Jak2 Tyrosine Kinase: A Potential Therapeutic Target for AT1 Receptor Mediated Cardiovascular Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
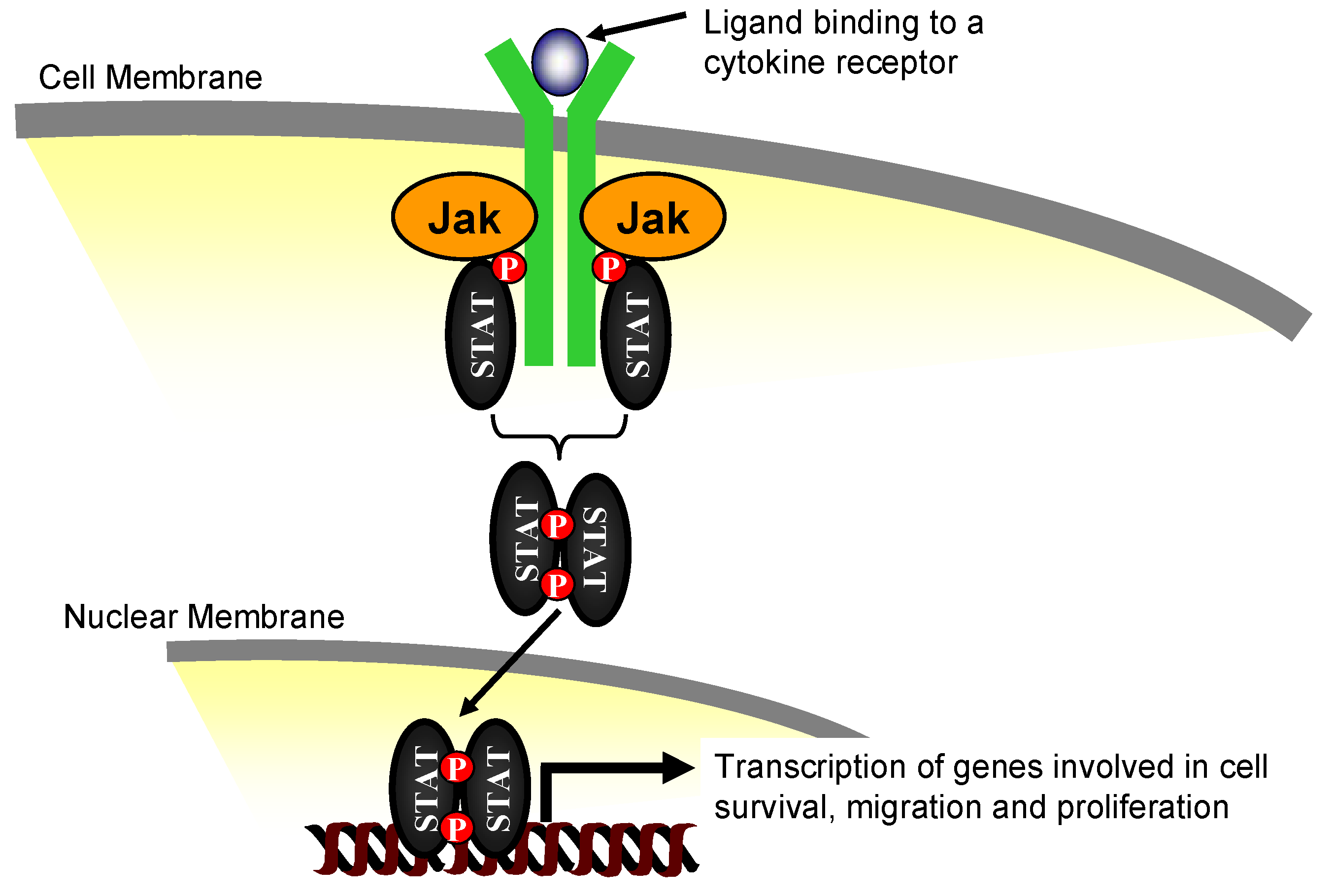
2. The Janus Kinase Family of Proteins
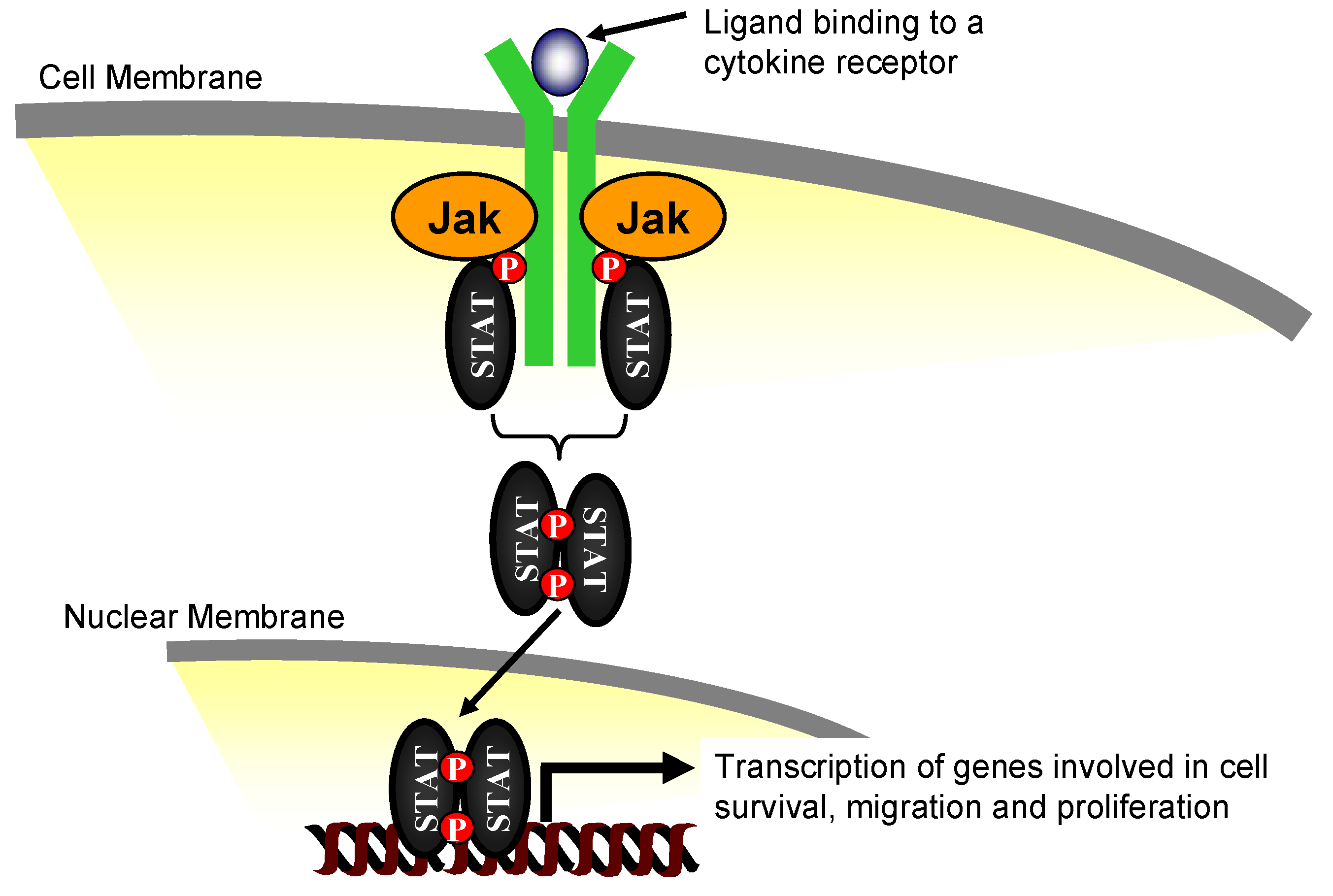
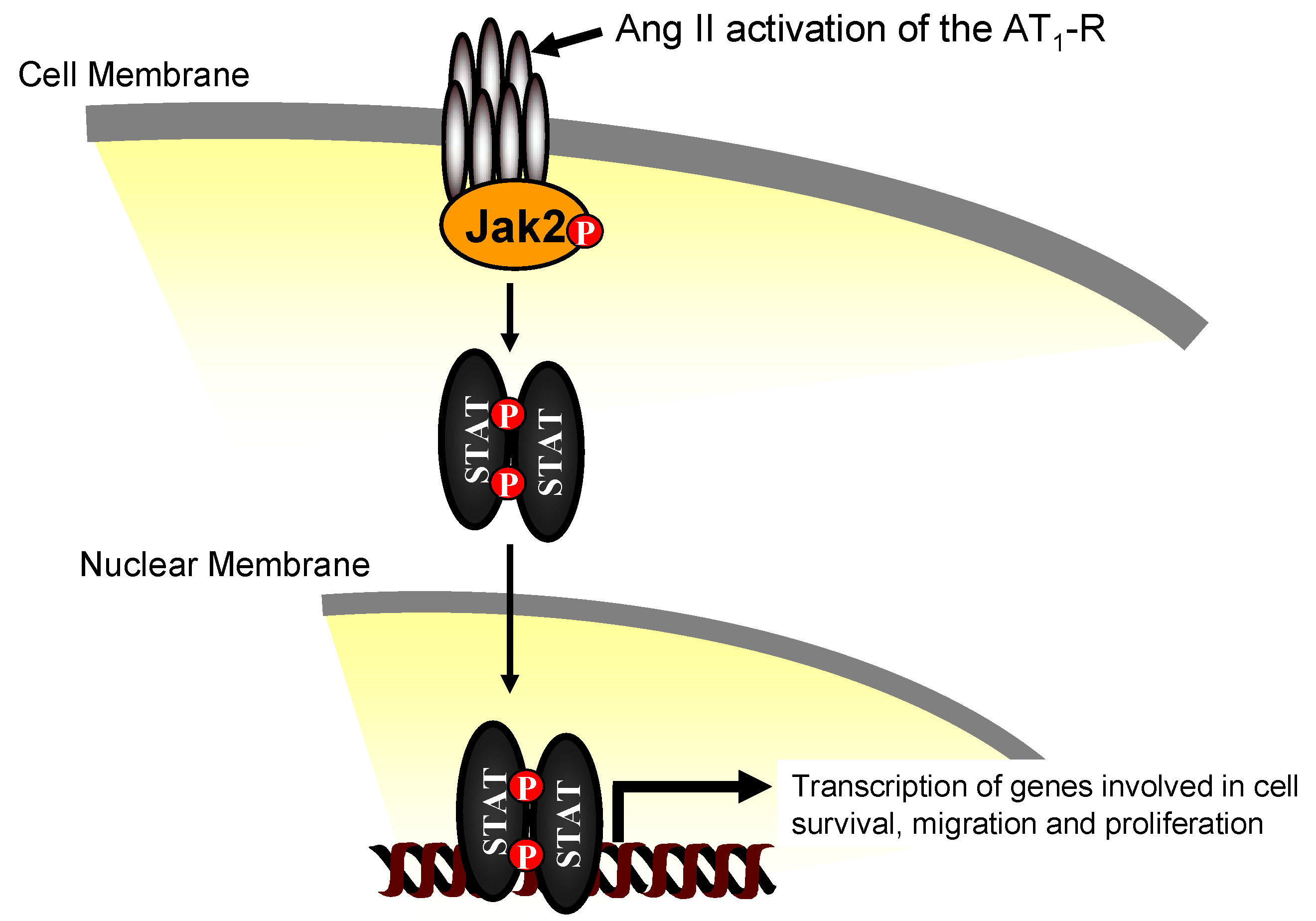
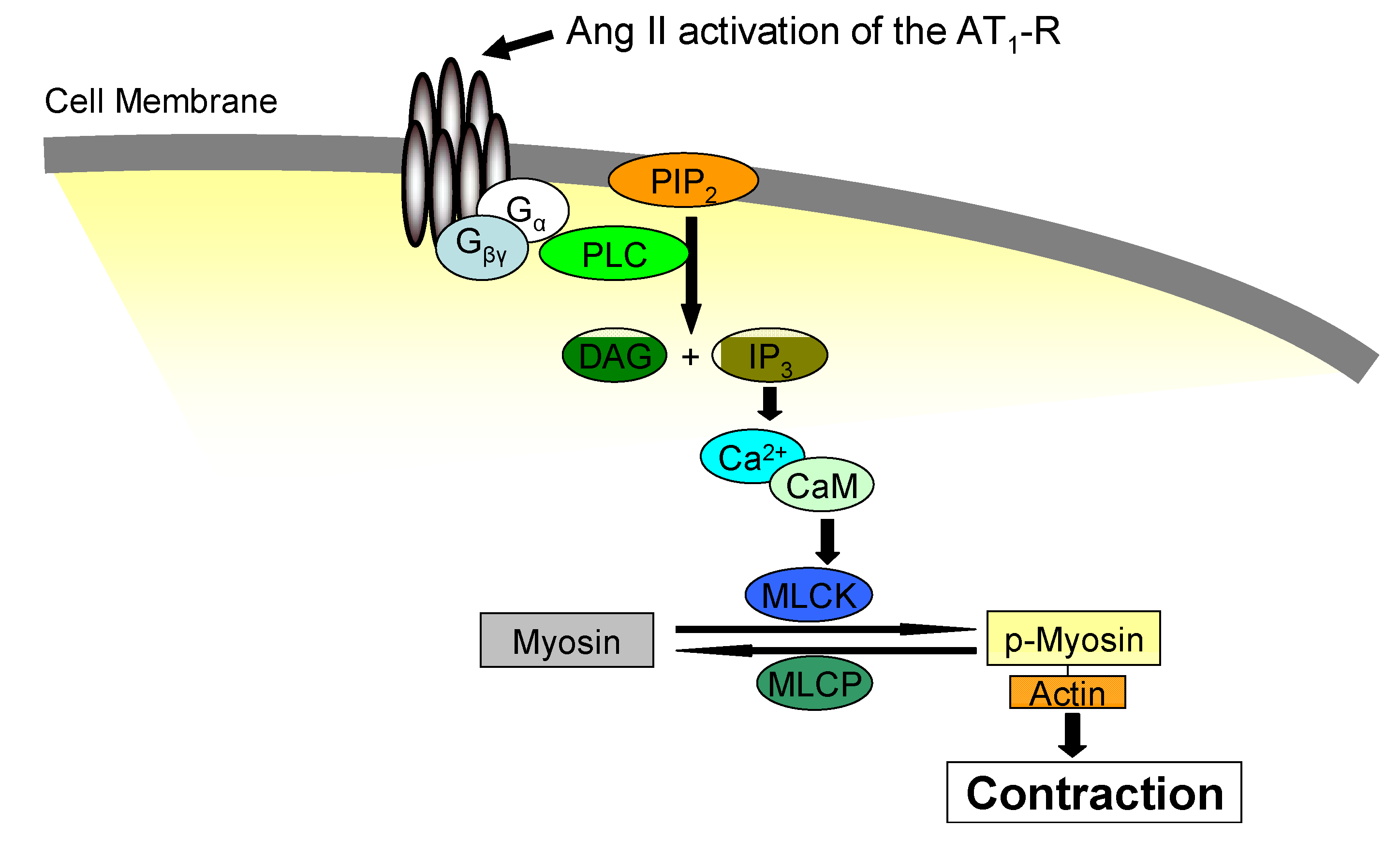
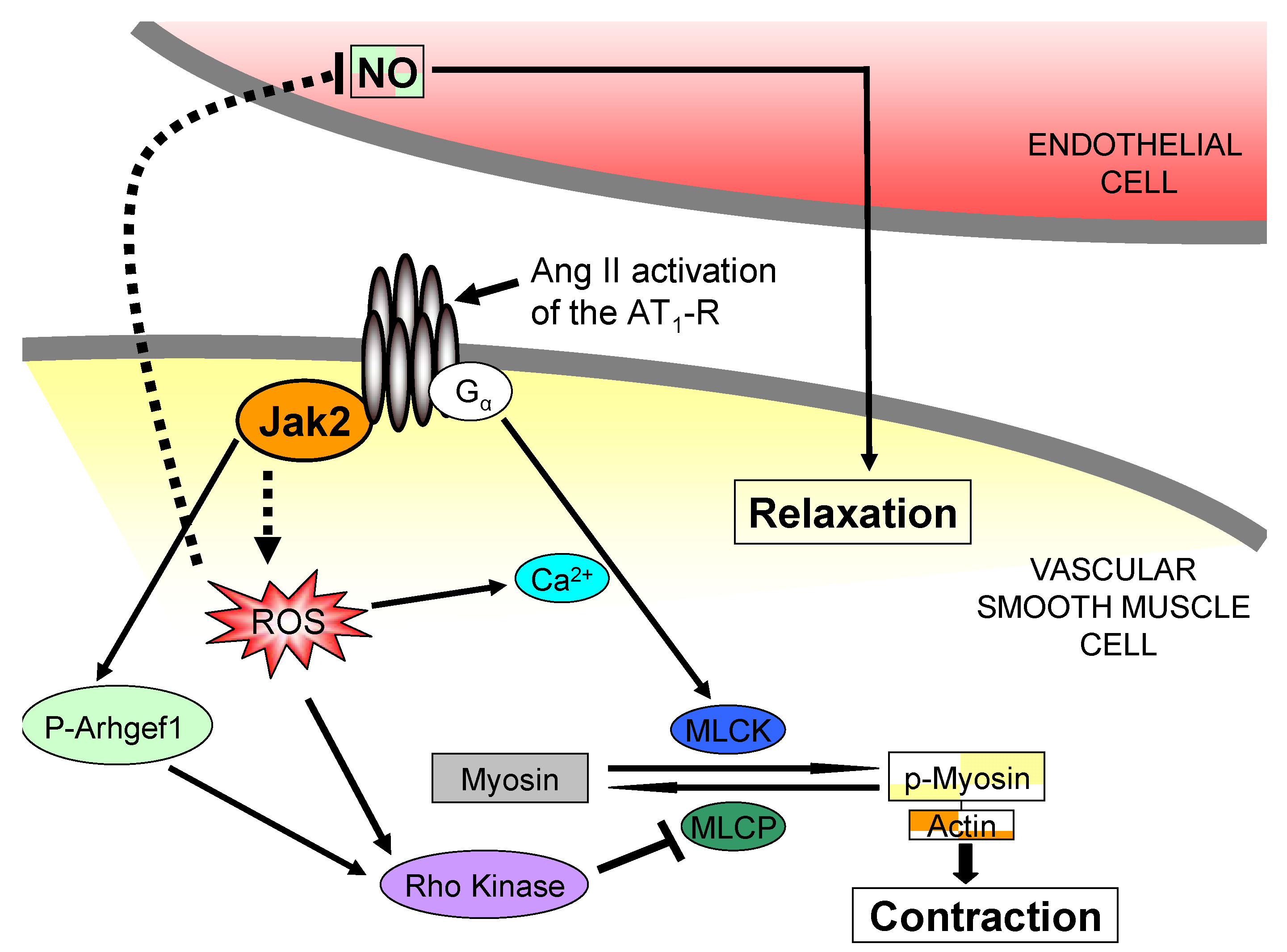
3. Jak2 in Angiotensin II-Induced Cardiovascular Disease

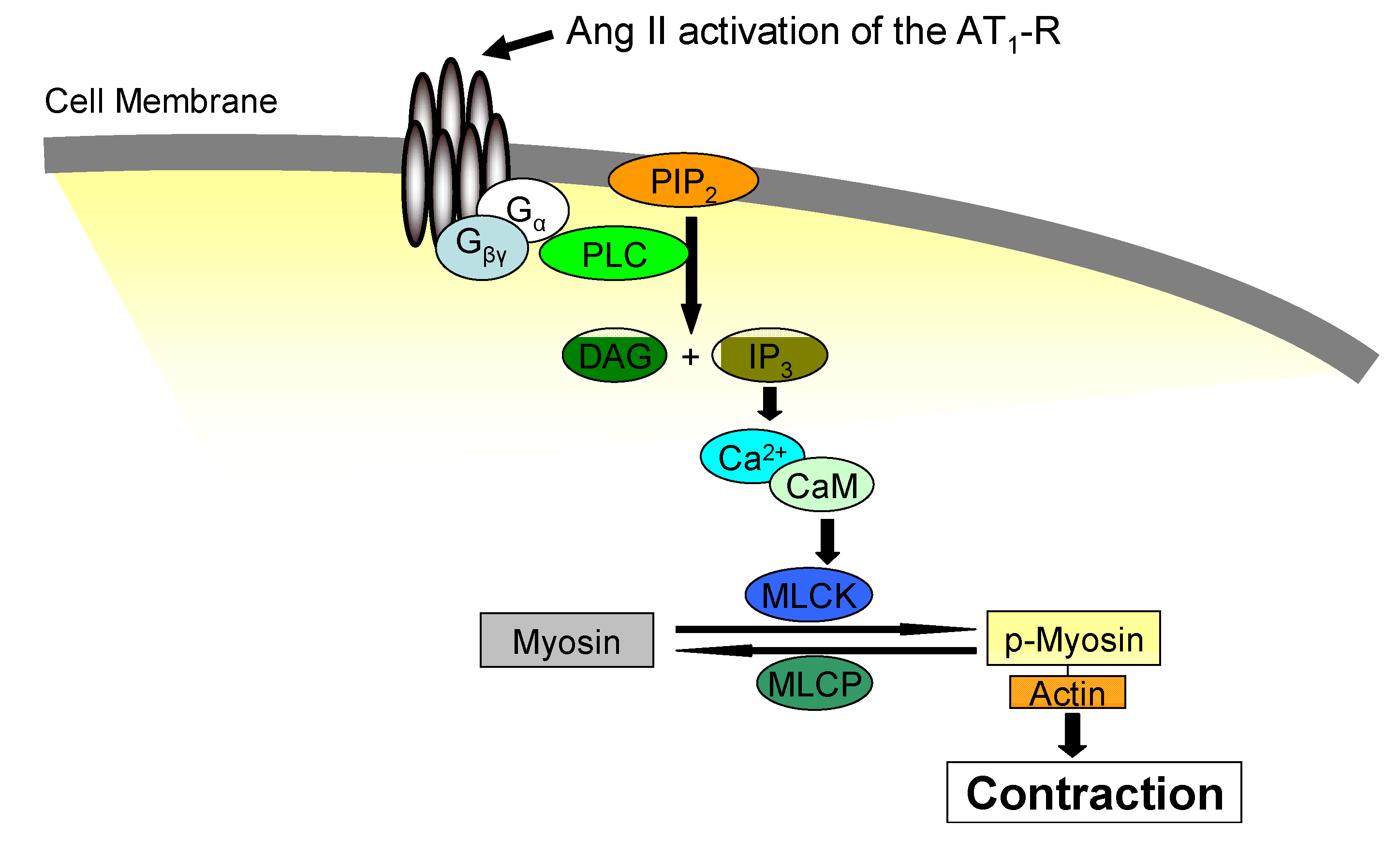
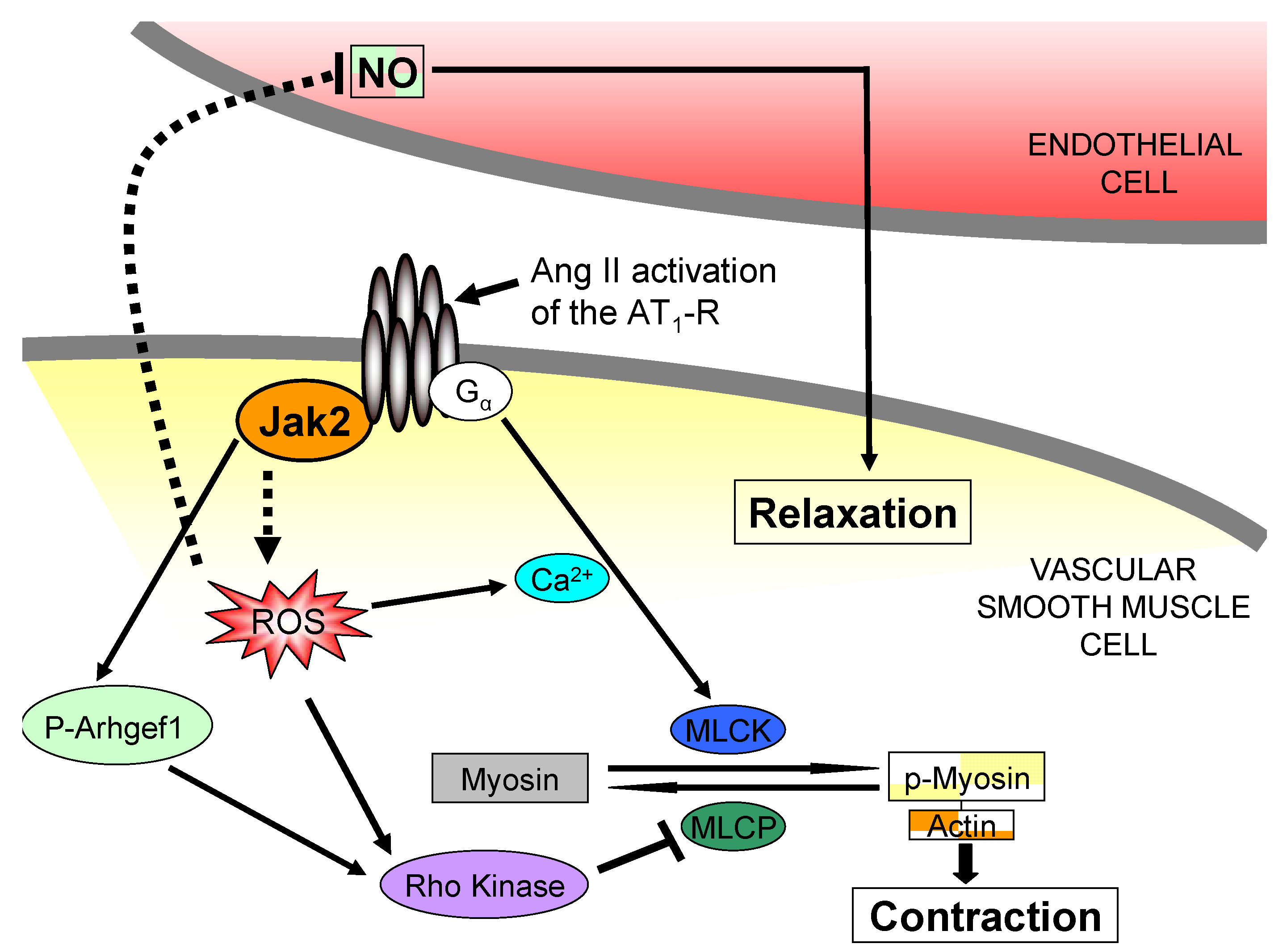
4. Pharmacological Jak2 Inhibition: An Emerging Therapeutic Strategy in Jak2-Mediated Diseases
5. Jak2 Inhibitors and Their Potential for Cardiovascular Disease Therapy
6. Conclusions
References
- Hansson, L.; Zanchetti, A.; Carruthers, S.G.; Dahlof, B.; Elmfeldt, D.; Julius, S.; Menard, J.; Rahn, K.H.; Wedel, H.; Westerling, S. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998, 351, 1755–1762. [Google Scholar]
- Duncia, J.V.; Carini, D.J.; Chiu, A.T.; Johnson, A.L.; Price, W.A.; Wong, P.C.; Wexler, R.R.; Timmermans, P.B. The discovery of DuP 753, a potent, orally active nonpeptide angiotensin II receptor antagonist. Med. Res. Rev. 1992, 12, 149–191. [Google Scholar]
- Chiu, A.T.; McCall, D.E.; Nguyen, T.T.; Carini, D.J.; Duncia, J.V.; Herblin, W.F.; Uyeda, R.T.; Wong, P.C.; Wexler, R.R.; Johnson, A.L.; et al. Discrimination of angiotensin II receptor subtypes by dithiothreitol. Eur. J. Pharmacol. 1989, 170, 117–118. [Google Scholar]
- Chiu, A.T.; McCall, D.E.; Price, W.A., Jr.; Wong, P.C.; Carini, D.J.; Duncia, J.V.; Wexler, R.R.; Yoo, S.E.; Johnson, A.L.; Timmermans, P.B. In vitro pharmacology of DuP 753. Am. J. Hypertens. 1991, 4, 282S–287S. [Google Scholar]
- Wong, P.C.; Price, W.A., Jr.; Chiu, A.T.; Duncia, J.V.; Carini, D.J.; Wexler, R.R.; Johnson, A.L.; Timmermans, P.B. In vivo pharmacology of DuP 753. Am. J. Hypertens. 1991, 4, 288S–298S. [Google Scholar]
- Matsubara, H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ. Res. 1998, 83, 1182–1191. [Google Scholar]
- Katz, A.M. Is angiotensin II a growth factor masquerading as a vasopressor? Heart Dis. Stroke 1992, 1, 151–154. [Google Scholar]
- Daemen, M.J.; Lombardi, D.M.; Bosman, F.T.; Schwartz, S.M. Angiotensin II induces smooth muscle cell proliferation in the normal and injured rat arterial wall. Circ. Res. 1991, 68, 450–456. [Google Scholar]
- Powell, J.S.; Clozel, J.P.; Muller, R.K.; Kuhn, H.; Hefti, F.; Hosang, M.; Baumgartner, H.R. Inhibitors of angiotensin-converting enzyme prevent myointimal proliferation after vascular injury. Science 1989, 245, 186–188. [Google Scholar]
- Chiu, A.T.; Roscoe, W.A.; McCall, D.E.; Timmermans, P.B. Angiotensin II-1 receptors mediate both vasoconstrictor and hypertrophic responses in rat aortic smooth muscle cells. Receptor 1991, 1, 133–140. [Google Scholar]
- Timmermans, P.B.; Wong, P.C.; Chiu, A.T.; Herblin, W.F.; Benfield, P.; Carini, D.J.; Lee, R.J.; Wexler, R.R.; Saye, J.A.; Smith, R.D. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 1993, 45, 205–251. [Google Scholar]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar]
- Xi, X.P.; Graf, K.; Goetze, S.; Fleck, E.; Hsueh, W.A.; Law, R.E. Central role of the MAPK pathway in ang II-mediated DNA synthesis and migration in rat vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 73–82. [Google Scholar]
- Berk, B.C.; Vekshtein, V.; Gordon, H.M.; Tsuda, T. Angiotensin II-stimulated protein synthesis in cultured vascular smooth muscle cells. Hypertension 1989, 13, 305–314. [Google Scholar]
- Fingerle, J.; Muller, R.M.; Kuhn, H.; Pech, M.; Baumgartner, H.R. Mechanism of inhibition of neointimal formation by the angiotensin-converting enzyme inhibitor cilazapril. A study in balloon catheter-injured rat carotid arteries. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1945–1950. [Google Scholar]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell. Physiol. 2007, 292, C82–C97. [Google Scholar]
- Su, E.J.; Lombardi, D.M.; Wiener, J.; Daemen, M.J.; Reidy, M.A.; Schwartz, S.M. Mitogenic effect of angiotensin II on rat carotid arteries and type II or III mesenteric microvessels but not type I mesenteric microvessels is mediated by endogenous basic fibroblast growth factor. Circ. Res. 1998, 82, 321–327. [Google Scholar]
- Griffin, S.A.; Brown, W.C.; MacPherson, F.; McGrath, J.C.; Wilson, V.G.; Korsgaard, N.; Mulvany, M.J.; Lever, A.F. Angiotensin II causes vascular hypertrophy in part by a non-pressor mechanism. Hypertension 1991, 17, 626–635. [Google Scholar]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E., 3rd; Silvennoinen, O.; O'Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar]
- Saharinen, P.; Vihinen, M.; Silvennoinen, O. Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol. Biol. Cell 2003, 14, 1448–1459. [Google Scholar]
- Luo, H.; Rose, P.; Barber, D.; Hanratty, W.P.; Lee, S.; Roberts, T.M.; D'Andrea, A.D.; Dearolf, C.R. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol. Cell. Biol. 1997, 17, 1562–1571. [Google Scholar]
- Kampa, D.; Burnside, J. Computational and functional analysis of the putative SH2 domain in Janus Kinases. Biochem. Biophys. Res. Commun. 2000, 278, 175–182. [Google Scholar]
- Girault, J.A.; Labesse, G.; Mornon, J.P.; Callebaut, I. Janus kinases and focal adhesion kinases play in the 4.1 band: a superfamily of band 4.1 domains important for cell structure and signal transduction. Mol. Med. 1998, 4, 751–769. [Google Scholar]
- Tanner, J.W.; Chen, W.; Young, R.L.; Longmore, G.D.; Shaw, A.S. The conserved box 1 motif of cytokine receptors is required for association with JAK kinases. J. Biol. Chem. 1995, 270, 6523–6530. [Google Scholar]
- Zhao, Y.; Wagner, F.; Frank, S.J.; Kraft, A.S. The amino-terminal portion of the JAK2 protein kinase is necessary for binding and phosphorylation of the granulocyte-macrophage colony-stimulating factor receptor beta c chain. J. Biol. Chem. 1995, 270, 13814–13818. [Google Scholar]
- Neubauer, H.; Cumano, A.; Muller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar]
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; Grosveld, G.; Ihle, J.N. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998, 93, 385–395. [Google Scholar]
- Bhat, G.J.; Thekkumkara, T.J.; Thomas, W.G.; Conrad, K.M.; Baker, K.M. Angiotensin II stimulates sis-inducing factor-like DNA binding activity. Evidence that the AT1A receptor activates transcription factor-Stat91 and/or a related protein. J. Biol. Chem. 1994, 269, 31443–31449. [Google Scholar]
- Marrero, M.B.; Schieffer, B.; Paxton, W.G.; Heerdt, L.; Berk, B.C.; Delafontaine, P.; Bernstein, K.E. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995, 375, 247–250. [Google Scholar]
- Marrero, M.B.; Schieffer, B.; Li, B.; Sun, J.; Harp, J.B.; Ling, B.N. Role of Janus kinase/signal transducer and activator of transcription and mitogen-activated protein kinase cascades in angiotensin II- and platelet-derived growth factor-induced vascular smooth muscle cell proliferation. J. Biol. Chem. 1997, 272, 24684–24690. [Google Scholar]
- Ali, M.S.; Sayeski, P.P.; Dirksen, L.B.; Hayzer, D.J.; Marrero, M.B.; Bernstein, K.E. Dependence on the motif YIPP for the physical association of Jak2 kinase with the intracellular carboxyl tail of the angiotensin II AT1 receptor. J. Biol. Chem. 1997, 272, 23382–23388. [Google Scholar]
- McWhinney, C.D.; Hunt, R.A.; Conrad, K.M.; Dostal, D.E.; Baker, K.M. The type I angiotensin II receptor couples to Stat1 and Stat3 activation through Jak2 kinase in neonatal rat cardiac myocytes. J. Mol. Cell. Cardiol. 1997, 29, 2513–2524. [Google Scholar]
- Dostal, D.E.; Hunt, R.A.; Kule, C.E.; Bhat, G.J.; Karoor, V.; McWhinney, C.D.; Baker, K.M. Molecular mechanisms of angiotensin II in modulating cardiac function: intracardiac effects and signal transduction pathways. J. Mol. Cell. Cardiol. 1997, 29, 2893–2902. [Google Scholar]
- Pan, J.; Fukuda, K.; Kodama, H.; Makino, S.; Takahashi, T.; Sano, M.; Hori, S.; Ogawa, S. Role of angiotensin II in activation of the JAK/STAT pathway induced by acute pressure overload in the rat heart. Circ. Res. 1997, 81, 611–617. [Google Scholar]
- Kodama, H.; Fukuda, K.; Pan, J.; Makino, S.; Sano, M.; Takahashi, T.; Hori, S.; Ogawa, S. Biphasic activation of the JAK/STAT pathway by angiotensin II in rat cardiomyocytes. Circ. Res. 1998, 82, 244–250. [Google Scholar]
- McWhinney, C.D.; Dostal, D.; Baker, K. Angiotensin II activates Stat5 through Jak2 kinase in cardiac myocytes. J. Mol. Cell. Cardiol. 1998, 30, 751–761. [Google Scholar]
- Frank, G.D.; Saito, S.; Motley, E.D.; Sasaki, T.; Ohba, M.; Kuroki, T.; Inagami, T.; Eguchi, S. Requirement of Ca(2+) and PKCdelta for Janus kinase 2 activation by angiotensin II: involvement of PYK2. Mol. Endocrinol. 2002, 16, 367–377. [Google Scholar]
