambai, B.; Rogge, C.E.; Stec, B.; Kulmacz, R.J. Role of Asn-382 and Thr-383 in activation and inactivation of human prostaglandin H synthase cyclooxygenase catalysis. J. Biol. Chem. 2004, 279, 4084–4092. [Google Scholar]
- Bambai, B.; Kulmacz, R.J. Prostaglandin H synthase. Effects of peroxidase cosubstrates on cyclooxygenase velocity. J. Biol. Chem. 2000, 275, 27608–27614. [Google Scholar] [PubMed]
- Wu, G.; Kulmacz, R.J.; Tsai, A.L. Cyclooxygenase inactivation kinetics during reaction of prostaglandin H synthase-1 with peroxide. Biochem. 2003, 42, 13772–13777. [Google Scholar]
- Wu, G.; Wei, C.; Kulmacz, R.J.; Osawa, Y.; Tsai, A.L. A mechanistic study of self-inactivation of the peroxidase activity in prostaglandin H synthase-1. J. Biol. Chem. 1999, 274, 9231–9237. [Google Scholar]
- Song, I.; Ball, T.M.; Smith, W.L. Different suicide inactivation processes for the peroxidase and cyclooxygenase activities of prostaglandin endoperoxide H synthase-1. Biochem. Biophys. Res. Commun. 2001, 289, 869–875. [Google Scholar]
- Kulmacz, R.J. Topography of prostaglandin H synthase. J. Biol. Chem. 1989, 264, 14136–14144. [Google Scholar]
- Kulmacz, R.J.; Palmer, G.; Tsai, A.L. Prostaglandin H synthase: Perturbation of the tyrosyl radical as a probe of anticyclooxygenase agents. Mol. Pharmacol. 1991, 40, 833–837. [Google Scholar]
- Lassmann, G.; Odenwaller, R.; Curtis, J.F.; DeGray, J.A.; Mason, R.P.; Marnett, L.J.; Eling, T.E. Electron spin resonance investigation of tyrosyl radicals of prostaglandin H synthase. Relation to enzyme catalysis. J. Biol. Chem. 1991, 266, 20045–20055. [Google Scholar] [PubMed]
- Goryanin, I.; Hodgman, T.C.; Selkov, E. Mathematical simulation and analysis of cellular metabolism and regulation. Bioinformatics 1999, 15, 749–758. [Google Scholar]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Invest. 2001, 107, 1339–1345. [Google Scholar]
- Sergeeva, M.G.; Varfolomeeva, A.T. Cascade of Arachidonic Acid; Public Education: Moscow, Russian, 2006. [Google Scholar]
- Hazelton, W.D.; Tien, J.H.; Donato, V.W.; Sparks, R.; Ulrich, C.M. Prostaglandin H synthases: Members of a class of quasi-linear threshold switches. Biochem. Pharmacol. 2004, 68, 423–432. [Google Scholar]
- Kulmacz, R.J.; Pendleton, R.B.; Lands, W.E. Interaction between peroxidase and cyclooxygenase activities in prostaglandin-endoperoxide synthase. Interpretation of reaction kinetics. J. Biol. Chem. 1994, 269, 5527–5536. [Google Scholar] [PubMed]
- Mbonye, U.R.; Wada, M.; Rieke, C.J.; Tang, H.Y.; Dewitt, D.L.; Smith, W.L. The 19-amino acid cassette of cyclooxygenase-2 mediates entry of the protein into the endoplasmic reticulum-associated degradation system. J. Biol. Chem. 2006, 281, 35770–35778. [Google Scholar]
- Mancini, J.A.; Riendea, D.; Falgueyret, J.P.; Vickers, P.J.; O'Neill, G.P. Arginine 120 of prostaglandin G/H synthase-1 is required for the inhibition by nonsteroidal anti-inflammatory drugs containing a carboxylic acid moiety. J. Biol. Chem. 1995, 270, 29372–29377. [Google Scholar]
- Gierse, J.K.; Hauser, S.D.; Creely, D.P.; Koboldt, C.; Rangwala, S.H.; Isakson, P.C.; Seibert, K. Expression and selective inhibition of the constitutive and inducible forms of human cyclo-oxygenase. Biochem. J. 1995, 305, 479–484. [Google Scholar]
- Chulada, P.C.; Langenbach, R. Differential inhibition of murine prostaglandin synthase-1 and -2 by nonsteroidal anti-inflammatory drugs using exogenous and endogenous sources of arachidonic acid. J. Pharmacol. Exp. Ther. 1997, 280, 606–613. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goltsov, A.; Lebedeva, G.; Humphery-Smith, I.; Goltsov, G.; Demin, O.; Goryanin, I. In Silico Screening of Nonsteroidal Anti-Inflammatory Drugs and Their Combined Action on Prostaglandin H Synthase-1. Pharmaceuticals 2010, 3, 2059-2081. https://doi.org/10.3390/ph3072059
Goltsov A, Lebedeva G, Humphery-Smith I, Goltsov G, Demin O, Goryanin I. In Silico Screening of Nonsteroidal Anti-Inflammatory Drugs and Their Combined Action on Prostaglandin H Synthase-1. Pharmaceuticals. 2010; 3(7):2059-2081. https://doi.org/10.3390/ph3072059
Chicago/Turabian StyleGoltsov, Alexey, Galina Lebedeva, Ian Humphery-Smith, Gregory Goltsov, Oleg Demin, and Igor Goryanin. 2010. "In Silico Screening of Nonsteroidal Anti-Inflammatory Drugs and Their Combined Action on Prostaglandin H Synthase-1" Pharmaceuticals 3, no. 7: 2059-2081. https://doi.org/10.3390/ph3072059