Allosteric Inhibitors of NMDA Receptor Functions

Abstract
:
1. Introduction
2. NMDA Receptor Modulators as Potential Therapies
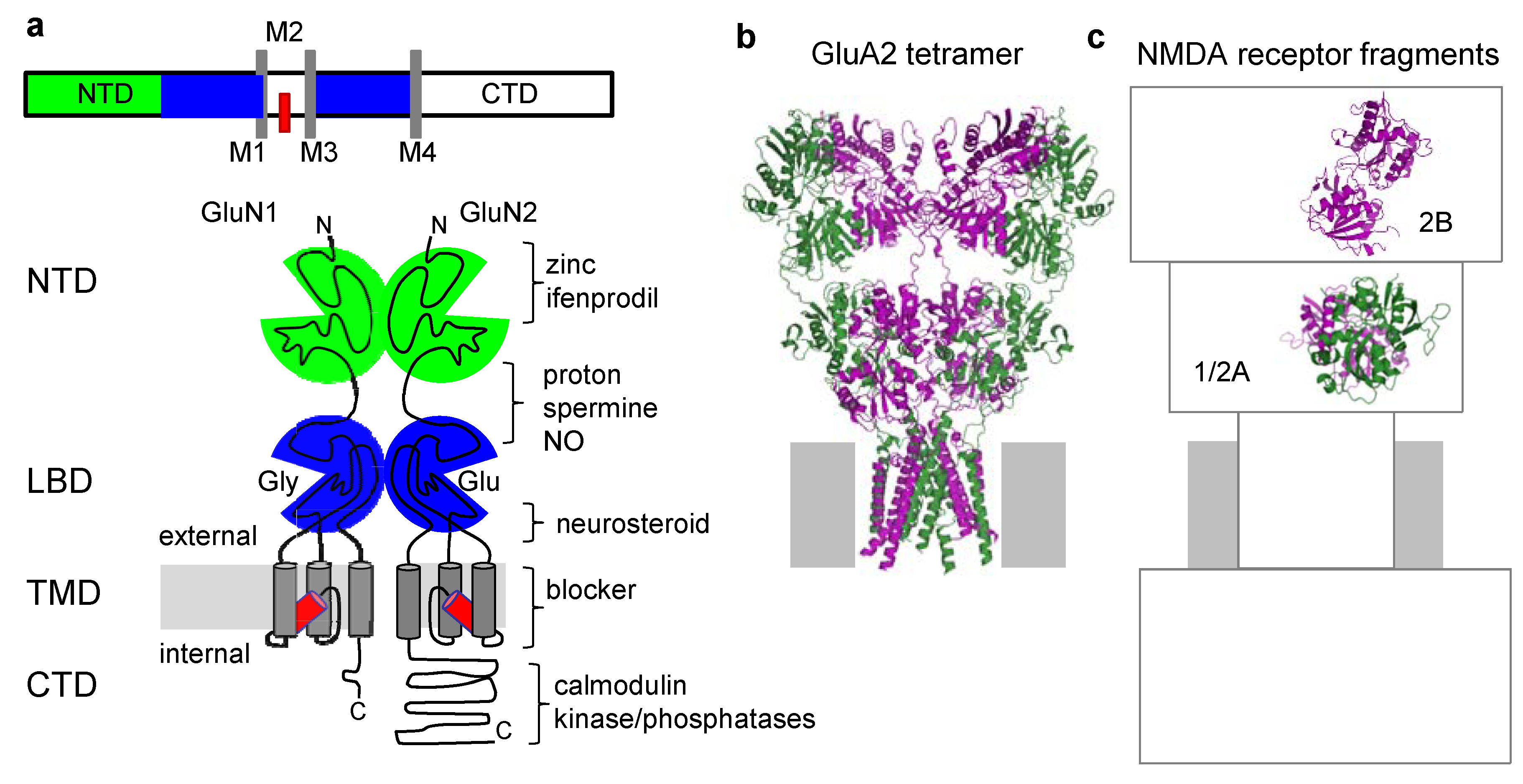
3. Structural Information of NMDA Receptor Allosteric Sites Is Limited
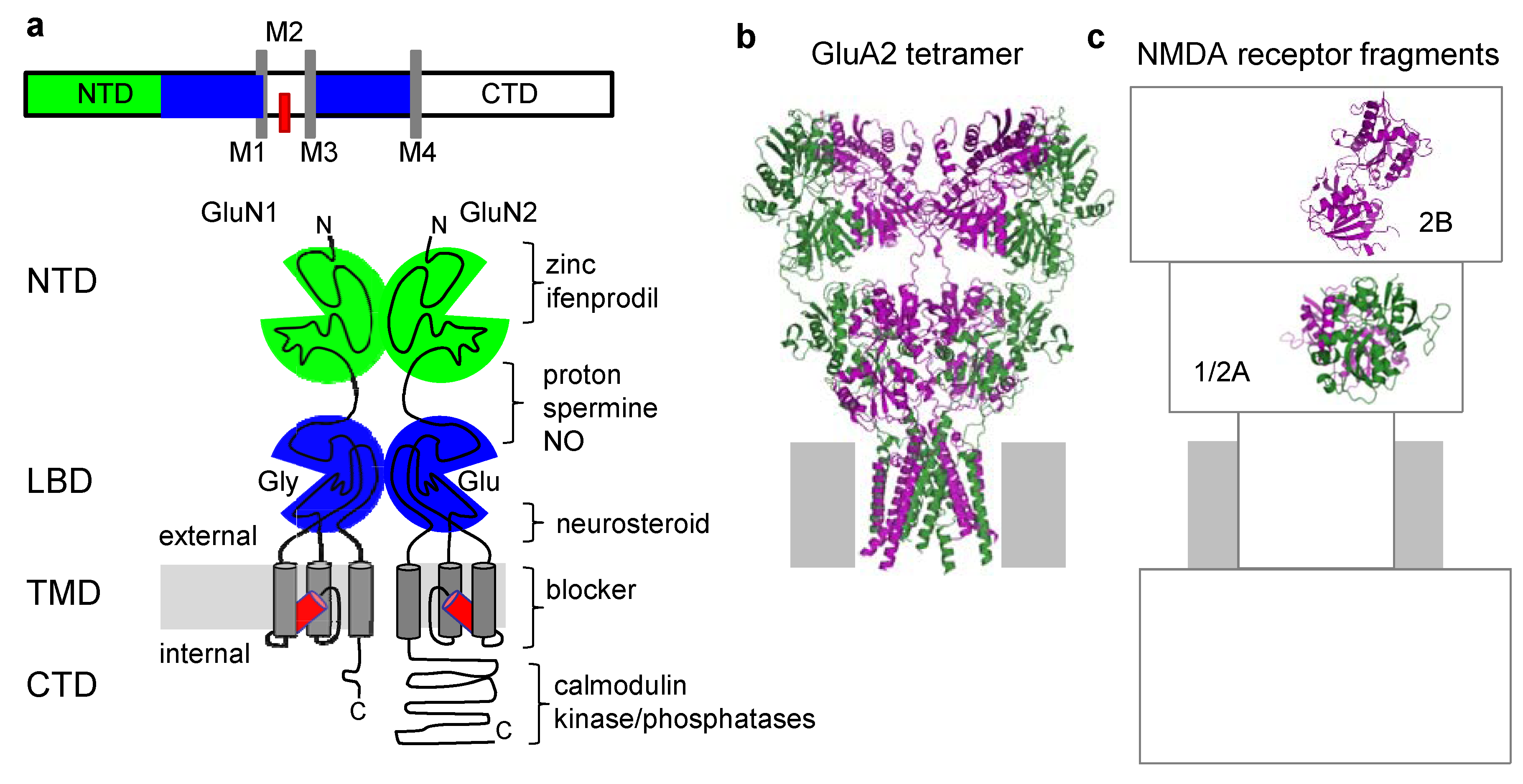

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Ligand | Pharmacological action | MMDB/PDB ID | Reference |
|---|---|---|---|---|
| GluN1-LBD | Glycine | co-agonist (neuromodulator) | 23627/1PB7 | [54,57] |
| co-agonist (neuromodulator) | 23628/1PB8 | [57] | ||
| partial agonist (synthetic) | 34203/1Y1Z | [58] | ||
| partial agonist (synthetic) | 34204/1Y20 | [58] | ||
| antagonist; (synthetic) | 34202/1Y1M | [58] | ||
| antagonist; (synthetic) | 24334/1PBQ | [57] | ||
| partial agonist; (synthetic) | 23629/1PB9 | [57] | ||
| GluN2A-LBD | Glutamate | agonist (neurotransmitter) | 36073/2A5S | [54] |
| GluN2B-NTD | Zinc | non competitive inhibitor (neuromodulator) | 78606/3JPY | [53] |
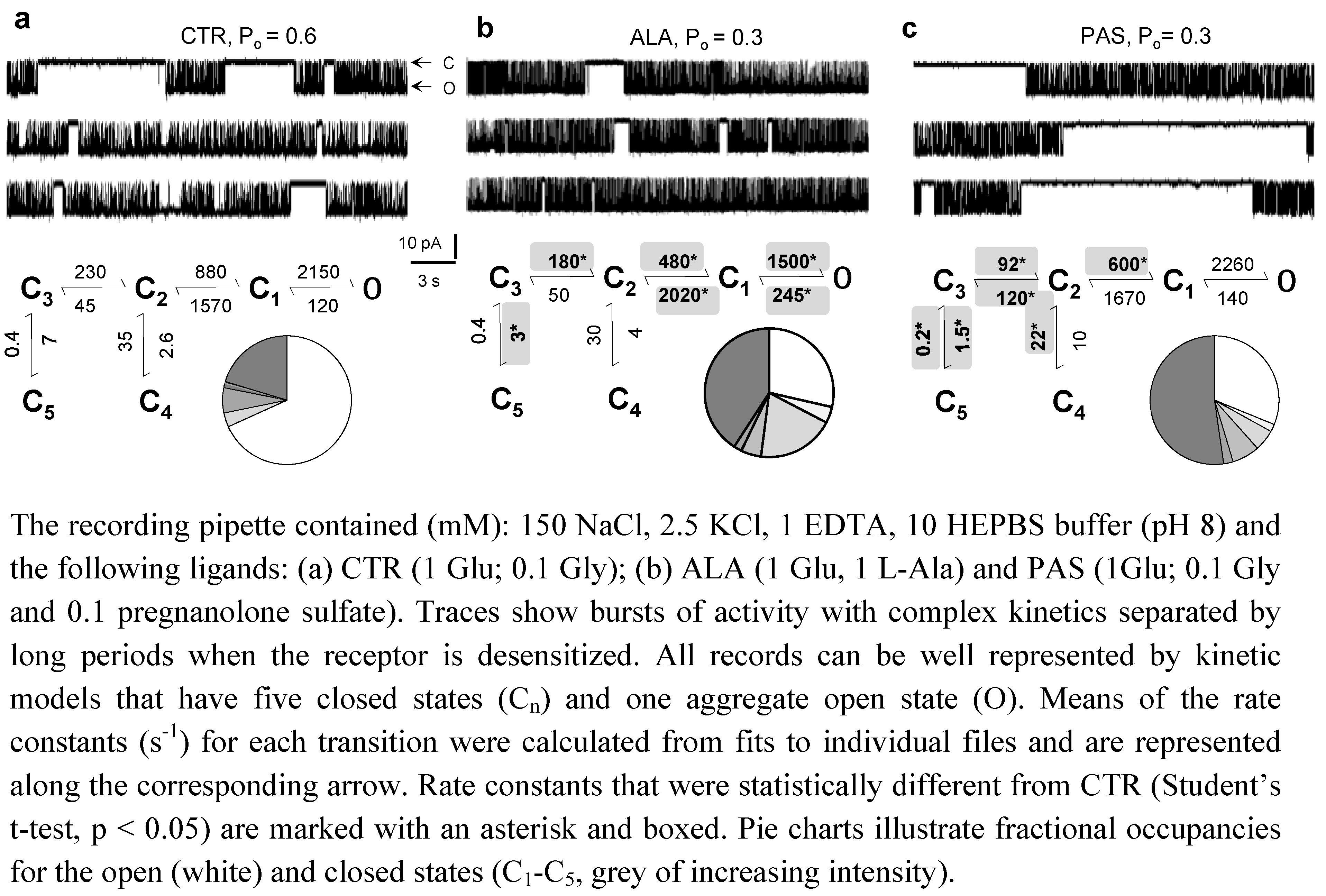
4. NMDA Receptors Have Multi-Step Gating Reactions
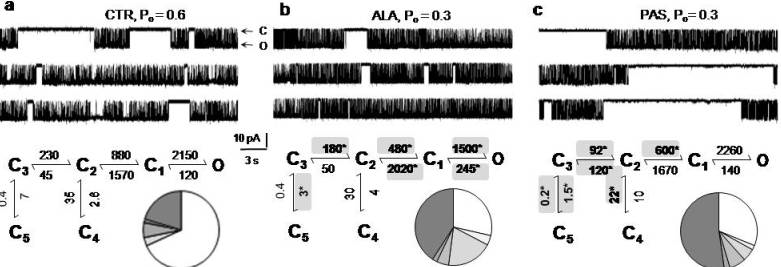
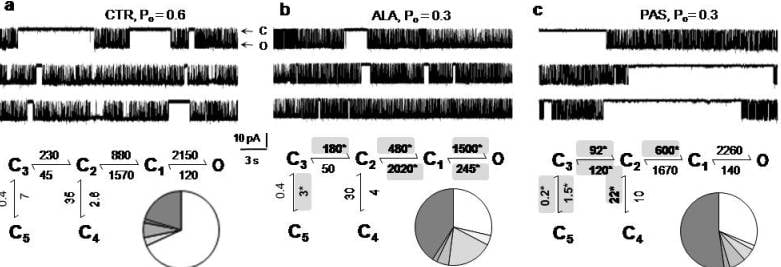
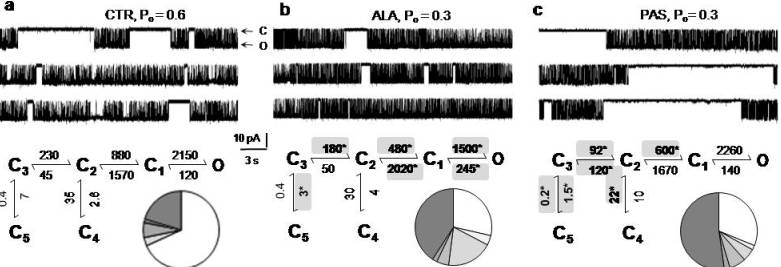
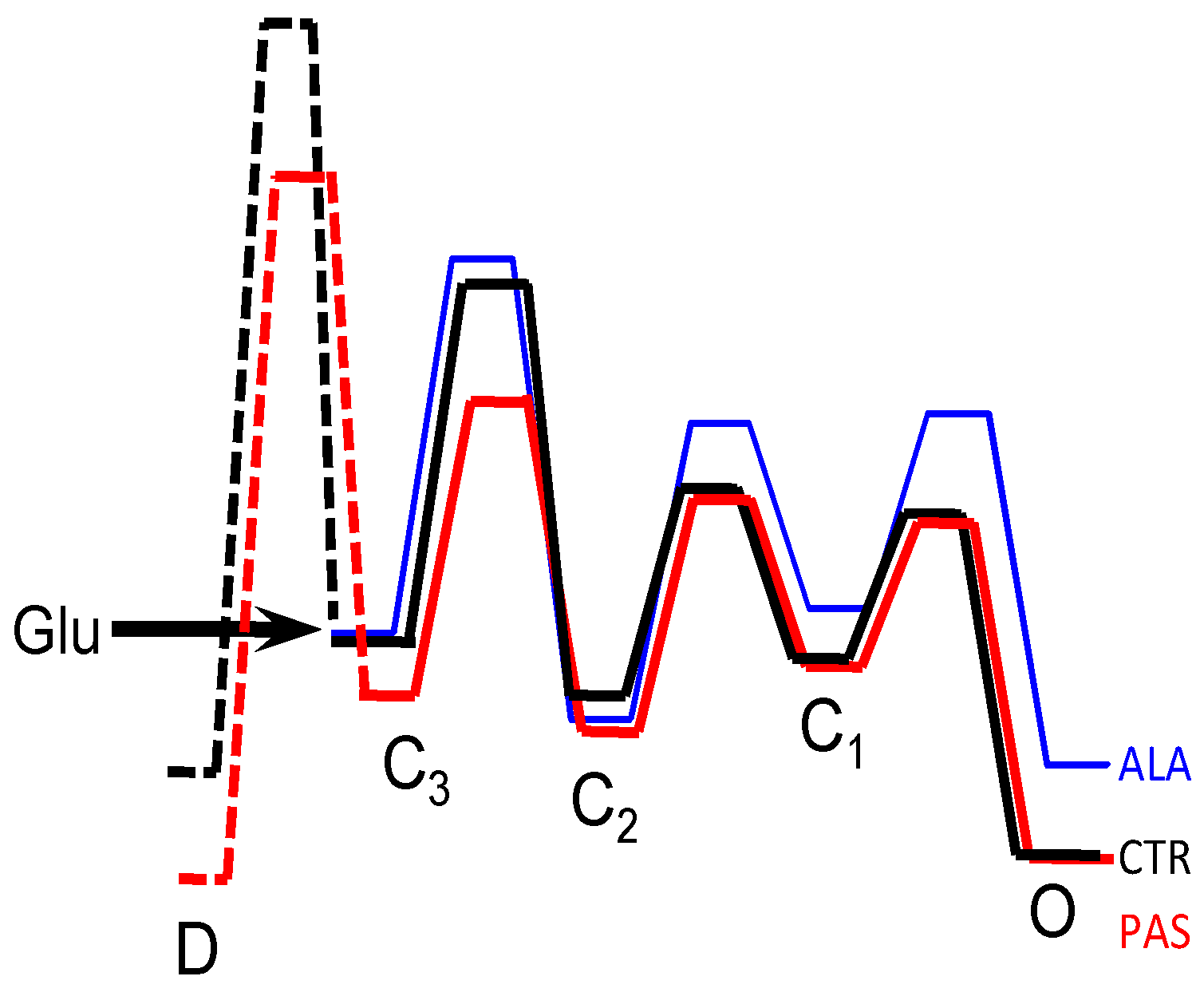
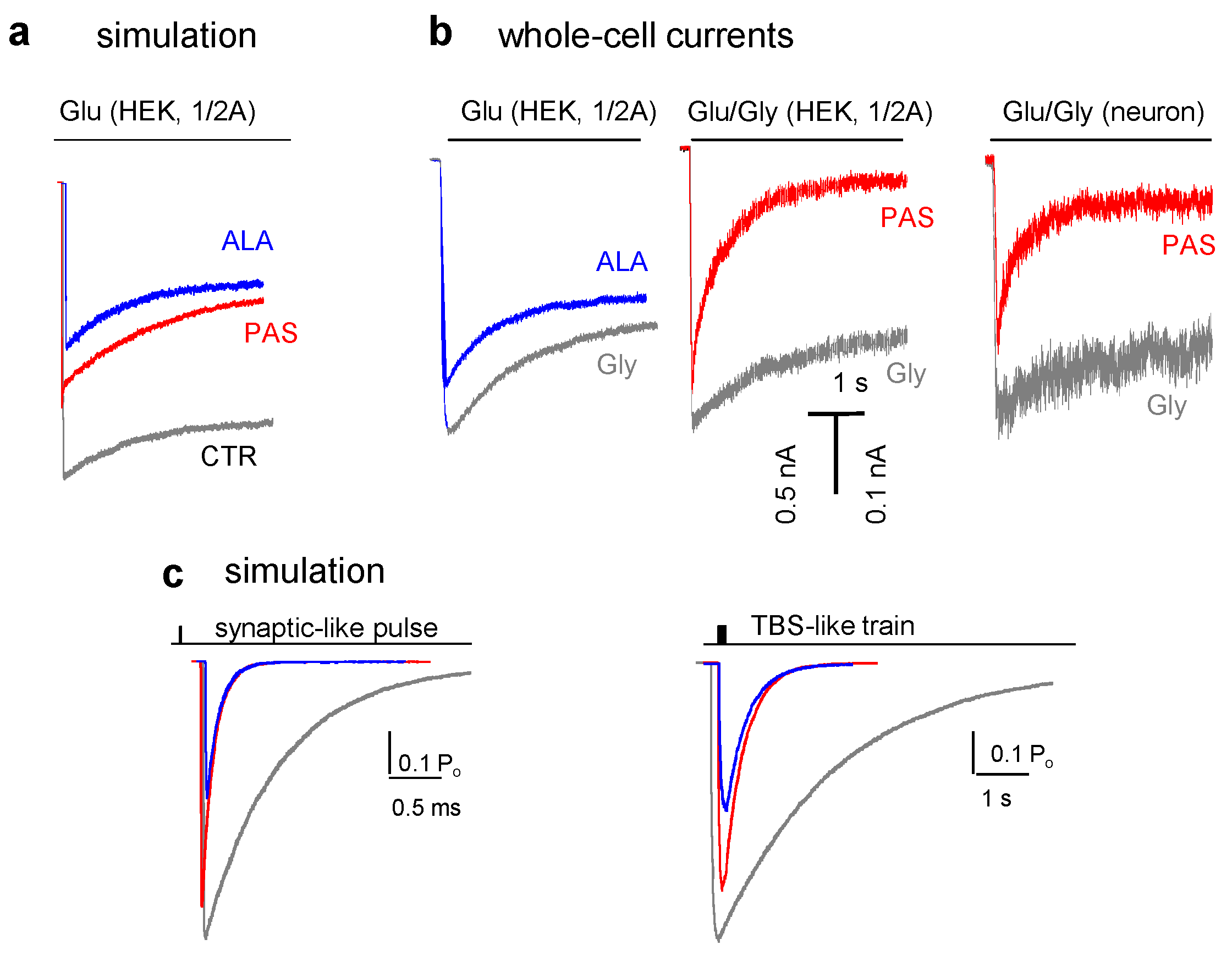
5. Modulators with Stimulus-Specific Effects
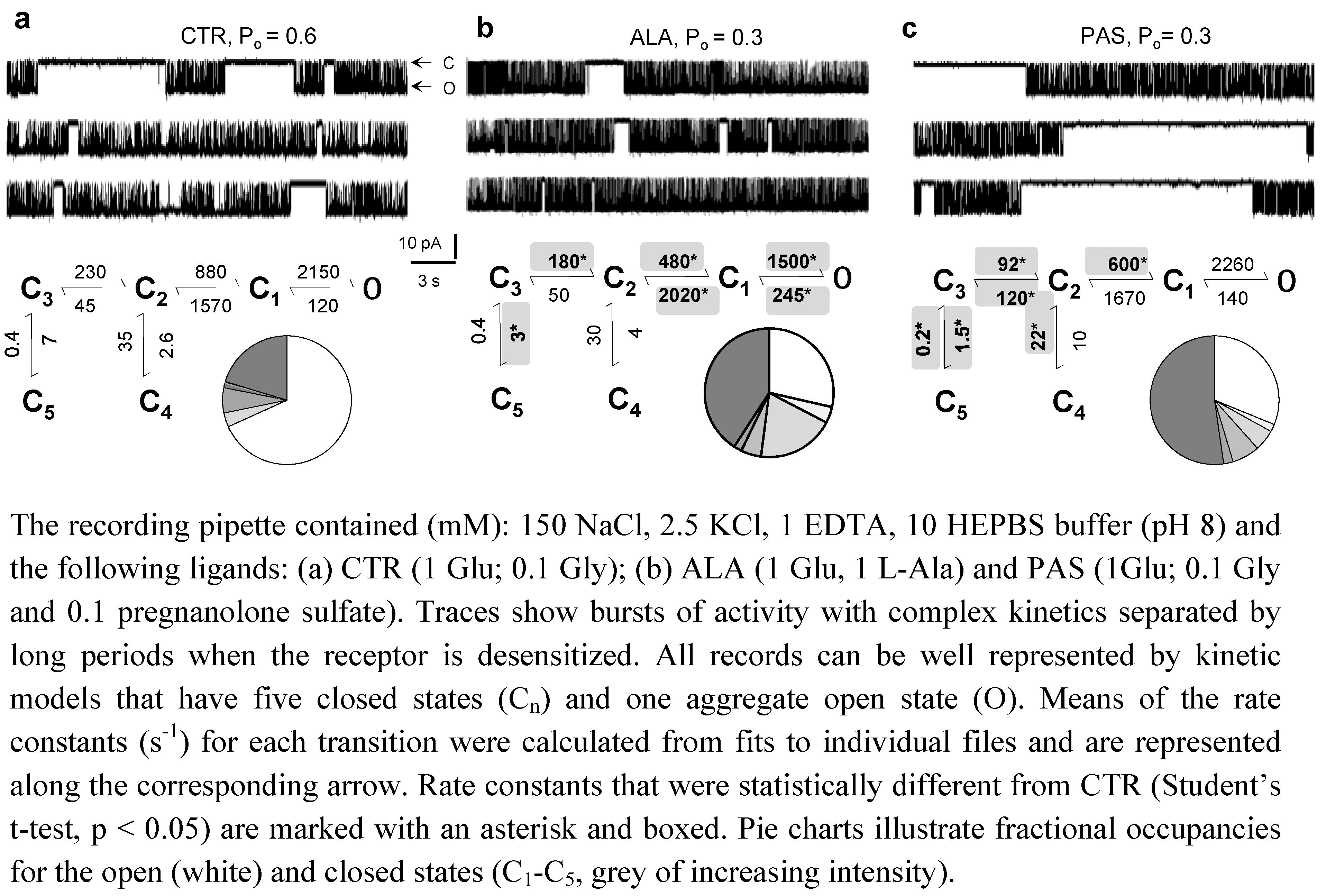
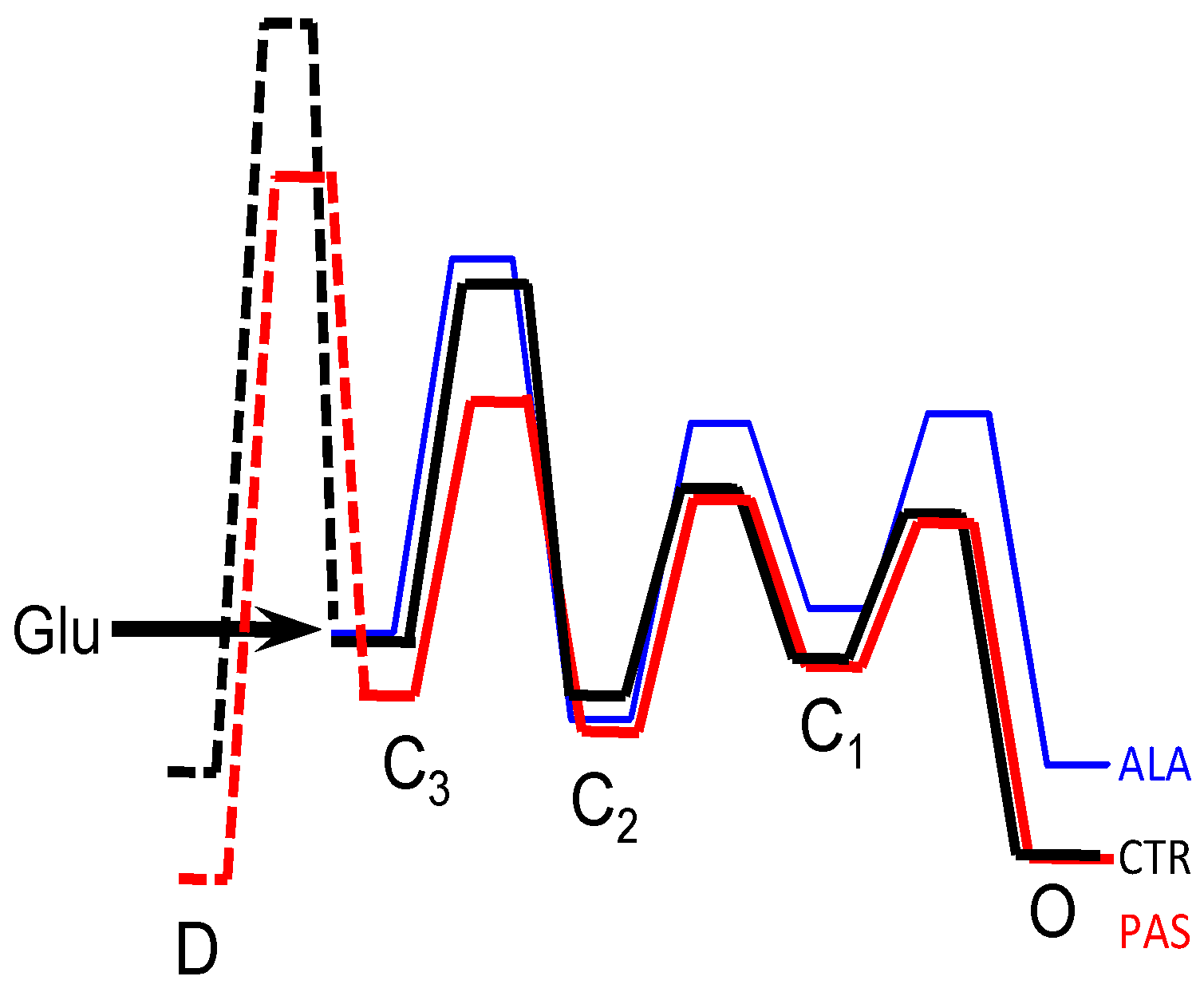
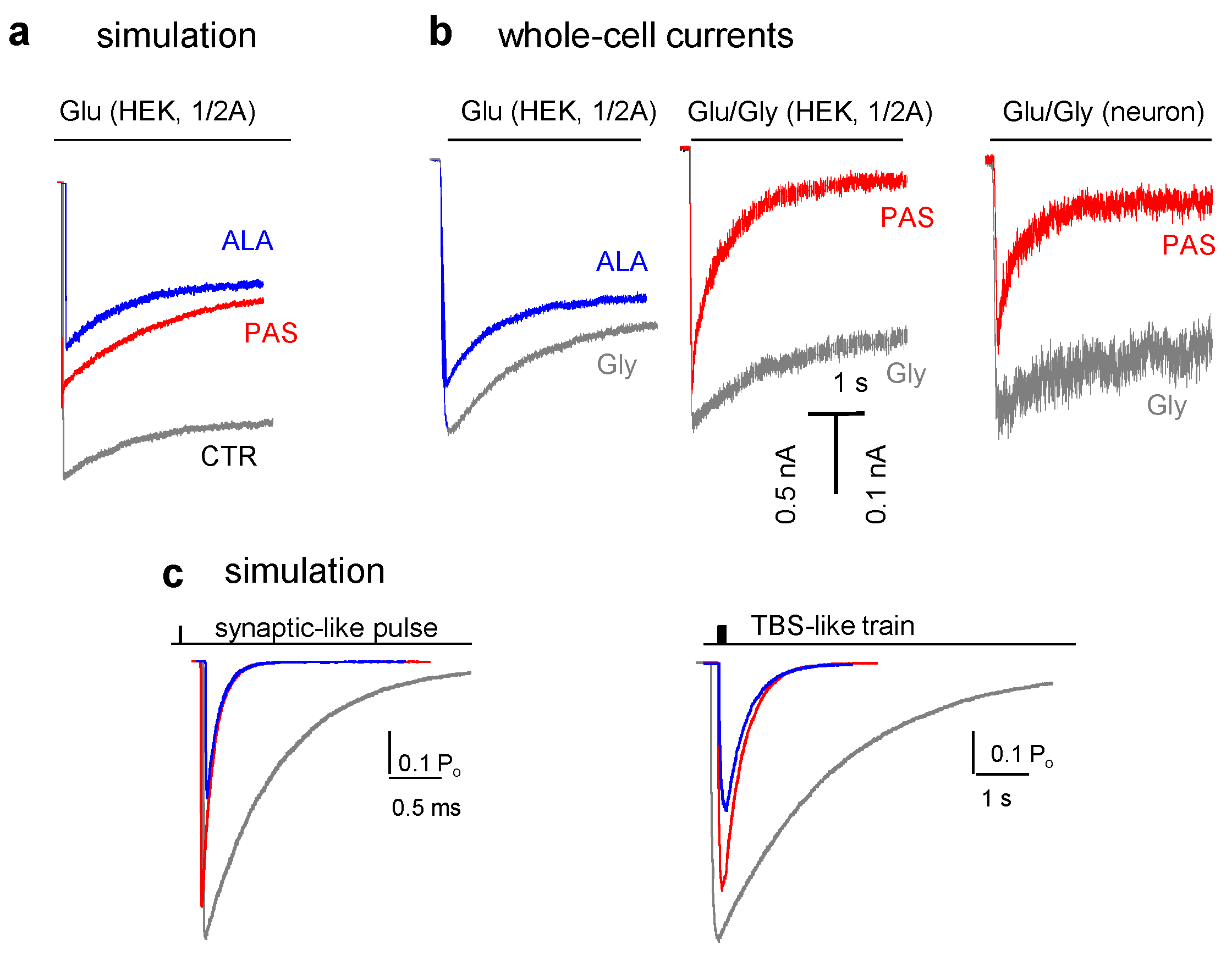
6. Conclusions
Abbreviations
| ACBC | aminocyclobutane-1-carboxylic acid, GluN1 partial agonist; |
| ACPC | 1-aminocyclopropane-1-carboxylic acid, GluN1 partial agonist; |
| ALA | l-alanine, GluN1 ligand; |
| AMPA | alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid; |
| APV | α-amino-5-phosphonopentanoic acid, GluN2 antagonist; |
| DCKA | 5,7-dichlorokynurenic acid, GluN1 antagonist; |
| DCS | d-cycloserine, GluN1 ligand; |
| PAS | pregnanolone sulfate, 3α-hydroxy-5β-pregnan-20-one sulfate |
Acknowledgements
References and Notes
- Kalia, L.V.; Kalia, S.K.; Salter, M.W. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008, 7, 742–755. [Google Scholar]
- McBain, C.J.; Mayer, M.L. N-methyl-D-aspartic acid receptor structure and function. Physiol. Rev. 1994, 74, 723–760. [Google Scholar]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar]
- Gardiner, K.J. Molecular basis of pharmacotherapies for cognition in Down syndrome. Trends Pharmacol. Sci. 2010, 31, 66–73. [Google Scholar]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar]
- Liu, X.J.; Gingrich, J.R.; Vargas-Caballero, M.; Dong, Y.N.; Sengar, A.; Beggs, S.; Wang, S.-H.; Ding, H.K.; Frankland, P.W.; Salter, M.W. Treatment of inflammatory and neuropathic pain by uncoupling Src from the NMDA receptor complex. Nat. Med. 2008, 14, 1325–1332. [Google Scholar]
- Kantrowitz, J.T.; Javitt, D.C. NMDA receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res. Bull. 2010, in press. [Google Scholar]
- Kalivas, P.W.; Lalumiere, R.T.; Knackstedt, L.; Shen, H. Glutamate transmission in addiction. Neuropharmacology 2009, 56 (Suppl. 1), 169–173. [Google Scholar]
- Cull-Candy, S.G.; Leszkiewicz, D.N. Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, 2004, re16. [Google Scholar]
- Yashiro, K.; Philpot, B.D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 2008, 55, 1081–1094. [Google Scholar]
- Tsai, G.E.; Lin, P.Y. Strategies to enhance NMDA receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr. Pharm Des 2010, 16, 522–537. [Google Scholar]
- Mittmann, T. Role of hippocampal NMDA receptors in a mouse model for fragile x mental retardation syndrome. J. Physiol. 2009, 587, 723. [Google Scholar]
- Pilpel, Y.; Kolleker, A.; Berberich, S.; Ginger, M.; Frick, A.; Mientjes, E.; Oostra, B.A.; Seeburg, P.H. Synaptic ionotropic glutamate receptors and plasticity are developmentally altered in the CA1 field of Fmr1 knockout mice. J. Physiol. 2009, 587, 787–804. [Google Scholar]
- Pittenger, C.; Sanacora, G.; Krystal, J.H. The NMDA receptor as a therapeutic target in major depressive disorder. CNS Neurol. Disord. Drug Targets 2007, 6, 101–115. [Google Scholar]
- Mehta, S.L.; Manhas, N.; Raghubir, R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 2007, 54, 34–66. [Google Scholar]
- Millecamps, M.; Centeno, M.V.; Berra, H.H.; Rudick, C.N.; Lavarello, S.; Tkatch, T.; Apkarian, A.V. d-Cycloserine reduces neuropathic pain behavior through limbic NMDA-mediated circuitry. Pain 2007, 132, 108–123. [Google Scholar]
- Gogas, K.R. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr. Opin. Pharmacol. 2006, 6, 68–74. [Google Scholar]
- Kemp, J.A.; McKernan, R.M. NMDA receptor pathways as drug targets. Nat. Neurosci. 2002, 5 Suppl, 1039–1042. [Google Scholar]