- Marrero, M.B.; Paxton, W.G.; Schieffer, B.; Ling, B.N.; Bernstein, K.E. Angiotensin II signalling events mediated by tyrosine phosphorylation. Cell. Signal. 1996, 8, 21–26. [Google Scholar]
- Venema, R.C.; Venema, V.J.; Eaton, D.C.; Marrero, M.B. Angiotensin II-induced tyrosine phosphorylation of signal transducers and activators of transcription 1 is regulated by Janus-activated kinase 2 and Fyn kinases and mitogen-activated protein kinase phosphatase 1. J. Biol. Chem. 1998, 273, 30795–30800. [Google Scholar]
- Prescott, M.F.; Webb, R.L.; Reidy, M.A. Angiotensin-converting enzyme inhibitor versus angiotensin II, AT1 receptor antagonist. Effects on smooth muscle cell migration and proliferation after balloon catheter injury. Am. J. Pathol. 1991, 139, 1291–1296. [Google Scholar]
- Rakugi, H.; Jacob, H.J.; Krieger, J.E.; Ingelfinger, J.R.; Pratt, R.E. Vascular injury induces angiotensinogen gene expression in the media and neointima. Circulation 1993, 87, 283–290. [Google Scholar]
- Rakugi, H.; Kim, D.K.; Krieger, J.E.; Wang, D.S.; Dzau, V.J.; Pratt, R.E. Induction of angiotensin converting enzyme in the neointima after vascular injury. Possible role in restenosis. J. Clin. Invest. 1994, 93, 339–346. [Google Scholar]
- Iwai, N.; Izumi, M.; Inagami, T.; Kinoshita, M. Induction of renin in medial smooth muscle cells by balloon injury. Hypertension 1997, 29, 1044–1050. [Google Scholar]
- Viswanathan, M.; Stromberg, C.; Seltzer, A.; Saavedra, J.M. Balloon angioplasty enhances the expression of angiotensin II AT1 receptors in neointima of rat aorta. J. Clin. Invest. 1992, 90, 1707–1712. [Google Scholar]
- Sandberg, E.M.; Ma, X.; VonDerLinden, D.; Godeny, M.D.; Sayeski, P.P. Jak2 tyrosine kinase mediates angiotensin II-dependent inactivation of ERK2 via induction of mitogen-activated protein kinase phosphatase 1. J. Biol. Chem. 2004, 279, 1956–1967. [Google Scholar]
- Madamanchi, N.R.; Li, S.; Patterson, C.; Runge, M.S. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 321–326. [Google Scholar]
- Gharavi, N.M.; Alva, J.A.; Mouillesseaux, K.P.; Lai, C.; Yeh, M.; Yeung, W.; Johnson, J.; Szeto, W.L.; Hong, L.; Fishbein, M.; Wei, L.; Pfeffer, L.M.; Berliner, J.A. Role of the Jak/STAT pathway in the regulation of interleukin-8 transcription by oxidized phospholipids in vitro and in atherosclerosis in vivo. J. Biol. Chem. 2007, 282, 31460–31468. [Google Scholar]
- Seki, Y.; Kai, H.; Shibata, R.; Nagata, T.; Yasukawa, H.; Yoshimura, A.; Imaizumi, T. Role of the JAK/STAT pathway in rat carotid artery remodeling after vascular injury. Circ. Res. 2000, 87, 12–18. [Google Scholar]
- Macrez-Lepretre, N.; Kalkbrenner, F.; Morel, J.L.; Schultz, G.; Mironneau, J. G protein heterotrimer Galpha13beta1gamma3 couples the angiotensin AT1A receptor to increases in cytoplasmic Ca2+ in rat portal vein myocytes. J. Biol. Chem. 1997, 272, 10095–10102. [Google Scholar]
