Functional Consequences of Mutations and Polymorphisms in the Coding Region of the PAF Acetylhydrolase (PAF-AH) Gene

Abstract
:1. Introduction
2. Naturally Occurring Genetic Alterations in the PAF-AH Gene
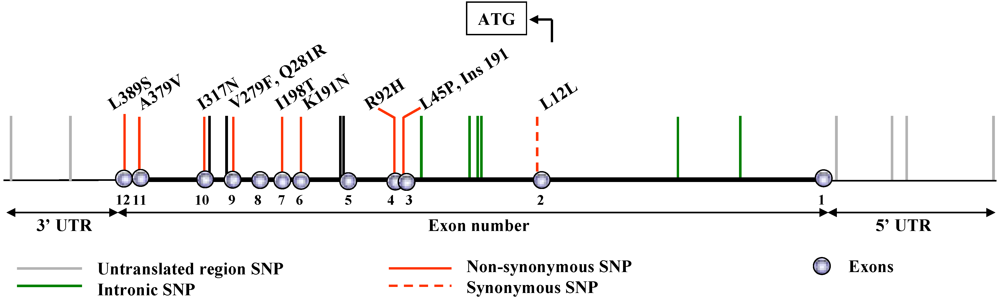
3. Inactivating Mutations
3.1. V279F (rs 16874954)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Clinical Status | n | Allele frequency, % | p | Authors [Ref.] |
|---|---|---|---|---|---|
| Japanese | Controls | 263 | 17.9 | Stafforini et al. [10] | |
| Severe asthma | 266 | 22.6 | 0.02 | ||
| Control adults | 188 | 21.0 | |||
| Japanese | Control children | 142 | 20.0 | Ito et al. [15] | |
| Asthmatic children | 118 | 31.0 | 0.007-0.004 | ||
| Japanese | Controls | 217 | 21.7 | N. Satoh et al. [16] | |
| Asthma | 279 | 18.6 | N/S | ||
| Japanese | Controls | 106 | 12.7 | Unno et al. [17] | |
| Abdominal aortic aneurysm | 131 | 21.4 | 0.012 | ||
| Japanese | Controls | 106 | 12.7 | Unno et al. [18] | |
| Femoropopliteal bypass | 50 | 21.4 | <0.001 | ||
| Japanese | Controls | 158 | 19.0 | Unno et al. [19] | |
| Peripheral artery occl. disease | 150 | 13.0 | 0.031 | ||
| Japanese | Controls | 114 | 13.0 | Unno et al. [20] | |
| Atherosclerotic occl. disease | 104 | 20.0 | <0.05 | ||
| Controls | 222 | 12.6 | |||
| Japanese | Non fam. dilated cardiomyop. | 122 | 21.3 | 0.003 | Ichihara et al. [21] |
| Controls | 1,684 | 17.0 | |||
| Japanese | Risk for atherosclerosis | 1,398 | 18.0 | <0.001 | Yamada et al. [22] |
| Atherosclerosis | 850 | 22.0 | 0.0019 | ||
| Japanese men | Controls | 452 | 12.7 | ||
| Japanese women | Myocardial infarction | 373 | 18.6 | 0.0002 | Yamada et al. [23] |
| Controls | 150 | 16.3 | |||
| Myocardial infarction | 81 | 25.9 | N/S | ||
| Controls | 284 | 16.0 | |||
| Japanese | Nonfamilial hypertrophic cardiomyopathy | 142 | 27.0 | 0.004 | Yamada et al. [24] |
| Korean | Controls | 670 | 14.0 | Jang et al. [4] | |
| CVD | 532 | 10.2 | 0.005 | ||
| Controls | 909 | 5.6 | |||
| Chinese Han | CHD | 808 | 5.0 | N/S | Hou et al. [5] |
| Myocardial infarction | 502 | 5.2 | |||
| Turkish | Controls | 128 | 1.3 | Sekuri et al. [25] | |
| Premature CAD | 115 | 0 | N/S | ||
| Taiwanese | Controls | 200 | 17.0 | Liu et al. [3] | |
| Myocardial infarction | 200 | 16.0 | N/S | ||
| Japanese | Controls | 134 | 14.2 | Hiramoto et al. [26] | |
| Stroke | 120 | 23.8 | 0.01 | K. Satoh [27] | |
| Controls | 270 | 15.6 | |||
| Japanese | Cerebral hemorrhage Hypertension | 99 | 24.2 | <0.01 | Yoshida et al. [28] |
| 138 | 19.9 | N/S | |||
| Chinese Han | Controls | 215 | 11.2 | Statistically | Zhang et al. [29] |
| Cerebral Infarction | 102 | 19.0 | significant | ||
| Japanese | Type 2 diabetes, 40-59 y/o | 50 | 14.0 | Statistically | Yamamoto et al. [30] |
| Type 2 diabetes, 60-79 y/o | 50 | 30.0 | significant | ||
| Japanese | Controls | 100 | 16.0 | Tanaka et al. [31] | |
| IgA nephropathy | 89 | 17.0 | N/S | ||
| Controls | 100 | 16.0 | |||
| Japanese | Steroid responsive nephrotic syndrome | 101 | 12.0 | N/S | Xu et al. [32] |
| Japanese | Controls | 100 | 16.0 | Xu et al. [33] | |
| Hemolytic uremic syndrome | 50 | 15.0 | N/S | ||
| Controls | 11 | 0 | |||
| Caucasian | Uncomplicated infection with E. coli O157:H7 | 52 | 0 | N/A | Smith et al. [14] |
| Hemolytic uremic syndrome | 15 | 0 | |||
| Japanese | Control | 108 | 17.0 | Nakamura et al. [34] | |
| Ulcerative colitis | 53 | 22.5 | N/S | ||
| Japanese | Control | 188 | 21.0 | Ohtsuki et al. [35] | |
| Schizophrenia | 191 | 19.0 | N/S | ||
| Controls | 213 | 14.8 | |||
| Japanese | Conventional MS | 151 | 12.6 | N/S | Osoegawa et al. [36] |
| Opticospinal MS | 65 | 16.9 | N/S | ||
| Japanese | Control | 106 | 15.6 | Minami et al. [37] | |
| Kawasaki disease | 76 | 13.2 | N/S | ||
| Caucasian | Randomly selected | 1,202 | 0 | N/A | Schnabel et al. [13] |
3.2. Q281R, I317N and Ins 191
3.3. Consequences associated with complete versus partial deficiency of PAF-AH
4. Other Non-Synonymous and Synonymous Amino Acid Substitutions
| Construct | Amino acid at position 92 | Amino acid at position 198 | Amino acid at position 379 | Km μM [Kruse et al., (42)] | Km, μM (our work) |
|---|---|---|---|---|---|
| Wild type | R | I | A | 7.0 | 14.5 |
| R92H | H | I | A | 9.0 | 12.0 |
| I198T | R | T | A | 42.0 | 24.0 |
| A379V | R | I | V | 14.0 | 14.0 |
| R92H-I198T | H | T | A | 11.0 | 28.9 |
| R92H-A379V | H | I | V | 8.0 | 12.0 |
| I198T-A379V | R | T | V | 50.0 | 30.0 |
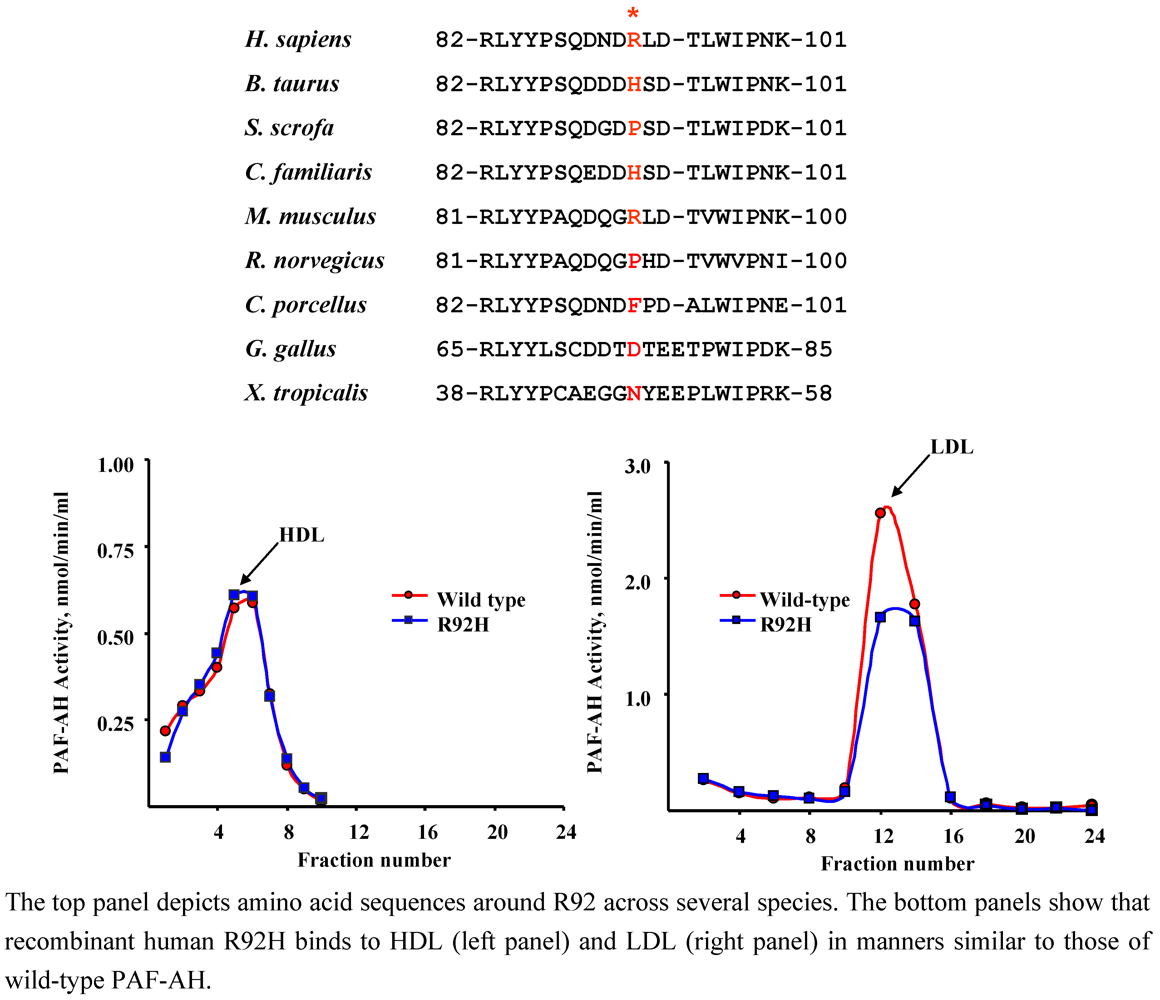
4.1. R92H (rs1805017)
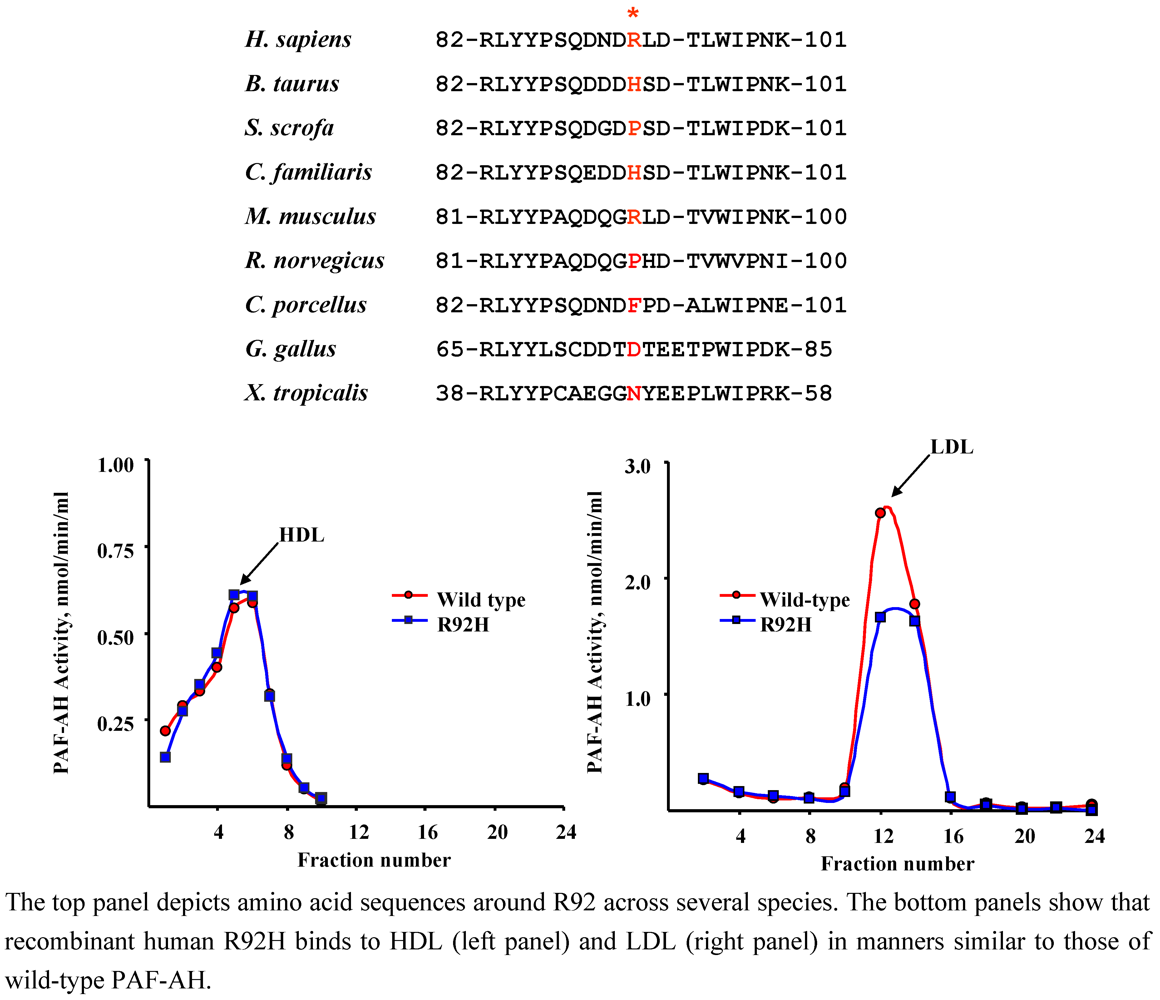
| Population | Clinical Status | n | Allele frequency | p | Authors [Ref.] |
|---|---|---|---|---|---|
| British | Controls | 150 | 23.2 | