A Novel Category of Anti-Hypertensive Drugs for Treating Salt-Sensitive Hypertension on the Basis of a New Development Concept

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Contents
1. Introduction
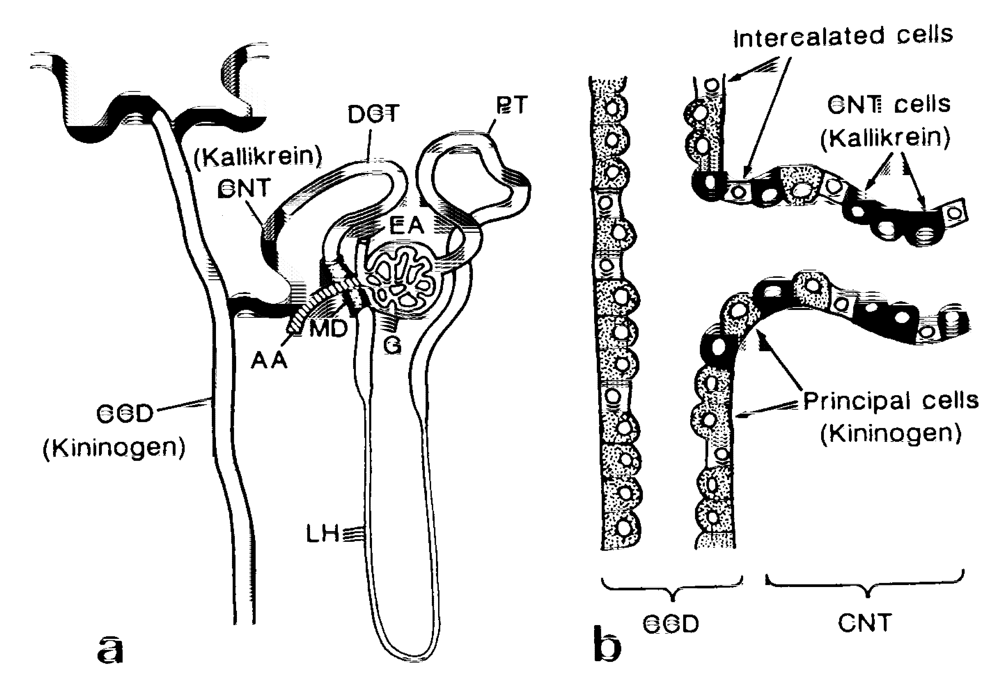
2. What Is the Renal Kallikrein-Kinin System (Renal KKS)? Is it Equipped for the Rectification of Excess Sodium Intake?
2.1. Renal Kallikrein and Low-Molecular-Weight Kininogen
2.2. Kallikrein Inhibitors
2.3. Kinins and Kinin Receptors
2.4. Renal Kininases and Their Selective Inhibitors

2.5. Stimuli for Kallikrein Secretion in the Kidney
2.5.1. Sodium
2.5.2. Sodium-Retaining Steroid Hormones
2.5.3. Potassium
2.5.4. ATP-Sensitive Potassium Channel Blockers

2.6. Roles of the Renal KKS in the Kidney
2.6.1. Vasodilating Action of BK in the Kidney
2.6.2. Diuresis and Natriuresis
3. Are Animals Deficient in the KKS Components Hypertensive?—Additional High Sodium Intake Is Necessary
3.1. BK B2 Receptor-Gene-Disrupted Mice
3.2. Tissue Kallikrein-Gene-Disrupted Mice
3.3. Kininogen-Deficient Rats
4. How Does an Excess Sodium Intake Cause Hypertension?
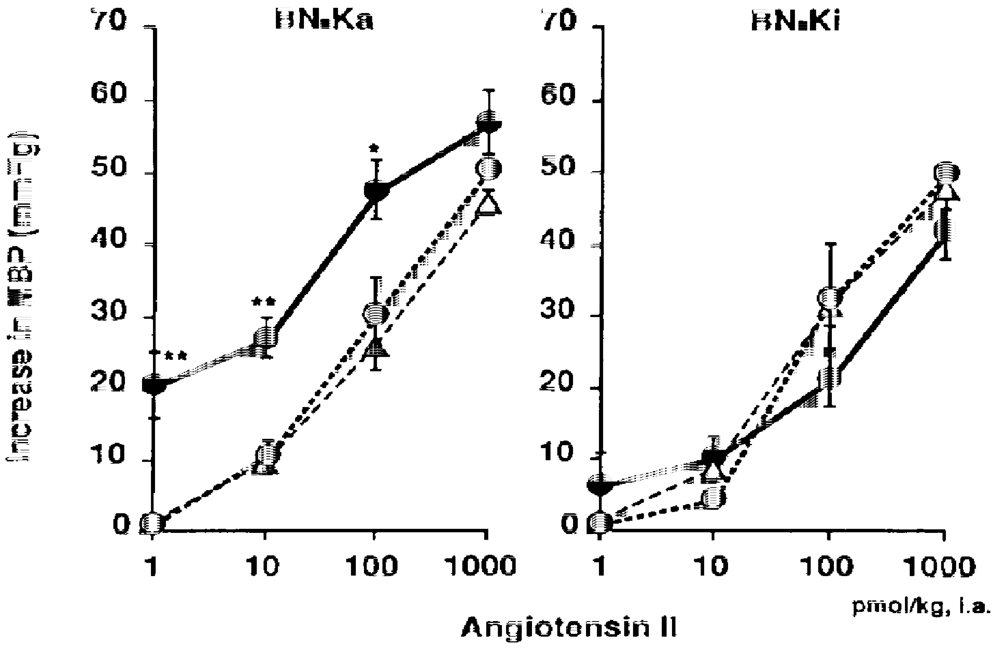
4.1. Kininogen-Deficient BN-Ka Rats Are Salt-Sensitive
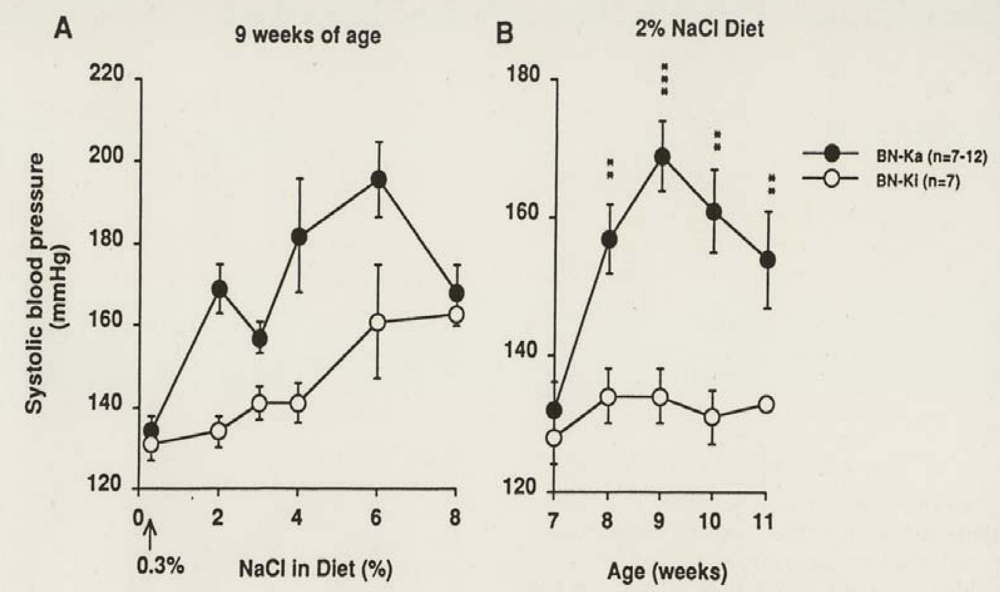
4.2. Sodium Accumulation in Cells, Particularly in Blood Vessels
4.3. Increased Sensitivity of Vascular Smooth Muscles to Angiotensin II and Norepinephrine after Sodium Accumulation