- Ushio-Fukai, M.; Griendling, K.K.; Akers, M.; Lyons, P.R.; Alexander, R.W. Temporal dispersion of activation of phospholipase C-beta1 and -gamma isoforms by angiotensin II in vascular smooth muscle cells. Role of alphaq/11, alpha12, and beta gamma G protein subunits. J. Biol. Chem. 1998, 273, 19772–19777. [Google Scholar]
- Rossier, M.F.; Capponi, A.M. Angiotensin II and calcium channels. Vitam Horm 2000, 60, 229–284. [Google Scholar]
- Penner, R.; Fasolato, C.; Hoth, M. Calcium influx and its control by calcium release. Curr. Opin. Neurobiol. 1993, 3, 368–374. [Google Scholar]
- Parekh, A.B. Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J. Physiol. 2003, 547, 333–348. [Google Scholar]
- Yan, C.; Kim, D.; Aizawa, T.; Berk, B.C. Functional interplay between angiotensin II and nitric oxide: cyclic GMP as a key mediator. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 26–36. [Google Scholar]
- Guilluy, C.; Bregeon, J.; Toumaniantz, G.; Rolli-Derkinderen, M.; Retailleau, K.; Loufrani, L.; Henrion, D.; Scalbert, E.; Bril, A.; Torres, R.M.; Offermanns, S.; Pacaud, P.; Loirand, G. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 2010, 16, 183–190. [Google Scholar] [Green Version]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar]
- Ohtsu, H.; Frank, G.D.; Utsunomiya, H.; Eguchi, S. Redox-dependent protein kinase regulation by angiotensin II: mechanistic insights and its pathophysiology. Antioxid. Redox Signal. 2005, 7, 1315–1326. [Google Scholar]
- Touyz, R.M. Reactive oxygen species and angiotensin II signaling in vascular cells - implications in cardiovascular disease. Braz. J. Med. Biol. Res. 2004, 37, 1263–1273. [Google Scholar]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Yin, Q.; Fujio, Y.; Walsh, K.; Griendling, K.K. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 22699–22704. [Google Scholar]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 1996, 97, 1916–1923. [Google Scholar]
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K.K. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension 1998, 32, 488–495. [Google Scholar]
- Ushio-Fukai, M.; Zafari, A.M.; Fukui, T.; Ishizaka, N.; Griendling, K.K. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J. Biol. Chem. 1996, 271, 23317–23321. [Google Scholar]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar]
- Zhang, H.; Schmeisser, A.; Garlichs, C.D.; Plotze, K.; Damme, U.; Mugge, A.; Daniel, W.G. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells: role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc. Res. 1999, 44, 215–222. [Google Scholar]
- Laursen, J.B.; Rajagopalan, S.; Galis, Z.; Tarpey, M.; Freeman, B.A.; Harrison, D.G. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation 1997, 95, 588–593. [Google Scholar]
- Harrison, D.G. Endothelial function and oxidant stress. Clin. Cardiol. 1997, 20, 11–17. [Google Scholar]