- Nowak, L.; Bregestovski, P.; Ascher, P.; Herbet, A.; Prochiantz, A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 1984, 307, 462–465. [Google Scholar]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261–263. [Google Scholar]
- Kawajiri, S.; Dingledine, R. Multiple structural determinants of voltage-dependent magnesium block in recombinant NMDA receptors. Neuropharmacology 1993, 32, 1203–1211. [Google Scholar]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of action of memantine. Curr. Opin Pharmacol 2006, 6, 61–67. [Google Scholar]
- Parsons, C.G.; Stöffler, A.; Danysz, W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system - too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723. [Google Scholar]
- Lipton, S.A. Pathologically activated therapeutics for neuroprotection. Nat Rev. Neurosci. 2007, 8, 803–808. [Google Scholar]
- Mony, L.; Kew, J.N.; Gunthorpe, M.J.; Paoletti, P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 2009, 157, 1301–1317. [Google Scholar]
- Kenakin, T. Allosteric Modulators: The New Generation of Receptor Antagonist. Mol. Interv. 2004, 4, 222–229. [Google Scholar]
- Huggins, D.J.; Grant, G.H. The function of the amino terminal domain in NMDA receptor modulation. J. Mol. Graph. Model 2005, 23, 381–388. [Google Scholar]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels:Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62. [Google Scholar]
- Blanpied, T.A.; Boeckman, F.A.; Aizenman, E.; Johnson, J.W. Trapping channel block of NMDA-activated responses by amantadine and memantine. J. NeuroPhysiol. 1997, 77, 309–323. [Google Scholar]
- Kampa, B.M.; Clements, J.; Jonas, P.; Stuart, G.J. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J. Physiol. (Lond) 2004, 556, 337–345. [Google Scholar]
- Vargas-Caballero, M.; Robinson, H.P.C. Fast and Slow Voltage-Dependent Dynamics of Magnesium Block in the NMDA Receptor: The Asymmetric Trapping Block Model. J. Neurosci. 2004, 24, 6171–6180. [Google Scholar]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. Amantadine Inhibits NMDA Receptors by Accelerating Channel Closure during Channel Block. J. Neurosci. 2005, 25, 3312–3322. [Google Scholar]
- Krupp, J.J.; Vissel, B.; Thomas, C.G.; Heinemann, S.F.; Westbrook, G.L. Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J. Neurosci. 1999, 19, 1165–1178. [Google Scholar]
- Zhang, S.; Ehlers, M.D.; Bernhardt, J.P.; Su, C.T.; Huganir, R.L. Calmodulin mediates calcium-dependent inactivation of N-methyl-D-aspartate receptors. Neuron 1998, 21, 443–453. [Google Scholar]
- Ehlers, M.D.; Zhang, S.; Bernhadt, J.P.; Huganir, R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell 1996, 84, 745–755. [Google Scholar]
- Tong, G.; Shepherd, D.; Jahr, C.E. Synaptic desensitization of NMDA receptors by calcineurin. Science 1995, 267, 1510–1512. [Google Scholar]
- Lieberman, D.N.; Mody, I. Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase. Nature 1994, 369, 235–239. [Google Scholar]
- Wang, Y.T.; Salter, M.W. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 1994, 369, 233–235. [Google Scholar]
- Clements, J.D.; Lester, R.A.; Tong, G.; Jahr, C.E.; Westbrook, G.L. The time course of glutamate in the synaptic cleft. Science 1992, 258, 1498–1501. [Google Scholar]
- Tong, G.; Jahr, C.E. Block of glutamate transporters potentiates postsynaptic excitation. Neuron 1994, 13, 1195–1203. [Google Scholar]
- Herman, M.A.; Jahr, C.E. Extracellular Glutamate Concentration in Hippocampal Slice. J. Neurosci. 2007, 27, 9736–9741. [Google Scholar]
- Gabernet, L.; Pauly-Evers, M.; Schwerdel, C.; Lentz, M.; Bluethmann, H.; Vogt, K.; Alberati, D.; Mohler, H.; Boison, D. Enhancement of the NMDA receptor function by reduction of glycine transporter-1 expression. Neurosci. Lett. 2005, 373, 79–84. [Google Scholar]
- Whitehead, K.J.; Pearce, S.M.; Walker, G.; Sundaram, H.; Hill, D.; Bowery, N.G. Positive N-methyl--aspartate receptor modulation by selective glycine transporter-1 inhibition in the rat dorsal spinal cord in vivo. Neuroscience 2004, 126, 381–390. [Google Scholar]
- Martina, M.; Gorfinkel, Y.; Halman, S.; Lowe, J.A.; Periyalwar, P.; Schmidt, C.J.; Bergeron, R. Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J. Physiol. (Lond) 2004, 557, 489–500. [Google Scholar]
- Yang, C.R.; Svensson, K.A. Allosteric modulation of NMDA receptor via elevation of brain glycine and d-serine: The therapeutic potentials for schizophrenia. Pharmacol. Therap. 2008, 120, 317–332. [Google Scholar]
- Shim, S.; Hammonds, M.; Kee, B. Potentiation of the NMDA receptor in the treatment of schizophrenia: focused on the glycine site. Eur. Arch.s.Psych. Clin. Neurosci. 2007, 258, 16–27. [Google Scholar]
- Tsai, G.E.; Yang, P.; Chang, Y.-C.; Chong, M.-Y. D-Alanine Added to Antipsychotics for the Treatment of Schizophrenia. Biol. Psych. 2006, 59, 230–234. [Google Scholar]
- Blanke, M.L.; VanDongen, A.M. Constitutive activation of the N-methyl-D-aspartate receptor via cleft-spanning disulfide bonds. J. Biol. Chem. 2008, 283, 21519–21529. [Google Scholar]
- Kussius, C.L.; Popescu, G.K. NMDA Receptors with Locked Glutamate-Binding Clefts Open with High Efficacy. J. Neurosci. 2010, 30, 12474–12479. [Google Scholar]
- Schorge, S.; Colquhoun, D. Studies of NMDA receptor function and stoichiometry with truncated and tandem subunits. J. Neurosci. 2003, 23, 1151–1158. [Google Scholar]
- Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 2009. [Google Scholar]
- Rambhadran, A.; Gonzalez, J.; Jayaraman, V. Subunit arrangement in NMDA receptors. J. Biol.Chem. 2010, 285, 15296–15301. [Google Scholar]
- Karakas, E.; Simorowski, N.; Furukawa, H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009, 28, 3910–3920. [Google Scholar]
- Furukawa, H.; Singh, S.K.; Mancusso, R.; Gouaux, E. Subunit arrangement and function in NMDA receptors. Nature 2005, 438, 185. [Google Scholar]
- Vicini, S.; Wang, J.F.; Li, J.H.; Zhu, W.J.; Wang, Y.H.; Luo, J.H.; Wolfe, B.B.; Grayson, D.R. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 1998, 79, 555–566. [Google Scholar] [PubMed]
- Monyer, H.; Sprengel, R.; Schoepfer, R.; Herb, A.; Higuchi, M.; Lomeli, H.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 1992, 256, 1217–1221. [Google Scholar]
- Furukawa, H.; Gouaux, E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003, 22, 2873–2885. [Google Scholar]
- Inanobe, A.; Furukawa, H.; Gouaux, E. Mechanism of partial agonist action at the NR1 subunit of NMDA receptors. Neuron 2005, 47, 71–84. [Google Scholar]
- Durand, G.M.; Bennett, M.V.; Zukin, R.S. Splice variants of the N-methyl-D-aspartate receptor NR1 identify domains involved in regulation by polyamines and protein kinase C. Proc. Natl. Acad. Sci. USA 1993, 90, 6731–6735. [Google Scholar]
- Choi, Y.B.; Tenneti, L.; Le, D.A.; Ortiz, J.; Bai, G.; Chen, H.S.; Lipton, S.A. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat. Neurosci. 2000, 3, 15–21. [Google Scholar]
- Jang, M.K.; Mierke, D.F.; Russek, S.J.; Farb, D.H. A steroid modulatory domain on NR2B controls N-methyl-D-aspartate receptor proton sensitivity. Proc. Natl. Acad. Sci. USA 2004, 101, 8198–8203. [Google Scholar]
- Spivak, V.; Lin, A.; Beebe, P.; Stoll, L.; Gentile, L. Identification of a neurosteroid binding site contained within the GluR2-S1S2 domain. Lipids 2004, 39, 811–819. [Google Scholar]
- Mott, D.D.; Doherty, J.J.; Zhang, S.; Washburn, M.S.; Fendley, M.J.; Lyuboslavsky, P.; Traynelis, S.F.; Dingledine, R. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat. Neurosci. 1998, 1, 659–667. [Google Scholar]
- Kashiwagi, K.; Fukuchi, J.; Chao, J.; Igarashi, K.; Williams, K. An aspartate residue in the extracellular loop of the N-methyl-D-aspartate receptor controls sensitivity to spermine and protons. Mol. Pharmacol. 1996, 49, 1131–1141. [Google Scholar]
- Erreger, K.; Dravid, S.M.; Banke, T.G.; Wyllie, D.J.; Traynelis, S.F. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J. Physiol. 2005, 563, 345–358. [Google Scholar]
- Dravid, S.M.; Prakash, A.; Traynelis, S.F. Activation of recombinant NR1/NR2C NMDA receptors. J. Physiol. 2008, 586, 4425–4439. [Google Scholar]
- Schorge, S.; Elenes, S.; Colquhoun, D. Maximum likelihood fitting of single channel NMDA activity with a mechanism composed of independent dimers of subunits. J. Physiol. 2005. [Google Scholar]
- Auerbach, A.; Zhou, Y. Gating reaction mechanisms for NMDA receptor channels. J. Neurosci. 2005, 25, 7914–7923. [Google Scholar]
- Popescu, G.; Auerbach, A. Modal gating of NMDA receptors and the shape of their synaptic response. Nat. Neurosci. 2003, 6, 476–483. [Google Scholar]
- Banke, T.G.; Traynelis, S.F. Activation of NR1/NR2B NMDA receptors. Nat. Neurosci. 2003, 6, 144–152. [Google Scholar]
- Amico-Ruvio, S.A.; Popescu, G.K. Stationary gating of GluN1/GluN2B receptors in intact membrane patches. Biophys. J. 2010, 98. in press. [Google Scholar]
- Popescu, G.; Auerbach, A. The NMDA receptor gating machine: lessons from single channels. Neuroscientist 2004, 10, 192–198. [Google Scholar]
- Erreger, K.; Traynelis, S.F. Zinc inhibition of rat NR1/NR2A N-methyl-D-aspartate receptors. J. Physiol. 2008, 586, 763–778. [Google Scholar]
- Banke, T.G.; Dravid, S.M.; Traynelis, S.F. Protons Trap NR1/NR2B NMDA Receptors in a Nonconducting State. J. Neurosci. 2005, 25, 42–51. [Google Scholar]
- Popescu, G.; Robert, A.; Howe, J.R.; Auerbach, A. Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature 2004, 430, 790–793. [Google Scholar]
- Zhang, W.; Howe, J.R.; Popescu, G.K. Distinct gating modes determine the biphasic relaxation of NMDA receptor currents. Nat. Neurosci. 2008, 11, 1373–1375. [Google Scholar]
- Kussius, C.L.; Popescu, G.K. Kinetic basis of partial agonism at NMDA receptors. Nat. Neurosci. 2009, 12, 1114–1120. [Google Scholar] [PubMed]
- Sheinin, A.; Shavit, S.; Benveniste, M. Subunit specificity and mechanism of action of NMDA partial agonist D-cycloserine. Neuropharmacology 2001, 41, 151–158. [Google Scholar]
- Kussius, C.L.; Kaur, N.; Popescu, G.K. Pregnanolone Sulfate Promotes Desensitization of Activated NMDA Receptors. J. Neurosci. 2009, 29, 6819–6827. [Google Scholar]
- Kussius, C.L.; Popescu, A.M.; Popescu, G.K. Agonist-specific gating of NMDA receptors. Channels 2010, 4, 1–5. [Google Scholar]
- Dravid, S.M.; Burger, P.B.; Prakash, A.; Geballe, M.T.; Yadav, R.; Le, P.; Vellano, K.; Snyder, J.P.; Traynelis, S.F. Structural determinants of D-cycloserine efficacy at the NR1/NR2C NMDA receptors. J. Neurosci. 2010, 30, 2741–2754. [Google Scholar]
- Popescu, G. Mechanism-based targeting of NMDA receptor functions. Cell Mol. Life Sci. 2005, 62, 2100–2111. [Google Scholar]
- Loftis, J.M.; Janowsky, A. The N-methyl-D-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol. Ther. 2003, 97, 55–85. [Google Scholar]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar]
- Davies, J.; Evans, R.H.; Francis, A.A.; Watkins, J.C. Excitatory amino acid receptors and synaptic excitation in the mammalian central nervous system. J. Physiol. (Paris) 1979, 75, 641–654. [Google Scholar]
- Kleckner, N.W.; Dingledine, R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 1988, 241, 835–837. [Google Scholar]
- Chen, P.E.; Geballe, M.T.; Katz, E.; Erreger, K.; Livesey, M.R.; O'Toole, K.K.; Le, P.; Lee, C.J.; Snyder, J.P.; Traynelis, S.F.; Wyllie, D.J. Modulation of glycine potency in rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J. Physiol. 2008, 586, 227–245. [Google Scholar]
- Park-Chung, M.; Wu, F.S.; Farb, D.H. 3 alpha-Hydroxy-5 beta-pregnan-20-one sulfate: a negative modulator of the NMDA-induced current in cultured neurons. Mol. Pharmacol. 1994, 46, 146–150. [Google Scholar]
- Sedlacek, M.; Korinek, M.; Petrovic, M.; Cais, O.; Adamusova, E.; Chodounska, H.; Vyklicky, L., Jr. Neurosteroid modulation of ionotropic glutamate receptors and excitatory synaptic transmission. Physiol. Res. 2008, 57 (Suppl. 3), S49–S57. [Google Scholar]
- Malayev, A.; Gibbs, T.T.; Farb, D.H. Inhibition of the NMDA response by pregnenolone sulphate reveals subtype selective modulation of NMDA receptors by sulphated steroids. Br. J. Pharmacol. 2002, 135, 901–909. [Google Scholar]
- Park-Chung, M.; Wu, F.S.; Purdy, R.H.; Malayev, A.A.; Gibbs, T.T.; Farb, D.H. Distinct sites for inverse modulation of N-methyl-D-aspartate receptors by sulfated steroids. Mol. Pharmacol. 1997, 52, 1113–1123. [Google Scholar]
- Petrovic, M.; Sedlacek, M.; Horak, M.; Chodounska, H.; Vyklicky, L., Jr. 20-oxo-5beta-pregnan-3alpha-yl sulfate is a use-dependent NMDA receptor inhibitor. J. Neurosci. 2005, 25, 8439–8450. [Google Scholar]
- Weaver, C.E., Jr.; Marek, P.; Park-Chung, M.; Tam, S.W.; Farb, D.H. Neuroprotective activity of a new class of steroidal inhibitors of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 10450–10454. [Google Scholar]
- Sather, W.; Johnson, J.W.; Henderson, G.; Ascher, P. Glycine-insensitive desensitization of NMDA responses in cultured mouse embryonic neurons. Neuron 1990, 4, 725–731. [Google Scholar]
- Lester, R.A.; Jahr, C.E. NMDA channel behavior depends on agonist affinity. J. Neurosci. 1992, 635–643. [Google Scholar]
- Tong, G.; Jahr, C.E. Regulation of glycine-insensitive desensitization of the NMDA receptor in outside-out patches. J. Neurophysiol. 1994, 72, 754–761. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Popescu, G.K.; Murthy, S.; Borschel, W.F. Allosteric Inhibitors of NMDA Receptor Functions. Pharmaceuticals 2010, 3, 3240-3257. https://doi.org/10.3390/ph3103240
Popescu GK, Murthy S, Borschel WF. Allosteric Inhibitors of NMDA Receptor Functions. Pharmaceuticals. 2010; 3(10):3240-3257. https://doi.org/10.3390/ph3103240
Chicago/Turabian StylePopescu, Gabriela K., Swetha Murthy, and William F. Borschel. 2010. "Allosteric Inhibitors of NMDA Receptor Functions" Pharmaceuticals 3, no. 10: 3240-3257. https://doi.org/10.3390/ph3103240
APA StylePopescu, G. K., Murthy, S., & Borschel, W. F. (2010). Allosteric Inhibitors of NMDA Receptor Functions. Pharmaceuticals, 3(10), 3240-3257. https://doi.org/10.3390/ph3103240