4.4. Sodium accumulation in cerebrospinal fluids and increased sympathetic discharge
5. Do Hypertensive Patients Secrete Less Urinary Kallikrein?
5.1. Hypertensive Patients
5.2. Salt-Sensitive Hypertension
5.3. Hypertensive Animal Models
5.3.1. DOCA-Salt Hypertension Model
5.3.2. Spontaneously Hypertensive Rats (SHR)
5.3.3. Dahl Salt-Sensitive Rats
5.3.4. Other Genetically and Experimentally Hypertensive Rats
6. Antihypertensive Effects of Renal Kallikrein Releasers and of Urinary Kininase Inhibitors on Salt-Induced Hypertensive Models
6.1. Agents, Which Accelerate Release of Renal Kallikrein

6.2. Inhibitors of Renal Kininase (Kinin-Degradation Enzyme)
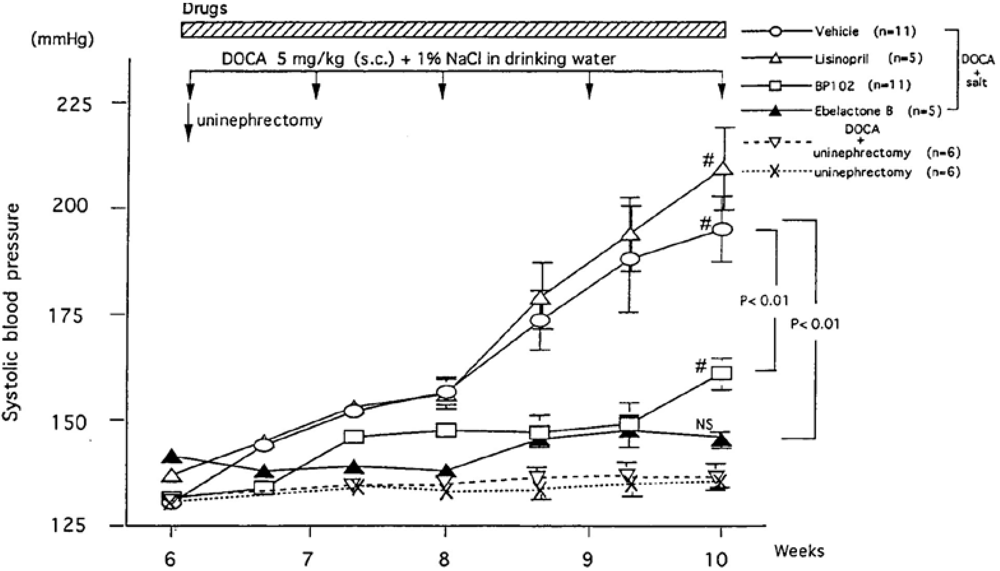
7. Roles of KKS on Cardiovascular Disorders and Angiogenesis and a Therapeutic Aspect of Tissue Kallikrein Gene Delivery
7.1. Cardiac Ischemia and ACE Inhibitors
7.2. Ischemic Preconditioning and Reperfusion of the Heart
7.3. Angiogenesis
7.4. Tissue Kallikrein-Gene Delivery
8. Proposal of a New Category of Anti-Hypertensive Drugs against the Salt-Sensitive Hypertension
Abbreviations:
| BP | blood pressure |
| BK | bradykinin |
| CD | collecting duct |
| CNT | connecting tubules |
| CPY | carboxypeptidase Y-like exopeptidase |
| SBP | systolic blood pressure |
Acknowledgements
References
- Baldwin, E. An Introduction to Comparative Biochemistry; 1949; Cambridge University Press: Cambridge, UK. [Google Scholar]
- Evans, D.; Piermarini, P.; Choe, K. The multifunctional fish gill: Dominant site of gas exchange, osmoregulation, acid-base regulation, and excretion of nitrogen waste. Physiol. Rev. 2005, 85, 97–177. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.B. Ion transport, osmoregulation, and acid-vase balance. In The Physiology of Fishes; Evans, D., Claiborne, J., Eds.; Raylor & Francis: Boca, London, New York, 2006. [Google Scholar]
- Frey, E.; Kraut, H.; Werle, E. Einleitung und Übersicht. In Das Kallikrein-Kinin-System und seine Inhibitoren; 1968; Ferdinand Enke Verlag: Stuttgart, Germany. [Google Scholar]
- Kraut, H.; Frey, E.; Werle, E. Der Nachweis eines Kreislaufhormons in der Pankreasdrüse. Hoppe-Seylers Z Physiol.Chem. 1930, 189, 97–106. [Google Scholar]
- Rocha e Silva, M.; Berald, W.; Rosenfeld, G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am. J. Physiol. 1949, 156, 261–273. [Google Scholar] [PubMed]
- Scicli, A.G.; Carretero, O.A. Renal kallikrein-kinin system. Kidney Int. 1986, 29, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Majima, M.; Katori, M. Approaches to the development of novel antihypertensive drugs: Crucial role of the renal kallikrein-kinin system. Trends Pharmacol. Sci. 1995, 16, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Katori, M.; Majima, M. Pivotal role of renal kallikrein-kinin system in the development of hypertension and approaches to new drugs based on this relationship. Jpn. J. Pharmacol. 1996, 70, 95–128. [Google Scholar] [CrossRef] [PubMed]
- Majima, M.; Katori, M.; Ogino, M.; Saito, M.; Sugimoto, K.; Adachi, K.; Sunahara, N.; Katoh, K.; Tatemachi, N.; Takei, Y. Failure of endogenous blood kinin levels elevated by captopril to induce hypotension in normotensive and hypertensive rats--An assay for kinin by a new ELISA. Biochem. Res. 1995, 17, 698–708. [Google Scholar]
- Vio, C.P.; Figueroa, C.D. Subcellular localization of renal kallikrein by ultrastructural immunochemistry. Kidney Int. 1985, 28, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, C.D.; Caorsi, I.; Vio, C.P. Visualization of renal kallikrein in luminal and basolateral membranes. J. Histochem. Cytochem. 1984, 32, 1238–1240. [Google Scholar] [PubMed]
- Figueroa, C.; MacIver, A.; Mackenzie, J.; Bhoola, K. Localization of immunoreactive kininogen and tissue kallikrein in the human nephron. Histochemistry 1988, 89, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Scicli, A.; Carretero, O.; Hampton, A.; Cortes, P.; Oza, M. Site of kininogenase secretion in dog nephron. Am. J. Physiol. 1976, 230, 533–537. [Google Scholar] [PubMed]
- Fujita, T.; Kumagai, Y.; Ikeda, Y.; Inamura, N.; Iwata, T.; Ogino, M.; Majima, M. Involvement of the renal kallikrein-kinin system in furosemide-induced natriuresis in rats. Jpn. J. Pharmacol. 2000, 84, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Chao, L. Biochemistry, regulation and potential function of kallistatin. Biol. Chem. Hoppe Seyler 1995, 376, 705–713. [Google Scholar] [PubMed]
- Yang, T.; Terada, Y.; Nonoguchi, H.; Tsujino, M.; Tomita, K.; Marumo, F. Distribution of kallikrein-binding protein mRNA in kidneys and difference between SHR and WKY . Am. J. Physiol. 1994, 267, F325–F330. [Google Scholar] [PubMed]
- Miwa, I.; Erdös, E.; Seki, T. Presence of three peptides in urinary kinin (substance Z). Life Science 1968, 7, 1339–1343. [Google Scholar] [CrossRef]
- Miwa, I.; Erdös, E.; Seki, T. Separation of peptide components of urinary kinin (substance Z). Proc. Soc. Exp. Biol. Med. 1969, 131, 768–772. [Google Scholar] [PubMed]
- Hial, V.; Keiser, H.; Pisano, J. Origin and content of methionyl-lysyl-bradykinin, lysyl-bradykinin and bradykinin in human urine. Biochem. Pharmacol. 1976, 25, 2499–2503. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, K.; Abe, K.; Miwa, I.; Furuyama, T.; Suzuki, C. Evidence for the renal origin of urinary kinin. Experientia 1964, 20, 396–397. [Google Scholar] [CrossRef] [PubMed]
- Abe, K. Urinary excretion of kinin in man with special reference to its origin. Tohoku J. Exp. Med. 1965, 87, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Nasjletti, A.; Colina-Chourio, J.; McGiff, J. Disappearance of bradykinin in the renal circulation of dogs: Effects of kininase inhibition. Circ. Res. 1975, 37, 59–65. [Google Scholar] [PubMed]
- Tomita, K.; Pisano, J. Binding of [3H]-bradykinin in isolated nephron segments of the rabbit . Am. J. Physiol. 1984, 246, F732–F737. [Google Scholar] [PubMed]
- Kauker, M. Bradykinin action on the efflux of luminal 23Na in the rat nephron. J. Pharmacol. Exp. Ther. 1980, 214, 119–123. [Google Scholar] [PubMed]
- Bachvarof, D.; Saint-Jacques, E.; Larrovee, J.; Levelsque, L.; Rioux, F.; Drapeau, G.; Marceau, F. Cloning and pharmacological characterization of the rabbit bradykinin B2 receptor. J. Pharmacol. Exp. Ther. 1995, 275, 1523–1530. [Google Scholar]
- Figueroa, C.; Gonzalez, C.; Grigoriev, S.; Abd Alla, S.; Hassemann, M.; Jarnagin, K.; Müller-Ester, W. Probing for the bradykinin B2 receptor in rat kidney by anti-peptide and anti-ligand antibodies. J. Histochem. Cytochem. 1995, 43, 137–148. [Google Scholar] [PubMed]
- Song, Q.; Wang, D.; Harley, R.; Chao, L.; Chao, J. Cellular localization of low-molecular-weight kininogen and bradykinin B2 receptor mRNA in human kidney . Am. J. Physiol. 1996, 270, F919–F926. [Google Scholar] [PubMed]
- Tomita, K.; Pisano, J.; Knepper, M. Control of sodium and potassium transport in the cortical collecting duct of the rat. J. Clin. Invest. 1985, 76, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Ardaillou, N.; Placier, S.; Zhao, J.; Baudouin, B.; Ardaillou, R. Characterization of B2-bradykinin receptors in rabbit principal cells of the collecting duct. Exp. Nephrol. 1998, 6, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Kose, H.; Boese, S.; Glanville, M.; Gray, M.; Brown, C.; Simmmons, N. Bradykinin regulation of salt transport across mouse inner medullary collecting duct epithelium involve activation of a Ca(2+)-dependent CL(-) conductance. Br. J. Pharmacol. 2000, 131, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Mukai, H.; Fitzgibbon, W.; Bozeman, G.; Margolius, H.; Ploth, D. Bradykinin B2 receptor antagonist increases chloride and water absorption in rat medullary collecting duct . Am. J. Physiol. 1996, 271, R352–R360. [Google Scholar] [PubMed]
- Kuribayashi, Y.; Majima, M.; Katori, M. Major kininases in rat urine are neutral endopeptidase and carboxypeptidase Y-like exopeptidse. Biomed. Res. 1993, 14, 191–201. [Google Scholar]
- Shima, M.; Seino, Y.; Torikai, S.; Imai, M. Intrarenal localization of degradation of atrial natriuretic peptide in isolated glomerular and cortical nephron segments. Life Sci. 1988, 43, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.; Hagler, H.; Buja, L.; Erdös, E. Ultrastructural localization of angiotensin I-converting enzyme (EC 3.4.15.1) and neutral metalloendopeptidase (EC 3.4.24.11) in the proximal tubule of the human kidney. Lab. Invest. 1988, 59, 789–797. [Google Scholar] [PubMed]
- Sakakibara, T.; Ura, N.; Shimamoto, K.; Ogata, H.; Ando, T.; Fukuyama, S.; Yamaguchi, Y.; Masuda, A.; Mori, Y.; Saito, S.; Ise, T.; Sasa, Y.; Yamauchi, K.; Iimura, O. Localization of neutral endopeptidase in the kidney determined by the stop-flow method . In Advances in Exerimental Medicine and Biolology; Abe, K., Moriya, H., Fujii, S., Eds.; Plenum Press: New York, NY, USA, 1989. [Google Scholar]
- Ogata, H.; Ura, N.; Shimamoto, K.; Sakakibara, T.; Ando, T.; Nishimiya, T.; Nakagawa, M.; Fukuyama, S.; Masuda, A.; Yamaguchi, Y.; Ise, T.; Saito, S.; Shiiki, M.; Uno, K.; Iimura, O. A sensitive method for differential determination of kininase I, II, and neutral endopeptidase (NEP) in human urine. In Advances in Exerimental Medicine and Biolology; Abe K.;, Moriya, H.; Fujii, S., Eds.; Plenum Press: New York, NY, 1989. [Google Scholar]