- Jernigan, N.L.; Walker, B.R.; Resta, T.C. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L515–L529. [Google Scholar]
- Gryglewski, R.J.; Palmer, R.M.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar]
- Rubanyi, G.M.; Vanhoutte, P.M. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am. J. Physiol. 1986, 250, H822–H827. [Google Scholar]
- Dzau, V.J. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension 2001, 37, 1047–1052. [Google Scholar]
- Jin, L.; Ying, Z.; Webb, R.C. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1495–H1500. [Google Scholar]
- Yasunari, K.; Kohno, M.; Kano, H.; Yokokawa, K.; Minami, M.; Yoshikawa, J. Antioxidants improve impaired insulin-mediated glucose uptake and prevent migration and proliferation of cultured rabbit coronary smooth muscle cells induced by high glucose. Circulation 1999, 99, 1370–1378. [Google Scholar]
- Walz, C.; Crowley, B.J.; Hudon, H.E.; Gramlich, J.L.; Neuberg, D.S.; Podar, K.; Griffin, J.D.; Sattler, M. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J. Biol. Chem. 2006, 281, 18177–18183. [Google Scholar]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar]
- Ihle, J.N. Cytokine receptor signalling. Nature 1995, 377, 591–594. [Google Scholar]
- Delhommeau, F.; Pisani, D.F.; James, C.; Casadevall, N.; Constantinescu, S.; Vainchenker, W. Oncogenic mechanisms in myeloproliferative disorders. Cell. Mol. Life Sci. 2006, 63, 2939–2953. [Google Scholar]
- Ma, X.; Vanasse, G.; Cartmel, B.; Wang, Y.; Selinger, H.A. Prevalence of polycythemia vera and essential thrombocythemia. Am. J. Hematol. 2008, 83, 359–362. [Google Scholar]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; Scott, M.A.; Erber, W.N.; Green, A.R. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; Villeval, J.L.; Constantinescu, S.N.; Casadevall, N.; Vainchenker, W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; Adelsperger, J.; Koo, S.; Lee, J.C.; Gabriel, S.; Mercher, T.; D'Andrea, A.; Frohling, S.; Dohner, K.; Marynen, P.; Vandenberghe, P.; Mesa, R.A.; Tefferi, A.; Griffin, J.D.; Eck, M.J.; Sellers, W.R.; Meyerson, M.; Golub, T.R.; Lee, S.J.; Gilliland, D.G. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar]
- Zhao, R.; Xing, S.; Li, Z.; Fu, X.; Li, Q.; Krantz, S.B.; Zhao, Z.J. Identification of an acquired JAK2 mutation in polycythemia vera. J. Biol. Chem. 2005, 280, 22788–22792. [Google Scholar]
- Jelinek, J.; Oki, Y.; Gharibyan, V.; Bueso-Ramos, C.; Prchal, J.T.; Verstovsek, S.; Beran, M.; Estey, E.; Kantarjian, H.M.; Issa, J.P. JAK2 mutation 1849G > T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood 2005, 106, 3370–3373. [Google Scholar]
- Jones, A.V.; Kreil, S.; Zoi, K.; Waghorn, K.; Curtis, C.; Zhang, L.; Score, J.; Seear, R.; Chase, A.J.; Grand, F.H.; White, H.; Zoi, C.; Loukopoulos, D.; Terpos, E.; Vervessou, E.C.; Schultheis, B.; Emig, M.; Ernst, T.; Lengfelder, E.; Hehlmann, R.; Hochhaus, A.; Oscier, D.; Silver, R.T.; Reiter, A.; Cross, N.C. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 2005, 106, 2162–2168. [Google Scholar]
- Levine, R.L.; Loriaux, M.; Huntly, B.J.; Loh, M.L.; Beran, M.; Stoffregen, E.; Berger, R.; Clark, J.J.; Willis, S.G.; Nguyen, K.T.; Flores, N.J.; Estey, E.; Gattermann, N.; Armstrong, S.; Look, A.T.; Griffin, J.D.; Bernard, O.A.; Heinrich, M.C.; Gilliland, D.G.; Druker, B.; Deininger, M.W. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 2005, 106, 3377–3379. [Google Scholar]
- Levine, R.L.; Pardanani, A.; Tefferi, A.; Gilliland, D.G. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 2007, 7, 673–683. [Google Scholar]
- Wernig, G.; Kharas, M.G.; Okabe, R.; Moore, S.A.; Leeman, D.S.; Cullen, D.E.; Gozo, M.; McDowell, E.P.; Levine, R.L.; Doukas, J.; Mak, C.C.; Noronha, G.; Martin, M.; Ko, Y.D.; Lee, B.H.; Soll, R.M.; Tefferi, A.; Hood, J.D.; Gilliland, D.G. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell 2008, 13, 311–320. [Google Scholar]
- Lipka, D.B.; Hoffmann, L.S.; Heidel, F.; Markova, B.; Blum, M.C.; Breitenbuecher, F.; Kasper, S.; Kindler, T.; Levine, R.L.; Huber, C.; Fischer, T. LS104, a non-ATP-competitive small-molecule inhibitor of JAK2, is potently inducing apoptosis in JAK2V617F-positive cells. Mol. Cancer Ther. 2008, 7, 1176–1184. [Google Scholar]
- Ferrajoli, A.; Faderl, S.; Van, Q.; Koch, P.; Harris, D.; Liu, Z.; Hazan-Halevy, I.; Wang, Y.; Kantarjian, H.M.; Priebe, W.; Estrov, Z. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007, 67, 11291–11299. [Google Scholar]
- Gozgit, J.M.; Bebernitz, G.; Patil, P.; Ye, M.; Parmentier, J.; Wu, J.; Su, N.; Wang, T.; Ioannidis, S.; Davies, A.; Huszar, D.; Zinda, M. Effects of the JAK2 inhibitor, AZ960, on Pim/BAD/BCL-xL survival signaling in the human JAK2 V617F cell line SET-2. J. Biol. Chem. 2008, 283, 32334–32343. [Google Scholar]
- Pardanani, A.; Lasho, T.; Smith, G.; Burns, C.J.; Fantino, E.; Tefferi, A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia 2009, 23, 1441–1445. [Google Scholar]
- Antonysamy, S.; Hirst, G.; Park, F.; Sprengeler, P.; Stappenbeck, F.; Steensma, R.; Wilson, M.; Wong, M. Fragment-based discovery of JAK-2 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 279–282. [Google Scholar]
- Sandberg, E.M.; Ma, X.; He, K.; Frank, S.J.; Ostrov, D.A.; Sayeski, P.P. Identification of 1,2,3,4,5,6-hexabromocyclohexane as a small molecule inhibitor of jak2 tyrosine kinase autophosphorylation. J. Med. Chem. 2005, 48, 2526–2533. [Google Scholar]
- Sayyah, J.; Magis, A.; Ostrov, D.A.; Allan, R.W.; Braylan, R.C.; Sayeski, P.P. Z3, a novel Jak2 tyrosine kinase small-molecule inhibitor that suppresses Jak2-mediated pathologic cell growth. Mol. Cancer Ther. 2008, 7, 2308–2318. [Google Scholar]
- Kiss, R.; Polgar, T.; Kirabo, A.; Sayyah, J.; Figueroa, N.C.; List, A.F.; Sokol, L.; Zuckerman, K.S.; Gali, M.; Bisht, K.S.; Sayeski, P.P.; Keseru, G.M. Identification of a novel inhibitor of JAK2 tyrosine kinase by structure-based virtual screening. Bioorg. Med. Chem. Lett. 2009, 19, 3598–3601. [Google Scholar]
- Pardanani, A.; Hood, J.; Lasho, T.; Levine, R.L.; Martin, M.B.; Noronha, G.; Finke, C.; Mak, C.C.; Mesa, R.; Zhu, H.; Soll, R.; Gilliland, D.G.; Tefferi, A. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 2007, 21, 1658–1668. [Google Scholar]
- Hexner, E.O.; Serdikoff, C.; Jan, M.; Swider, C.R.; Robinson, C.; Yang, S.; Angeles, T.; Emerson, S.G.; Carroll, M.; Ruggeri, B.; Dobrzanski, P. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood 2008, 111, 5663–5671. [Google Scholar]
- Tyner, J.W.; Bumm, T.G.; Deininger, J.; Wood, L.; Aichberger, K.J.; Loriaux, M.M.; Druker, B.J.; Burns, C.J.; Fantino, E.; Deininger, M.W. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010, 115, 5232–5240. [Google Scholar]
- Verstovsek, S. Therapeutic potential of JAK2 inhibitors. Hematology Am. Soc. Hematol. Educ. Program. 2009, 636–642. [Google Scholar]
- Geron, I.; Abrahamsson, A.E.; Barroga, C.F.; Kavalerchik, E.; Gotlib, J.; Hood, J.D.; Durocher, J.; Mak, C.C.; Noronha, G.; Soll, R.M.; Tefferi, A.; Kaushansky, K.; Jamieson, C.H. Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell 2008, 13, 321–330. [Google Scholar]
- Staessen, J.A.; Wang, J.; Bianchi, G.; Birkenhager, W.H. Essential hypertension. Lancet 2003, 361, 1629–1641. [Google Scholar]
- Acelajado, M.C.; Calhoun, D.A.; Oparil, S. Reduction of blood pressure in patients with treatment-resistant hypertension. Expert Opin. Pharmacother. 2009, 10, 2959–2971. [Google Scholar]
- Nesbitt, S.D. Overcoming therapeutic inertia in patients with hypertension. Postgrad. Med. 2010, 122, 118–124. [Google Scholar]
- Meydan, N.; Grunberger, T.; Dadi, H.; Shahar, M.; Arpaia, E.; Lapidot, Z.; Leeder, J.S.; Freedman, M.; Cohen, A.; Gazit, A.; Levitzki, A.; Roifman, C.M. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 1996, 379, 645–648. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kirabo, A.; Sayeski, P.P. Jak2 Tyrosine Kinase: A Potential Therapeutic Target for AT1 Receptor Mediated Cardiovascular Disease. Pharmaceuticals 2010, 3, 3478-3493. https://doi.org/10.3390/ph3113478
Kirabo A, Sayeski PP. Jak2 Tyrosine Kinase: A Potential Therapeutic Target for AT1 Receptor Mediated Cardiovascular Disease. Pharmaceuticals. 2010; 3(11):3478-3493. https://doi.org/10.3390/ph3113478
Chicago/Turabian StyleKirabo, Annet, and Peter P. Sayeski. 2010. "Jak2 Tyrosine Kinase: A Potential Therapeutic Target for AT1 Receptor Mediated Cardiovascular Disease" Pharmaceuticals 3, no. 11: 3478-3493. https://doi.org/10.3390/ph3113478
APA StyleKirabo, A., & Sayeski, P. P. (2010). Jak2 Tyrosine Kinase: A Potential Therapeutic Target for AT1 Receptor Mediated Cardiovascular Disease. Pharmaceuticals, 3(11), 3478-3493. https://doi.org/10.3390/ph3113478