- Skidgel, R.; Davis, R.; Erdös, E. Purification of a human urinary carboxypeptidase (kininase) distinct from carboxypeptidase A, B, or N. Anal. Biochem. 1984, 140, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Erdös, E. Some old and some new ideas on kinin metabolism . J. Cardiovasc. Pharmacol. 1990, 15 (Suppl.), 520–524. [Google Scholar]
- Wilks, S. Prolyl endopeptidase. Life Sci. 1983, 33, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
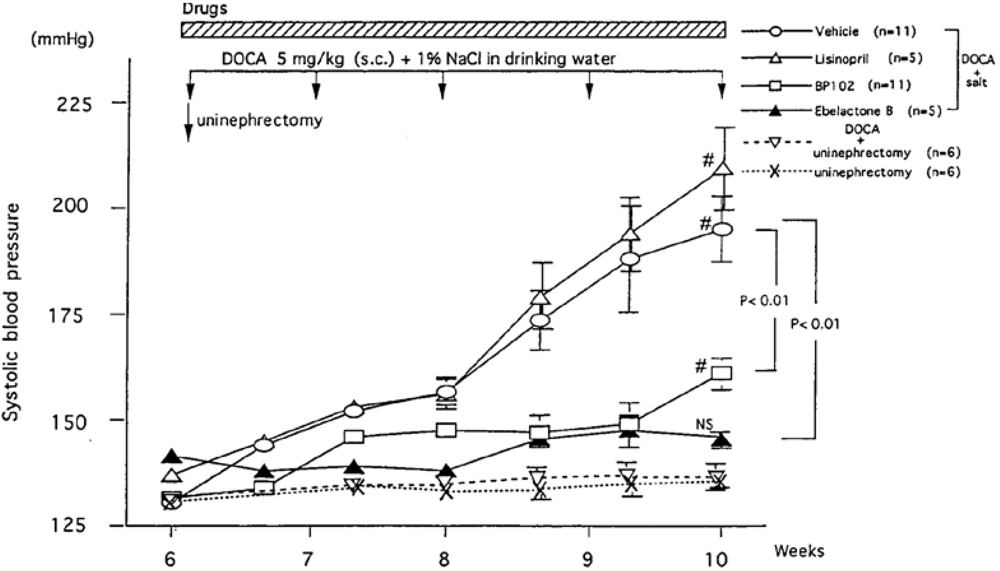
- Majima, M.; Ikeda, Y.; Kuribayashi, Y.; Mizogami, S.; Katori, M.; Aoyagi, T. Ebelactone B, an inhibitor of urinary carboxypeptidase Y-like kininase, prevents the development of deoxycorticosterone acetate-salt hypertension in rats. Eur. J. Pharmacol. 1995, 284, 1–11. [Google Scholar] [CrossRef]
- Majima, M.; Shima, C.; Saito, M.; Kuribayashi, Y.; Katori, M.; Aoyagi, T. Poststatin, a novel inhibitor of bradykinin-degrading enzymes in rat urine. Eur. J. Pharmacol. 1993, 232, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, H.; Aoyagi, T.; Uotani, K.; Hamada, M.; Takeuchi, Y.; Takahashi, S. Ebelactone, an inhibitor of esterase, produced by actinomysetes. J. Antibiot. (Tokyo) 1980, 33, 1594–1596. [Google Scholar] [PubMed]
- Uotani, K.; Magasawa, H.; Kondo, S.; Aoyagi, T.; Umezawa, H. Structural studies on ebelactone A and B, esterase inhibitors produced by actinomycetes. J. Antibiot. (Tokyo) 1982, 35, 1495–1499. [Google Scholar] [PubMed]
- Aoyagi, T.; Nagai, M.; Ogawa, K.; Kojima, F.; Okada, M.; Ikeda, T.; Hamada, M.; Takeuchi, T. Poststatin, a new inhibitor of prolyl endopeptidase, produced by streptomyces viridochromogenes MH534-30F3. I. Taxonomy, production, isolation, physico-chemical properties and biological activities. J. Antibiot. (Tokyo) 1991, 44, 949–955. [Google Scholar] [PubMed]
- Nagai, M.; Ogawa, K.; Muraoka, Y.; Naganawa, H.; Aoyagi, T.; Takeuchi, T. Poststatin, a new inhibitor of prolyl endopeptidase, produced by Streptomyces viridochromogenes MH534–30F3. II. Structure determination and inhibitory activities. J. Antibiot. (Tokyo) 1991, 44, 956–961. [Google Scholar] [PubMed]
- Satoh, U.; Kadota, Y.; Oheda, Y.; Kuwahara, J.; Aikawa, S.; Matsuzawa, F.; Doi, H.; Aoyagi, T.; Sakuraba, H.; Itoh, K. Microbial serine carboxypeptidase inhibitors--Comparative analysis of actions on homologous enzymes derived from man, yeast and wheat. J. Antibiot. (Tokyo) 2004, 57, 316–325. [Google Scholar] [PubMed]
- Majima, M.; Kuribayashi, Y.; Ikeda, Y.; Adachi, K.; Kato, H.; Katori, M.; Aoyagi, T. Diuretic and natriuretic effect of ebelactone B in anesthetized rats by inhibition of a urinary carboxypeptidase Y-like kininase. Jpn. J. Pharmacol. 1994, 65, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, I.; Majima, M.; Fujita, T.; Okumura, T.; Kumagai, Y.; Tomita, N.; Morishita, R.; Higaki, J.; Ogiwara, T. In vivo transfer of antisense oligonucleotide against urinary kininase blunts deoxycorticosterone acetate-salt hypertension in rats. Br. J. Pharmacol. 2000, 131, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Adetuyibi, A.; Mills, I. Relation between urinary kallikrein and renal function, hypertension, and excretion of sodium and water in man . Lancet 1972, ii, 203–207. [Google Scholar] [CrossRef]
- Margolius, H.; Horowitz, D.; Gellar, R.; Allexander, R.; Gill, J.; Pisano, J.; Keiser, H. Urinary kallikrein excretion in normal man. Relationships to sodium intake and sodium-retaining steroids. Circ. Res. 1974, 35, 812–819. [Google Scholar] [PubMed]
- Seino, J.; Abe, K.; Otuska, Y.; Saito, T.; Irokawa, N. Urinary kallikrein excretion and sodium metabolism in hypertensive patients. Tohoku J. Exp. Med. 1975, 116, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Zinner, S.H.; Margolius, H.S.; Rosner, B.; Keiser, H.R.; Rass, E.H. Familial aggregation of urinary kallikrein concentration in childhood: Relation to blood pressure, race and urinary electrolytes. Am. J. Epidemiol. 1976, 104, 124–132. [Google Scholar] [PubMed]
- Levy, S.B.; Lilly, J.J.; Frigon, R.P.; Stone, L.A. Urinary kallikrein and plasma renin activity as determinants of renal blood flow. J. Clin. Invest. 1977, 60, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Seino, J.; Otsuka, Y.; Yoshinaga, K. Urinary kallikrein excretion and sodium metabolism in human hypertension. In Chemistry and Biology of the Kallikrein Kinin System in Health and Disease; Pisano, J., Austen, K.F., Eds.; Forgarty Internal Center Proceedings No.27: Washington, DC, USA, 1977; pp. 411–414. [Google Scholar]
- Geller, R.; Margolius, H.; Pisano, J.; Keiser, H. Effects of mineral corticoids, altered sodium intake and adrenalectomy on urinary kallikrein. Circ. Res. 1972, 31, 857–861. [Google Scholar] [PubMed]
- Bascands, J.; Cirolami, J.-P.; Pecher, C.; Monatti, J.P.; Manuel, Y.; Suc, J. Compared effects of a low and a high sodium diet on the renal and urinary concentration and activity of kallikrein in normal rats. J. Hypertens. 1987, 5, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Omata, K.; Carretero, O.A.; Itoh, S.; Scicli, A.G. Active and inactive kallikrein in rabbit connecting tubules and urine during low and normal sodium intake. Kidney Int. 1983, 24, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Margolius, H.; Geller, K.; Pisano, J.; Sjoerdsma, A. Altered urinary kallikrein excretion in human hypertension . Lancet 1971, ii, 1063–1965. [Google Scholar] [CrossRef]
- Lechi, A.; Covi, G.; Lechi, C.; Mantero, F.; Scuro, I. Urinary kallikrein excretion in Bartter's Syndrome. J. Clin. Endocrinol. Metab. 1976, 43, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Seino, M.; Abe, K.; Sakurai, Y.; Irokawa, N.; Yasujima, M.; Chiba, S.; Otsuka, Y.; Yoshinaga, K. Effect of spironolactone on urinary kallikrein excretion in patients with essential hypertension and in primary aldosteronism. Tohoku J. Exp. Med. 1977, 121, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, A. Urinary kallikrein determination and its physiological role in human kidney. J. Urol. 1971, 62, 507–518. [Google Scholar]
- Vio, C.P.; Figueroa, C.D. Evidence for a stimulatory effect of high potassium diet on renal kallikrein. Kidney Int. 1987, 31, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Chao, L.; Chao, J. Potassium supplement upregulates the expression of renal kallikrein and bradykinin B2 receptor in SHR . Am. J. Physiol.-Renal Physiol. 1999, 276, F476–F484. [Google Scholar]
- Suzuki, T.; Katori, M.; Fujita, T.; Kumagai, Y.; Majima, M. Involvement of the renal kallikrein-kinin system in K+-induced diuresis and natriuresis in anesthetized rats. Eur. J. Pharmacol. 2000, 399, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Hayashi, I.; Kumagai, Y.; Imamura, N.; Majima, M. Early increase in renal kallikrein excretion on administration of potassium or ATP-sensitive potassium channel blockers in rats. Br. J. Pharmacol. 1999, 128, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Himathongkam, T.; Duly, R.; William, G. Potassium aldosterone-renin interrelationships. J. Clin. Endocrinol. Metab. 1975, 41, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, I. A secretary mechanism of renal kallikrein by a high potassium ion; a possible involvement of ATP-sensitive potassium channel. Immunopharmacology 1999, 44, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Hayashi, I.; Fujita, T.; Cha, S.; Endou, H.; Higashihara, M.; Majima, M. Potassium-induced increase in renal kallikrein secretion is attenuated in dissected renal connecting tubules of young spontaneously hypertensive rats. Int. Immunopharmacology 2002, 2, 1957–1964. [Google Scholar] [CrossRef]
- Wang, T.; Wang, W.; Klein-Robbenhaar, G.; Giebisch, G. Effects of glyburide on renal tubule transport and potassium-channel activity. Ren. Physiol. Biochem. 1995, 18, 169–182. [Google Scholar] [PubMed]
- Kamata, Y.; Fujita, T.; Kato, T.; Hayashi, I.; Kurosaka, M.; Katori, M.; Fujita, T.; Majima, M. An ATP-sensitive potassium channel blocker suppresses sodium-induced hypertension through increased secretion of urinary kallikrein. Hypertens. Res. 2009, 32, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Perricone, S.; Humphrey, S.; Skaletzky, L.; Graham, B.; Zandt, R. Synthesis and diuretic activity of alkyl- and arylguanidine analogs of N,N'-dyclohexyl-4-morpholinecarboxamidine in rats and dogs. J. Med. Chem. 1994, 28, 3693–3700. [Google Scholar] [CrossRef]
- Meisheri, K.; Humphrey, S.; Khan, S.; Cipkus-Dubrey, L.; Smith, M. 4-morpholinecarboximidine-N-1-adamantyl-N'-cyclohexylhydrochloride (U-37883A): pharmacological characterization of a novel antagonist of vascular ATP-sensitive K+ channel openers. J. Pharmacol. Exp. Ther. 1993, 266, 655–665. [Google Scholar] [PubMed]
- Guillemare, E.; Honore, E.; De Weille, J.; Fosset, M.; Lazdunski, M.; Meisheri, K. Functional receptors in Xenopus oocytes for U-37883A, a novel ATP-sensitive K+ channel blocker: Comparison with rat insulinoma cells. Mol. Pharmacol. 1994, 46, 139–145. [Google Scholar] [PubMed]
- Wang, T.; Wang, W.; Klein-Robbenhaar, G.; Giebisch, G. Effects of a novel KATP channel blocker on renal tubule function and K channel activity. J. Pharmacol. Exp. Ther. 1995, 273, 1382–1389. [Google Scholar] [PubMed]
- Stanton, B. Characterization of apical and basolateral membrane conductance of rat inner medullary collecting duct . Am. J. Physiol. 1989, 256, F862–F868. [Google Scholar] [PubMed]
- Giebisch, G. Renal potassium channels: An overview. Kidney Int. 1995, 48, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Nakamura, R. Function of distal convoluted and connecting tubules studied by isolated nephron fragments. Kidney Int. 1982, 21, 5849–5856. [Google Scholar]
- Wright, F.; Giebisch, G. Renal potassium: Contributions of individual nephron segments and populations . Am. J. Physiol. 1978, 235, F515–F527. [Google Scholar] [PubMed]
- Muto, S. Potassium transport in the mammalian collecting duct. Physiol. Rev. 2001, 81, 85–116. [Google Scholar] [PubMed]
- Ward, P.E.; Margolius, H.S. Renal and urinary kallikrein. In Bradykinin Kallidin and Kallikrein. Handbook of Experimental Pharmacology; Erdös, E.G., Wilde, A.F., Eds.; Springer-Verlag: Berlin, Heidelberg, New York, 1979. [Google Scholar]
- Levinskey, N.G. The renal kallikrein-kinin system. Circ. Res. 1979, 44, 441–451. [Google Scholar] [PubMed]
- Carretero, O.A.; Scicli, A.G. The renal kallikrein-kinin system . Am. J. Physiol. 1980, 238, F247–F255. [Google Scholar] [PubMed]
- Mayfield, R.K.; Margolius, H.S. Renal kallikrein-kinin system. Relation to renal function and blood pressure. Am. J. Nephrol. 1983, 3, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Margolius, H.S. Tissue kallikreins and kinins: Regulation and roles in hypertensive and diabetic diseases. Annu. Rev. Pharmacol. 1989, 29, 343–364. [Google Scholar]
- Carretero, O.A.; Scicli, A.G. Kinins as regulators of blood flow and blood pressure. In Hypertension: Pathology, Diagnosis, and Management; 1990; Raven Press: New York, NY, USA. [Google Scholar]
- Margolius, H.S. Kallikreins and kinins. Some unanswered questions about system characteristics and roles in human disease. Hypertension 1995, 26, 221–229. [Google Scholar] [PubMed]
- Katori, M.; Majima, M. The renal kallikrein-kinin system: Its role as a safety valve for excess sodium intake, and its attenuation as a possible etiologic factor in salt-sensitive hypertension. Crit. Rev. Clin. Lab. Sci. 2003, 40, 43–115. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Melmon, K.; Gillepsie, L.; Bartter, F. Bradykinin and renal function in normal man: Effects of adrenergic blockade. Am. J. Physiol. 1965, 209, 844–848. [Google Scholar] [PubMed]
- Bönner, G.; Preis, S.; Schunk, U.; Toussaint, C.; Kaufmann, W. Haemodynamic effects of bradykinin on systemic and pulmonary circulation in healthy and hypertensive humans . J. Cardiovasc. Pharmacol. 1990, 15 (Suppl. 6), 546–556. [Google Scholar]
- Nakano, J. Effects of synthetic bradykinin on the cardiovascular system. Arch. Int. Pharmacodyn. Ther. 1965, 157, 1–13. [Google Scholar] [PubMed]
- Dollery, C.; Goldberg, L.; Pentecost, B. Effects of intrarenal infusions of bradykinin and acetylcholine on renal blood flow in man. Clin. Sci. 1965, 29, 433–441. [Google Scholar] [PubMed]
- Stein, J.; Ferris, T.; Huprich, J.; Smith, Y.; Osgood, R. Effect of renal vasodilatation on the distribution of cortical blood flow in the kidney of the dog. J. Clin. Invest. 1971, 50, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- MaGiff, J.; Itskovitz, H.; Terragno, N. The actions of bradykinin and eledoisin in the canine isolated kidney: Relation to prostaglandins. Clin. Sci. Mol. Med. 1975, 49, 125–131. [Google Scholar] [PubMed]
- Nasjletti, A.; Colina-Choourio, J. Interaction of mineral corticoids, renal prostaglandins, and the renal kallikrein-kinin system. Fed. Proc. 1976, 35, 59–65. [Google Scholar]
- Heller, J.; Kramer, H.; Horacek, V. The effect of kinin and prostaglandin inhibitors on the renal response to angiotensin-converting inhibition: A micropuncture study in the dog. Pflugers Archiv. 1994, 427, 219–224. [Google Scholar] [CrossRef]
- Roman, R.; Kadunski, M.; Sicli, A.; Carretero, O.A. Influence of kinins and angiotensin II on the regulation of papillary blood flow . Am. J. Physiol. 1988, 255, F690–F698. [Google Scholar] [PubMed]
- Zimmerman, B.; Raich, P.; Vavrek, R.; Stewart, J. Bradykinin contribution to renal blood flow effect of angiotensin converting enzyme inhibitor in the conscious sodium-restrict dog. Circ. Res. 1990, 66, 234–240. [Google Scholar] [PubMed]
- Kon, V.; Fogo, A.; Ichikawa, I. Bradykinin causes selective efferent arteriolar dilatation during angiotensin I converting enzyme inhibition. Kidney Int. 1993, 66, 545–550. [Google Scholar] [CrossRef]
- Mattson, D.; Roman, R. Role of kinins and angiotensin in the renal hemodynamic response to captopril . Am. J. Physiol. 1991, 260, F670–F679. [Google Scholar] [PubMed]
- Hajj-ali, A.; Zimmerman, B. Kinin contribution to renal vasodilator effect of captopril in rabbit. Hypertension 1991, 17, 504–509. [Google Scholar] [PubMed]
- Hajj-ali, A.; Zimmerman, B. Enhanced blood pressure and renal hemodynamic effect of chronic versus acute lisinopril administration in the rabbit. J. Pharmacol. Exp. Ther. 1992, 263, 158–162. [Google Scholar] [PubMed]
- Komers, R.; Cooper, M. Acute renal hemodynamic effect of ACE inhibition in diabetic hyperfiltration: Role of kinins . Am. J. Physiol. 1995, 268, F588–F594. [Google Scholar] [PubMed]
- Chen, K.; Zimmerman, B. Comparison of renal hemodynamic effect of captopril: Possible role of kinins. J. Pharmacol. Exp. Ther. 1994, 270, 491–497. [Google Scholar] [PubMed]
- Dworkin, L.; Brenner, B. The renal circulation. In The kidney; Brenner, B.M., Ed.; WB Saunders Company: Philadelphia, London, Toronto, Montreal, Sydney, Tokyo, 1996. [Google Scholar]
- Cowley, A.; Mattson, D.; Lu, H.; Roman, R. The renal medulla and hypertension. Hypertension. 1995, 25, 663–673. [Google Scholar] [PubMed]
- Mattson, D.; Cowley, A. Kinin actions on renal papillary blood flow and sodium excretion. Hypertension. 1993, 21, 961–965. [Google Scholar] [PubMed]
- Fenoy, F.; Scicli, A.; Carretero, O.; Roman, R. Effect of an angiotensin II and a kinin receptor antagonist on the renal hemodynamic response to captopril. Hypertension 1991, 17, 1038–1044. [Google Scholar] [PubMed]
- Lu, H.; Roman, R.; Mattson, D.; Cowley, A. Renal medullary interstitial infusion of diltiazem alters sodium and water excretion in rats . Am. J. Physiol. 1992, 263, R1064–R1070. [Google Scholar] [PubMed]
- Mattson, D.; Roman, R.; Cowley, A. Role of nitric oxide in renal papillary blood flow and sodium excretion. Hypertension 1992, 19, 766–769. [Google Scholar] [PubMed]
- Webster, M.; Gilmore, J. Influence of kallidin-10 on renal function. Am. J. Physiol. 1964, 206, 714–718. [Google Scholar] [PubMed]
- Barracough, M.; Mills, I. Effects of bradykinin on renal function. Clin. Sci. 1965, 28, 199–206. [Google Scholar]
- Tomita, K.; Pisano, J.; Brug, M.; Knepper, M. Effects of vasopressin and bradykinin on anion transport by the rat cortical collecting duct Evidence for an electroneutral sodium chloride transport pathway. J. Clin. Invest. 1986, 77, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Borkowski, J.; Rasom, R.; Seabrook, G.; Trumbauer, G.; Chen, H.; Hill, R.; Strader, C.; Hess, J. Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin action in smooth muscle and neuron. J. Biol. Chem. 1995, 270, 13706–13710. [Google Scholar] [CrossRef] [PubMed]
- Cavla, C.; Tordiras, M.; Iliescu, R.; Saul, V.; Gross, V.; Pilz, B.; Chai, G.; Merino, V.; Pesquero, J.; Baltatu, O.; Bader, M. Mice deficient for both kinin receptors are normotensive and protected from endotoxin-induced hypotension. FASEB J. 2007, 21, 1689–1698. [Google Scholar] [PubMed]
- Alfie, M.E.; Yang, X.P.; Hess, F.; Carretero, O.A. Salt-sensitive hypertension in bradykinin B2 receptor knockout mice. Biochem. Biophys. Res. Commun. 1996, 224, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Alfie, M.E.; Sigmon, D.H.; Pomposiello, S.I.; Carretero, O.A. Effect of high salt intake in mutant mice lacking bradykinin-B2 receptors. Hypertension 1997, 29, 483–487. [Google Scholar] [PubMed]
- Madeddu, P.; Varoni, M.; Palomba, D.; Emanueli, C.; Demontis, M.P.; Gilorioso, N.; Dessl-Fulgheri, P.; Sarzani, R.; Anaia, V. Cardiovascular phenotype of a mouse strain with disruption of bradykinin B2-receptor gene. Circulation 1997, 96, 3570–3578. [Google Scholar] [PubMed]
- Cervenka, L.; Harrison-Bernard, L.; Dipps, S.; Primrose, G.; Imig, J.; El-Dahr, S. Early onset salt-sensitive hypertension in bradykinin B(2) receptor null mice. Hypertension 1999, 34, 176–180. [Google Scholar] [PubMed]
- Mila, A.; Gross, V.; Plehm, R.; De Silva, J.J.; Bader, M.; Luft, F. Normal blood pressure and renal function in mice lacking the bradykinin B(2) receptor. Hypertension 2001, 37, 1473–1479. [Google Scholar] [PubMed]
- Gainer, J.; Brown, N.; Bachvarova, M.; Bastien, L.; Maltais, I.; Marceau, F.; Bachvarov, D. Altered frequency of a promoter polymorphism of the kinin B2 receptor gene in hypertensive African-Americans. Am. J. Hypertens. 2000, 13, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Meneton, P.; Bloch-Faure, M.; Ruetten, H.; Huang, W.; Bergaya, S.; Ceiler, D.; Martins, I.; Salmon, G.; Boulanger, C.; Nussberger, J.; Crozatier, B.; Gasc, J.; Heudes, D.; Bruneval, P.; Doetschman, T.; Menard, J.; Alhenc-Gelas, F. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc. Nat. Acad. Sci. 2001, 98, 2634–2639. [Google Scholar] [CrossRef]
- Griol-Charhbili, V.; Sbbah, L.; Colucci, J.; Vincent, M.; Baudrie, V.; Laude, D.; Elghozi, J.; Bruneval, P.; Picard, N.; Meneton, P.; Alhenc-Gelas, F.; Richer, C. Tissue kallikrein deficiency and renovascular hypertension in the mouse . Am. J. Physiol. Integral Comp. Physiol. 2009, 296, R1385–R1391. [Google Scholar]
- Picard, N.; Eladari, D.; El Moghrabi, S.; Planes, C.; Bourgeois, S.; Houillier, P.; Wang, Q.; Burnier, M.; Deschenes, G.; Knepper, M.; Meneton, P.; Chambrey, R. Defective ENaC processing and function in tissue kallikrein-deficient mice. J. Biol. Chem. 2008, 283, 4602–4611. [Google Scholar] [CrossRef] [PubMed]
- Slim, R.; Torremocha, F.; Marceau, T.; Pizard, A.; Hunt, S.; Vuagnat, A.; Williams, G.; Gauthier, F.; Jeunemiitre, X.; Alhenc-Gelas, F. Loss-of-function polymorphism of the human kallikrein gene with reduced urinary kallikrein activity. J. Am. Assoc. Nephrol. 2002, 13, 968–976. [Google Scholar]
- Madeddu, P.; Varoni, M.; Pinna-Parpaglia, P.; Glorioso, N.; Anania, V. Urinary kallikrein: A marker of blood pressure sensitivity to salt. Kidney Int. 1996, 49, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, P.; Varioni, M.; Chao, J.; Soimson, J.; Glorioso, N.; Anania, V. Kallikrein-kinin system and blood pressure sensitivity to salt. Hypertension 1997, 20, 471–477. [Google Scholar]
- Madeddu, P.; Vio, C.; Straino, S.; Salis, M.; Milia, A.; Emanuelli, C. Renal phenotype of low kallikrein rats. Kidney Int. 2001, 59, 2233–2242. [Google Scholar] [PubMed]
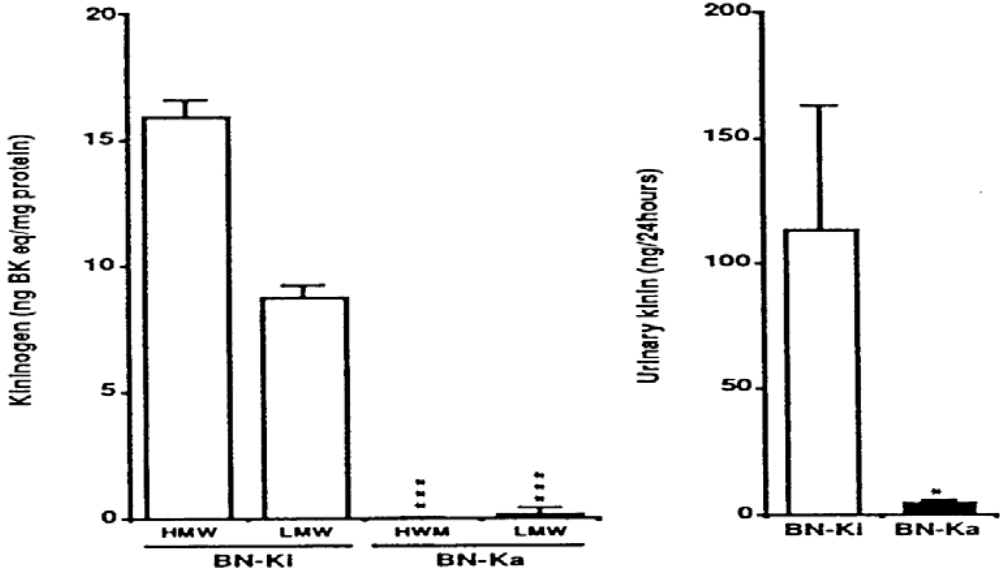
- Damas, J.; Adams, A. Congenital deficiency in plasma kallikrein and kininogen in the Brown Norway rat. Experientia 1980, 36, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Oh-ishi, S.; Satou, K.; I, H.; Yamazaki, K.; Nakano, T. Differences in prekallikrein and high molecular kininogen levels in two strains of Brown Norway rat (Kitasato strain and Katholiek strain). Thromb. Res. 1982, 28, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Oh-ishi, S.; Hayashi, I.; Hayasshi, M.; Yamaki, K.; Nagano, T.; Utsunomiya, I.; Nagashima, Y. Evidence for a role of the plasma kallikrein-kinin system in acute inflammation; Reduced exudation during carrageenin- and kaolin- pleurisies in kininogen-deficient rats. Agents Actions 1986, 18, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Majima, M.; Katori, M.; Hanazuka, z.; Mizogami, S.; Nakano, T.; Nakao, Y.; Mikami, R.; Uryu, H.; Okamura, R.; Mohsin, S.; Oh-ishi, S. Suppression of rat deoxycoricosterone-salt hypertension by kallikrein-kinin system. Hypertension 1991, 17, 806–813. [Google Scholar] [PubMed]
- Yamasu, S.; Oh-ishi, S.; Hayashi, I.; Hayashi, M.; Yamaki, K.; Nakano, T.; Sunhara, N. Differentiation of kinin fractions in ureter urine and bladder urine of normal and kininogen deficient rats. J. Pharamacodyn. 1989, 12, 287–292. [Google Scholar]
- Hayashi, I.; Hoshiko, S.; Manabe, O.; Oh-ishi, S. A point mutation of Alanine163 to threonine is responsible for the defective secretion of high molecular weight kininogen by the liver of Brown Norway Katholiek rats. J. Biol. Chem. 1993, 268, 17219–17224. [Google Scholar] [PubMed]
- Rhaleb, N.-E.; Yang, X.P.; Nanba, M.; Shesely, E.G.; Carretero, O.A. Effect of chronic blockade of the kallikrein-kinin system on the development of hypertension in rats. Hypertension 2001, 37, 121–128. [Google Scholar] [PubMed]
- Lattion, A.; Baussant, T.; Alhenc-Gelas, F.; Seidah, N.; Corvol, P.; Soubrier, F. The high-molecular-mass kininogen deficient rat expresses all kininogen mRNA species, but does not export the high-molecular-mass kininogen synthesized. FEBS Lett. 1988, 239, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Majima, M.; Adachi, K.; Kuribayashi, Y.; Mizogami, S.; Katori, M. Increase in vascular sensitivity to angiotensin II and epinephrine after four-day infusion of 0.3 M sodium chloride in conscious kininogen-deficient Brown Norway Katholiek rats . Jpn. J. Pharmacol. 1995, 69, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Bagdasarian, A.; Talamo, R.; Scott, C.; Seavey, M.; Guimaraes, J.; Pierce, J.; Kaplan, A. Williams trait: Human kininogen deficiency with diminished levels of plasminogen proactivator and prekallikrein associated with abnormalities of the Hageman factor-dependent pathways. J. Clin. Invest. 1975, 56, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, M.; Varet, B.; Levey, J.-P. A hitherto undescribed plasma acting at the contact phase of blood coagulation (Flaujeac factor): Case report and coagulation studies. Blood 1975, 46, 761–768. [Google Scholar] [PubMed]
- Wuepper, K.; Miller, R.; Lacombe, M. Flaujeac trait: Deficiency of human plasma kininogen. J. Clin. Invest. 1975, 56, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, V.; Glueck, H.; Miller, M.; Movat, H.; Habal, F. Kininogen deficiency in Fitzgerald trait: Role of high molecular weight kininogen in clotting and fibrinolysis. J. Lab. Clin. Invest. 1976, 87, 327–337. [Google Scholar]
- Hayashi, H.; Koya, H.; Kitajima, K.; Kimura, I. Coagulation factor deficiency apparently related to the Fitzgerald trait: The first case in Japan. Acta Med. Okayama 1978, 32, 81–83. [Google Scholar] [PubMed]
- Oh-ishi, S.; Ueno, A.; Ucida, Y.; Katori, M.; Hayashi, H.; Koya, H.; Kitajima, K.; Kimura, I. Abnormalities in the contact activation through factor XII in Fujiwara trait: A deficiency in both high and low molecular weight kininogen with low level of prekallikrein. Tohoku J. Exp. Med. 1981, 133, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Oh-ishi, S.; Hayashi, I.; Utsunomiya, I.; Hayashi, M.; Yamaki, K.; Yamasu, A.; Nakano, T. Role of kallikrein-kinin system in acute inflammation: Studies on high- and low-molecular weight kininogen-deficient rats (B/N-Katholiek strain). Agents Actions 1987, 21, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Majima, M.; Yoshida, O.; Mihara, H.; Muto, T.; Mizogami, S.; Kuribayashi, Y.; Katori, M.; Oh-ishi, S. High sensitivity to salt in kininogen-deficient Brown Norway Katholiek rats. Hypertension 1993, 22, 705–714. [Google Scholar] [PubMed]
- Coleman, T.; Manning, R.J.; Norman, R.J.; Granger, H.; Guyton, A. The role of salt in experimental and human hypertension. Am. J. Med. Sci. 1972, 264, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Majima, M.; Mizogami, S.; Kuribayashi, Y.; Katori, M.; Oh-ishi, S. Hypertension induced by a nonpressor dose of angiotensin II in kininogen-deficient rats. Hypertension 1994, 24, 111–119. [Google Scholar] [PubMed]
- Berk, B.; Vallega, G.; Muslin, A.; Gordon, H.; Canessa, M.; Alexander, R. Spontaneously hypertensive rat vascular smooth muscle cells in culture exhibit increased growth and Na+/H+ exchange. J. Clin. Invest. 1989, 83, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Takeda, K.; Okajima, H.; Takahashi, H.; Yoshimura, M.; Nakagawa, M.; Itiji, H. Pressor responses to intracisternal injection of hypertonic NaCl in rats. J. Cardiovasc. Pharmacol. 1984, 6, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Eagan, B. Neurogenic mechanism of initiating essential hypertension . Am. J. Hypertens. 1989, 2, 357S–362S. [Google Scholar] [PubMed]
- Elliot, A.H.; Nuzum, F.R. The urinary excretion of a depressor substance (kallikrein of Frey and Kraut) in arterial hypertension. Endocrinology 1934, 18, 462–474. [Google Scholar] [CrossRef]
- Carretero, O.; Oza, N.; Schork, A. Renal kallikrein, plasma renin, and plasma aldosterone in renal hypertension. Acta Physiolog. Latin Am. 1974, 24, 448–452. [Google Scholar]
- Horwitz, D.; Margolius, H.S.; Keiser, H.R. Effects of dietary potassium and race on urinary excretion of kallikrein and aldosterone in man. J. Clin. Endocrinol. Metab. 1978, 47, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Yasujima, M.; Irokawa, N.; Seino, M.; Chiba, S.; Sakurai, Y.; Sato, M.; Imai, M.; SAito, K.; Ito, T.; Haryuuama, T.; Otsuka, Y.; Yoshinaga, K. The role of intrarenal vasoactive substances in the pathogenesis of essential hypertension . Clin. Sci. Mol. Med. Suppl. 1978, 4, 363s–366s. [Google Scholar]
- Lechi, A.; Covi, G.; Lechi, C.; Corgati, A.; Arosio, E.; Zatti, M.; Scuro, L. Urinary kallikrein excretion and plasma renin activity in patient with essential hypertension and primary aldosteronism. Clin. Sci. Mol. Med. 1978, 55, 51–55. [Google Scholar] [PubMed]
- Keiser, H. The kallikrein-kinin system in essential hypertension. Clin. Exp. Hypertens. 1979, 2, 675–691. [Google Scholar] [CrossRef]
- Mersey, J.; Williams, G.; Emanual, R.; Dlunhy, R.; Wong, P.; Moore, T. Plasma bradykinin levels and urinary kallikrein excretion in normal renin essential hypertension. J. Clin. Endocrinol. Metab. 1979, 48, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, K.; Ura, N.; Tanaka, S.; Ogasawara, A.; Nakao, T.; Nakanhashi, Y.; Chao, J.; Margolius, H.; IImura, O. Excretion of human urinary kallikrein quantity measured by a direct radioimmunoassay of human urinary kallikrein in patients with essential hypertension and secondary hypertensive diseases. Jpn. Circ. J. 1981, 45, 1092–1097. [Google Scholar]
- Ura, N.; Shimamoto, K.; Nakao, T.; Ogaswara, A.; Tanaka, S.; Mita, T.; Nishimiya, T.; Iimura, O. The excretion of human urinary kallikrein quantity and activity in normal and low renin subgroups of essential hypertension. Clin. Exp. Hypertens. A. 1983, 5, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Favre, L.; Jornot, L.; Riondel, A.; Vallotton, M. Urinary excretion of renal prostaglandins, kallikrein, vasopressin and aldosterone in essential hypertension. Clin. Exp. Hypertens. A. 1985, 7, 1663–1679. [Google Scholar] [CrossRef] [PubMed]
- Carretero, O.A.; Scicli, A.G. The kallikrein-kinin system in human and in experimental hypertension . Klin. Wochenschr. 1978, 56 (Suppl. 1), 113–125. [Google Scholar] [CrossRef] [PubMed]
- Zinner, S.H.; Margolius, H.S.; Rosner, B.; Kass, E.H. Stability of blood pressure rank and urinary kallikrein concentration in childhood: An eight-year follow-up. Circulation 1978, 58, 908–915. [Google Scholar] [PubMed]
- Williams, R.R.; Hunt, S.C.; Hoplins, P.; Wu, L.; Hasstedt, S.J.; Berry, T.D.; Barlow, G.K.; Stults, B.M.; Schumacher, M.C.; Ludwig, E.H.; Elbein, S.C.; Wilson, D.E.; Lifton, R.P.; Lalouel, J.M. Genetic basis of familial dyslipidemia and hypertension: 15-years results from Utah . Am. J. Hypertens. 1993, 6, 319S–327S. [Google Scholar] [PubMed]
- Weinberger, M.; Miller, J.; Luft, F.; Grim, C.; Fineberg, N. Definitions and characteristics of sodium sensitivity and resistance of blood pressure . Hypertension 1986, 8 (6 Pt. 2), 127–134. [Google Scholar]
- Elliott, P.; Mamot, M.; Dyer, A.; Joossens, J.; Kesteloot, H.; Stamler, R.; Stamler, J.; Rose, G. The INTERSALT study: Main results, conclusions and some implications. Clin. Exp. Hypertens. A. 1989, 11, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Delea, C.; Bartter, F.; Smith, H. The effect of high-sodium and low-sodium intake on blood pressure and other related variables in human subjects with idopathic hypertension. Am. J. Med. 1978, 64, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M. Salt sensitivity; does it play an important role in the pathogenesis and treatment of hypertension? Curr. Opin. Nephrol. Hypertens. 1996, 5, 205–208. [Google Scholar] [CrossRef]
- Weinberger, M.; Fineberg, N. Sodium and volume sensitivity of blood pressure. Age and pressure change over time. Hypertension 1991, 18, 67–71. [Google Scholar] [PubMed]
- Katori, M.; Majima, M. A missing link between a high salt intake and blood pressure increase. J. Pharmacol. Sci. 2006, 100, 370–390. [Google Scholar] [CrossRef] [PubMed]
- Katori, M.; Majima, M. Are all individuals equally sensitive in the blood pressure to high salt intake? Acta Physiol. Hung. 2008, 95, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Bönner, G.; Thieven, B.; Rütten, H.; Chrosch, R.; Krone, W. Renal kallikrein is a determinant of salt sensitivity . J. Hypertens. 1993, 11 (Suppl. 5), S210–211. [Google Scholar]
- Ferri, C.; Bellini, C.; Carlomagno, A.; Perrone, A.; Santucci, A. Urinary kallikrein and salt sensitivity in essential hypertensive males. Kidney Int. 1994, 46, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Katori, M.; Majima, M.; Mohsin, S.S.J.; Hanazuka, M.; Mizogami, S.; Oh-ishi, S. Essential role of kallikrein-kinin system in suppression of blood pressure rise during developmental stage of hypertension induced by deoxycorticosterone acetate–salt in rats . Agents Actions Suppl. 1992, 38 (Pt.3), 235–242. [Google Scholar] [PubMed]
- Okamoto, K.; Aoki, K. Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J. 1963, 27, 282–293. [Google Scholar]
- Keiser, H.R.; Geller, K.; Margolius, H.S.; Pisano, J.J. Urinary kallikrein in hypertensive models. Fed. Proc. 1976, 35, 199–202. [Google Scholar] [PubMed]
- Ader, J.-L.; Pollock, D.; Butterfield, M.; Arendshorst, W. Abnormalities in kallikrein excretion in spontaneously hypertensive rats . Am. J. Physiol. 1985, 248, F396–F403. [Google Scholar] [PubMed]
- Praddaude, F.; Tran-Van, T.; Ader, J.-L. Renal kallikrein activity in rats developing spontaneous hypertension. Clin. Sci. 1989, 76, 311–315. [Google Scholar] [PubMed]
- Ader, J.-L.; Tran-Van, T.; Praddaude, F. Reduced urinary kallikrein activity in rats developing spontaneous hypertension . Am. J. Physiol. 1987, 252, F964–F969. [Google Scholar] [PubMed]
- Mohsin, S.S.J.; Majima, M.; Katori, M.; Sharma, J.N. Important suppressive roles of the kallikrein-kinin system during the developmental stage of hypertension in spontaneously hypertensive rats. Asia Pac. J. Pharmacol. 1992, 7, 73–82. [Google Scholar]
- Dilley, J.; Stier, C.; Ardenshorst, W. Abnormalities in glomerular function in rats developing spontaneous hypertension . Am. J. Physiol. 1984, 246, F12–F20. [Google Scholar] [PubMed]
- Gellar, R.; Margolius, H.S.; Pisano, J.; Keiser, H.R. Effects of chronic excess salt ingestion. Evidence that genetic factors play an important role in susceptibility to experimental hypertension. Circ. Res. 1975, 36 (Suppl. I.). [Google Scholar]
- Figueroa, C.D.; Bhoola, K.; Maclever, A.; Mckenzie, J. An ontogenic study of renal tissue kallikrein in Okamoto spontaneously hypertensive rats: Comparison with human hypertensive nephropathy. Nephrol. Dial. Transplant. 1992, 7, 516–525. [Google Scholar] [PubMed]
- Dahl, L.; Heine, M.; Tassinari, L. Effects of chronic excess salt ingestion. Evidence that genetic factors play an important role in susceptibility to experimental hypertension. J. Exp. Med. 1962, 115, 1173–1190. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.; Knudsen, K.; Heine, M.; Leiti, G. Effects of chronic excess salt ingestion. Genetic influence on the development of salt hypertension in parabiotic rats: Evidence for a humoral factor. J. Exp. Med. 1967, 126, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Iwai, J.; Knudsen, K.; Dahl, L.; Heine, M.; Leiti, G. Genetic influence on the development of renal hypertension in parabiotic rats. Evidence for humoral factor. J. Exp. Med. 1969, 129, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.; Iwai, J.; Heine, M.; Leiti, G.; Dahl, L. Genetic influence on the development of renoprival hypertension in parabioteic rats. Evidence that a humoral hypertensionogenic factor is produced in kidney tissue of hypertension-prone rats. J. Exp. Med. 1969, 130, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.; Heine, M.; Tassinari, L. Effects of chronic excess salt ingestion. Role of genetic factors in both DOCA-salt and renal hypertension. J. Exp. Med. 1963, 118, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.; Heine, M.; Tassinari, L. Effects of chronic excess salt ingestion. Further demonstration that genetic factors influence the development of hypertension; evidence from experimental hypertension due to cortisone and to adrenal regeneration. J. Exp. Med. 1965, 122, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Churchill, P.; Churchill, M.; Bidani, A.; Rabito, S. Kallikrein excretion in Dahl salt–sensitive and salt-resistant rats with native and transplanted kidney . Am. J. Physiol. 1995, 269, F710–717. [Google Scholar] [PubMed]
- Arbeit, L.; Serra, S. Decreased total and active urinary kallikrein in normotensive Dahl salt susceptible rats. Kidney Int. 1985, 28, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Rapp, J.; Tan, S.; Margolius, H.S. Plasma mineral corticoids, plasma renin, and urinary kallikrein in salt-sensitive and salt-resistant rats. Endocrinol. Res. Comm. 1978, 5, 35–41. [Google Scholar] [CrossRef]
- Sustarsic, D.; McPartland, R.; Rapp, J. Developmental patterns of blood pressure and urinary protein, kallikrein, and prostaglandin E2 in Dahl salt-hypertension-susceptible rats. J. Lab. Clin. Med. 1981, 98, 599–606. [Google Scholar] [PubMed]
- Rapp, J.; Joseph, M.; McPartland, R. Proteins binding to kallikrein and esterase A2 in the urine of salt-sensitive and salt-resistant rats. Hypertension 1982, 4, 545–555. [Google Scholar] [PubMed]
- McPartland, R.; Rapp, J.; Sustarsic, D. Effects of dexamethasone on excretion of urinary kallikrein and urinary protein in Dahl salt-sensitive and salt-resistant rats. Endocrine Res. Com. 1981, 8, 145–153. [Google Scholar] [CrossRef]
- Rapp, J.; McPartland, R.; Sustarsic, D. Anomalous response of urinary kallikrein to deoxycorticosterone in Dahl salt-sensitive rats. Hypertension 1982, 4, 20–26. [Google Scholar]
- Rapp, J.; McPartland, R.; Batten, C. Isoelectric focusing patterns of urinary kallikrein in Dahl salt-hypertension susceptible and resistant rats. Hypertension 1984, 6, 519–525. [Google Scholar] [PubMed]
- Carretero, O.A.; Polomski, C.; Hampton, A.; Scicli, A.G. Urinary kallikrein, plasma renin and aldosterone in New Zealand genetically hypertensive (GH) rats . Clin. Exp. Pharmacol. Physiol. 1976, 3 (Suppl.), 55–59. [Google Scholar]
- Gilboa, N.; Rudofsky, U.; Margo, A. Urinary and renal kallikrein in hypertensive fawn-hooded (FH/Wjd) rats. Lab. Invest. 1984, 50, 72–78. [Google Scholar] [PubMed]
- Bianchi, G.; Fox, U.; OImbasciati, E. The development of a new strain of spontaneously hypertensive rats. Life Sci. 1974, 14, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, G.; Bianchi, G.; Croxatto, H.R. Urinary kallikrein excretion in a spontaneously hypertensive strain of rats. Proc. Natl. Acad. Sci. USA 1975, 149, 983–986. [Google Scholar]
- Whelton, P. Potassium and blood pressure. In Hypertension Primer; 2008; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA. [Google Scholar]
- Ito, H.; Majima, M.; Nakajima, S.; Hayashi, I.; Katori, M.; Izumi, T. Effect of prolonged administration of a urinary kininase inhibitor, ebelactone B on the development of deoxycorticosterone acetate-salt hypertension in rats. Br. J. Pharmacol. 1999, 126, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Giros, B.; Gros, C.; Schwartz, J.; Danvy, D.; Plaquevent, J.; Duhamel, L.; Duhamel, P.; Vlaiculescu, A.; Costentin, J.; Lecomte, J. Enantiomers of thiorphan and acetorphan: Correlation between enkephalinase inhibitor, protection of endogenous enkaphalins and behavioral affects. J. Pharmacol. Exp. Ther. 1987, 243, 666–673. [Google Scholar] [PubMed]
- Lecomte, J.; Baumer, P.; Lim, C.; Duchier, J.; Cournot, A.; Dussaule, J.-C.; Ardaillou, R.; Gros, C.; Chaigon, B.; Souque, A.; Schwartz, J. Steroselective protection of exogenous and endogenous atrial natriuretic factors by enkephalinase inhibitors in mice and humans. Eur. J. Pharmacol. 1990, 170, 65–73. [Google Scholar] [CrossRef]
- Ertl, G.; RA, K.; de Graeff, P.; Kingma, J.; Wesseling, H.; de Langen, C. Captopril reduced purine loss and reperfusion arrhythmias in the rats heart after coronary artery occlusion. Europ. J. Pharmacol. 1984, 100, 113–117. [Google Scholar] [CrossRef]
- Linz, W.; Schoelkens, B.; Han, Y. Beneficial effects of converting enzyme inhibitor, ramipril, in ischemic rat hearts. J. Cardiovasc. Pharmacol. 1986, 8 (Suppl.). [Google Scholar]
- Schölkens, B.; Linz, W.; Konig, W. Effects of the angiotensin converting enzyme inhibitor, ramipril, in isolated ischemic rat heart are abolished by a bradykinin antagonist . J. Hypertens. 1988, 6 (Suppl.), S25–S28. [Google Scholar]
- Linz, W.; Wiemer, G.; Gohlke, P.; Unger, T.; Schölkens, B. Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol. Rev. 1995, 47, 25–49. [Google Scholar] [PubMed]
- Dendorfer, A.; Wlofrum, S.; Dominiak, P. Pharmacology and cardiovascular implications of the kinin-kallikrein system. Jpn. J. Pharmacol. 1999, 79, 403–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, V.; Sharov, V.; Sigmon, D.; Sabbath, H.; Carretero, O.A. Paracrine systems in the cardioprotective effect of angiotensin-converting enzyme inhibitors on myocardial ischemia/reperfusion injury in rats. Hypertension 1996, 27, 7–13. [Google Scholar] [PubMed]
- Linz, W.; Wiemer, G.; Schaper, J.; Zimmermann, R.; Nagasawa, K.; Gohlke, P.; Unger, T.; Schölkens, B. Angiotensin converting enzyme inhibitors, left ventricular hypertrophy and fibrosis. Mol. Biochem. Parasitol. 1995, 147, 89–97. [Google Scholar]
- Yayama, K.; Matsuoka, S.; Nagaoka, M.; Shimizu, E.; Takano, M.; Okamoto, H. Down-regulation of bradykinin B2-receptor mRNA in the heart in pressure-overload cardiac hypertrophy in the rats. Biochem. Pharmacol. 2003, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.; Bader, M.; Presquero, J.; Tschope, C.; Groningen, E.; van Gilst, W.; Buikema, H. Increased kallikrein expression protects against cardiac ischemia. FASEB J. 2000, 14, 1861–1863. [Google Scholar] [PubMed]
- Schölkens, B.; Linz, W. Bradykinin-mediated metabolic effects in isolated perfused rat hearts. Agents Actions 1992, 38, 36–42. [Google Scholar]
- Ito, H.; Hayashi, I.; Izumi, T.; Majima, M. Bradykinin inhibits development of myocardial infarction through B2 receptor signaling by increment of regional blood flow around the ischemic lesion in rats. Br. J. Pharmacol. 2003, 138, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.; Jennings, R.; Reier, K. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [PubMed]
- Yellon, D.; Alkhulaifi, A.; Browne, E.; Pugsley, W. Ischemic preconditioning limits infarct size in the rat heart. Cardiovasc. Res. 1992, 26, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Downey, J. Preconditioning: State of the art myocardial protection. Cardiovasc. Res. 1993, 27, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Linz, W.; Wiemer, G.; Schölkens, B. Role of kinins in the pathophysiology of myocardial ischemia. In vitro and in vivo studies. Diabetes 1996, 45, S51–S58. [Google Scholar] [PubMed]
- Yayama, K.; Nagaoka, M.; Takano, M.; Okamoto, H. Expression of kininogen, kallikrein and kinin receptors genes by rat cardiomyocytes. Biochim. Biophys. Acta. 2000, 1495, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Brew, E.; Mitchel, M.; Rhering, T.; Gamboni-Robertson, F.; McIntyre, R.J.; Harken, A.; Banerrjee, A. Role of bradykinin in cardiac functional protection after global ischemia-reperfusion in rat heart . Am. J. Physiol. 1995, 269, H1370–H1378. [Google Scholar] [PubMed]
- Vegh, A.; Szekeres, L.; Parratt, J. Protective role of preconditioning of the ischaemic myocardium involve cyclo-oxygenase products. Cardiovasc. Res. 1990, 24, 1020–1023. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Liu, Y.; Uang, X.; Ardell, J.; Cohen, M.; Downey, J. Role of bradykinin in protection of ischemic preconditioning in rabbit hearts. Circ. Res. 1995, 77, 611–621. [Google Scholar] [PubMed]
- Yang, X.; Liu, Y.; Sccli, G.; Webb, C.; Carretero, O. Role of kinins in the cardioprotective effect of preconditioning: Study of myocardial ischemia/reperfusion injury in B2 kinin receptor knockout mice and kininogen deficient rats. Hypertension 1997, 30, 735–740. [Google Scholar] [PubMed]
- Schultz, R.; Post, H.; Vahlhaus, C.; Heusch, G. Ischemic preconditioning in pigs: A graded phenomenon. Its relation to adenosine and bradykinin. Circulation 1998, 98, 1022–1029. [Google Scholar] [PubMed]
- Pan, H.-L.; Chen, S.-R.; Scicli, G.; Carretero, O.A. Cardiac interstitial bradykinin release during ischemia is enhanced by ischemic preconditioning . Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H116–H121. [Google Scholar] [PubMed]
- Griol-Charhbili, V.; Messadi-Laribi, E.; Bascands, J.-L.; Heudes, D.; Menton, P.; Giudecelli, J.-F.; Alhenc-Gelas, F.; Richer, C. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. FASEB J. 2005, 19, 1172–1174. [Google Scholar] [PubMed]
- Deutsch, E.; Berger, M.; Kussmaul, W.; Hirshfeld, J.J.; Herrmann, H.; Laskey, W. Adaptation to ischemia during percutaneous transluminal coronary angioplastry. Clinical, hemodynamic, and metabolic features. Circulation 1990, 82, 2044–2051. [Google Scholar] [PubMed]
- Yellon, D.; Alkhulaifi, A.; pulgsley, W. Preconditioning the human myocardium. Lancet 1993, 342, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Lessar, M.; Stoddard, M.; Manchikalapudi, S.; Bolli, R. Bradykinin-induced preconditioning in patients undergoing coronary angioplasty. J. Am. Coll. Cardiol. 1999, 34, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Schölkens, B. Kinins in the cardiovascular system. Immunopharmacology. 1996, 33, 209–2126. [Google Scholar] [CrossRef] [PubMed]
- Linz, W.; Wiemer, G.; Schölkens, B. Beneficial effects of bradykinin on myocardial energy metabolism and infarct size . Am. J. Cardiol. 1997, 80, 118A–123A. [Google Scholar] [CrossRef] [PubMed]
- Mclean, P.; Perretti, M.; Ahluwalia, A. Kinin B(1) receptors and cardiovascular system: Regulation of expression and function. Cardiovasc. Res. 2000, 48, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Cohen, M.; Behrends, M.; Downey, J.; Heusch, G. Signal transduction of ischemic preconditioning. Cardiovasc. Res. 2001, 52, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Messadi-Laribi, E.; Griol-Charhbili, V.; Gaies, E.; Vincent, M.; Heudes, D.; Menton, P.; Alhenc-Gelas, F.; Richer, C. Cardioprotection and kallikrein-kinin system in acute myocardial ischemia in mice. Clin. Exp. Pharmacol. Physiol. 2008, 35, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Bledsoe, G.; Yin, H.; Chao, L. The tissue kallikrein-kinin system protects against cardiovascular and renal diseases and ischemic stroke independently of blood pressure reduction. Biol. Chem. 2008, 387, 665–675. [Google Scholar] [CrossRef]
- Stone, O.; Richer, C.; Emanuelli, C.; van Weel, V.; Quax, P.; Katare, R.; Krankel, N.; Campagnolo, P.; Barcelos, L.; Soirgusa, M.; Sla-Newby, G.; Baldessari, D.; Mione, M.; Vincent, M.; Benest, A.; Al Haj Zen, A.; Gonzalez, J.; Bates, D.; Alhenc-Gelas, F.; Madeddu, P. Critical role of tissue kallikrein in vessel formation and maturation: Implications for therapeutic revascularization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Farhy, R.; Carretero, O.; Scicli, A. Role of kinins and nitric oxide in the effects of angiotensin converting enzyme inhibitors on neointima formation. Circ. Res. 1993, 72, 1202–1210. [Google Scholar] [PubMed]
- Murakami, H.; Yayama, K.; Miao, R.; Wang, C.; Chao, J.; Chao, L. Kallikrein gene delivery inhibits vascular smooth muscle cell growth and neointima formation in the rat artery after balloon angioplasty. Hypertension 1999, 34, 164–170. [Google Scholar] [PubMed]
- Emanueli, C.; Bonaria Salis, M.; Figueroa, C.D.; Chao, J.; Chao, L.; Capogrossi, M.; Madeddu, P. Participation of kinins in the captopril-induced inhibition of intimal hyperplasia caused by interruption of carotid blood flow in the mouse. Br. J. Pharmacol. 2000, 130, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Kränkel, N.; Katare, R.; Siragusa, M.; Barcelps, L.; Cpmpagnolo, P.; Mangialardi, G.; Fortunato, O.; Spinetti, G.; Tran, N.; Zacharowski, K.; Wngelini, G.; Emanueli, C.; Madeddu, P. Role of kinin B2 receptor signaling in the recruitment of circulating progenitor cells with neovascularization potential. Circ. Res. 2008, 103, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, I.; Amano, H.; Yoshida, S.; Kamata, K.; Inukai, M.; Fujita, T.; Kumagai, Y.; Furudate, S.; Majima, M. Suppressed angiogenesis in kininogen-deficiencies. Lab. Invest. 2002, 82, 871–880. [Google Scholar] [PubMed]
- Ishihara, K.; Hayashi, I.; Yamashina, S.; Majima, M. A potential role of bradykinin in angiogenesis and growth of S-180 mouse tumors. Jpn. J. Pharmacol. 2001, 87, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; kamata, M.; Hayashi, I.; Yamashina, S.; Majima, M. Role of bradykinin in vascular permeability and angiogenesis in solid tumor. Int. Immunopharmacol. 2002, 2, 499–509. [Google Scholar] [CrossRef]
- Ikeda, T.; Hayashi, I.; Kamoshita, E.; Yamazaki, A.; Endo, H.; Ishihara, K.; Yamashina, S.; Tsutsumi, Y.; Matsubara, H.; Majima, M. Host stromal bradykinin B2 receptor signaling facilitates tumor-associated angiogenesis and tumor growth. Cancer Res. 2004, 64, 5178–5185. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Colman, R. Two faces of high-molecular-weight kininogen (HK) in angiogenesis: Bradykinin turns it on and cleaved HK (HKa) turns it off. J. Thromb. Haemost. 2005, 3, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Schmuckler, N.; Bakdash, N.; Yoder, M.; Overall, C.; Coleman, R. Cleaved high molecular weight kininogen inhibits tube formation of endothelial pregenitor cells via suppression of matrix metalloproteinase 2. J. Thromb. Haemost. 2009, 8, 185–193. [Google Scholar] [CrossRef]
- Yao, Y.; Yin, H.; Shen, B.; Smith, R.J.; Liu, Y.; Chao, L.; Chao, J. Tissue kallikrein promotes neovascularization and improves cardiac function by the Akt-glycogen synthase kinase-3beta pathway. Circ. Res. 2008, 103, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Madeddu, P. Targeting kinin receptors for the treatment of tissue ischemia. Trends Pharmacol. Sci. 2001, 22, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Borgono, C.; Diamandis, E. The emerging roles of human kallikreins in cancer. Nat. Rev. Cancer 2004, 4, 876–890. [Google Scholar] [CrossRef]
- Madeddu, P. Therapeutic angiogenesis and vasculogenesis for tissue regeneration. Exp. Physiol. 2005, 90, 315–326. [Google Scholar] [CrossRef] [PubMed]
- McCrae, K.; Donate, F.; Merkulov, S.; Sun, D.; Qi, X.; Shaw, D. Inhibition of angiogenesis by cleaved high molecular weight kininogen (HKa) and HKa domain 5. Curr. Cancer Drug Targets 2005, 5, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Bledsoe, G.; Yin, H.; Chao, L. The tissue kallikrein-kinin system protects against cardiovascular and renal disease and ischemic stroke independently of blood pressure. Biol. Chem. 2006, 387, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Schulthesis, H.; Tschöpe, C. New perspective on the tissue kallikrein-kinin system in myocardial infarction: Role of angiogenesis and cardiac regeneration. Int. Immunopharmacol. 2008, 8, 148–154. [Google Scholar] [CrossRef]
- Chao, J.; Chao, L. Kallikrein gene therapy: A new strategy for hypertensive diseases. Immunopharmacology 1997, 36, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Chao, L. Experimental kallikrein gene therapy in hypertension, cardiovascular and renal diseases. Pharmacol. Res. 1997, 35, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Chao, J.; Chao, L. Muscle delivery of human kallikrein gene reduced blood pressure in hypertensive rats. Hypertension 1965, 25, 715–719. [Google Scholar]
- Chao, J.; Chao, L. Functional analysis of human tissue kallikrein in transgenic mouse models. Hypertension 1996, 27, 491–494. [Google Scholar] [PubMed]
- Wang, C.; Chao, L.; Chao, J. Direct gene delivery of human tissue kallikrein reduced blood pressure in spontaneously hypertensive rats. J. Clin. Invest. 1995, 95, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhang, J.; Chao, L.; Chao, J. Gene therapy in hypertension: Adenovirus-mediated kallikrein gene delivery in hypertensive rats. Hum. Gene Ther. 1997, 8, 1753–1761. [Google Scholar] [CrossRef]
- Chao, J.; Zhang, J.; Lin, K.; Chao, L. Adenovirus-mediated kallikrein gene delivery reverses salt-induced renal injury in Dahl salt-sensitive rats. Kidney Int. 1998, 54, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Zhang, J.; Lin, K.; Chao, L. Human kallikrein gene delivery attenuates hypertension, cardiac hypertrophy, and renal injury in Dahl salt-sensitive rats. Hum. Gene Ther. 1998, 9, 21–31. [Google Scholar] [CrossRef]
- Yayama, K.; Wang, C.; Chao, L.; Chao, J. kallikrein gene delivery attenuates hypertension and cardiac hypertrophy and enhances renal function in Goldblatt hypertensive rats. Hypertension 1998, 31, 1104–1110. [Google Scholar] [PubMed]
- Dobrzynski, E.; Yoshida, H.; Chao, J.; Chao, L. Adenovirus-mediated kallikrein gene delivery attenuates hypertension and protect against renal injury in deoxycorticosterone-salt rats. Immunopharmacology 1999, 44, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Bledsoe, G.; Chao, L.; Chao, J. Kallikrein gene transfer reduced renal fibrosis, hypertrophy, and proliferation in DOCA-salt hypertensive rats . Am. J. Physiol. Renal Physiol. 2005, 289, F622–F631. [Google Scholar] [CrossRef] [PubMed]
- Wolf, W.; Yoshida, H.; Agata, J.; Chao, L.; Chao, J. Human tissue kallikrein gene delivery attenuates hypertension, renal injury, and cardiac remodeling in chronic renal failure. Kidney Int. 2000, 58, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Deng, J.; Wang, T.; Zhao, C.; Xu, X.; Wang, P.; Voltz, J.; Edin, M.; Xiao, X.; Caho, L.; Chao, J.; Zhang, X.; Zeldin, D.; Wang, D. Tissue kallikrein reverses insulin resistance and attenuates nephropathy in diabetic rats by activation of phophatidylinositol 3-kinase/protein kinase B and adenosine 5'-monophosphate-activated protein kinase signaling pathways. Endocrinology 2007, 148, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Zacheo, A.; Minasi, A.; Chao, J.; Chao, L.; Salis, M.; Stcca, T; Straino, S.; Capogrossi, M.; Madeddu, P. Adenovirus-mediated human tissue kallikrein gene delivery induces angiogenesis in normoperfused skeletal muscle . Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2379–2385. [Google Scholar] [PubMed]
- Emanueli, C.; Salis, M.; Pinna, A.; Stacca, T.; Milia, A.; Spano, A.; Chao, J.; Chao, L.; Sciola, L.; Madeddu, P. Prevention of diabetes-induced microangiopathy by human tissue kallikrein gene transfer. Circulation 2002, 106, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Stallone, J.; Liang, Y.; Chen, L.; Wang, D.; Chao, L. Kallistatin is a potent new vasodilator. J. Clin. Invest. 1997, 100, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Miao, R.; Chen, L.; Chao, L. Novel roles of kallistatin, a specific tissue kallikrein inhibitors, in vascular remodeling. Biol. Chem. 2001, 382, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Simson, J.; Wang, J.; Chao, J.; Chao, L. Histopathology of lymphatic tissues in transgenic mice expressing human tissue kallikrein. Lab. Invest. 1994, 71, 680–687. [Google Scholar] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Katori, M.; Majima, M. A Novel Category of Anti-Hypertensive Drugs for Treating Salt-Sensitive Hypertension on the Basis of a New Development Concept. Pharmaceuticals 2010, 3, 59-109. https://doi.org/10.3390/ph3010059
Katori M, Majima M. A Novel Category of Anti-Hypertensive Drugs for Treating Salt-Sensitive Hypertension on the Basis of a New Development Concept. Pharmaceuticals. 2010; 3(1):59-109. https://doi.org/10.3390/ph3010059
Chicago/Turabian StyleKatori, Makoto, and Masataka Majima. 2010. "A Novel Category of Anti-Hypertensive Drugs for Treating Salt-Sensitive Hypertension on the Basis of a New Development Concept" Pharmaceuticals 3, no. 1: 59-109. https://doi.org/10.3390/ph3010059
APA StyleKatori, M., & Majima, M. (2010). A Novel Category of Anti-Hypertensive Drugs for Treating Salt-Sensitive Hypertension on the Basis of a New Development Concept. Pharmaceuticals, 3(1), 59-109. https://doi.org/10.3390/ph3010059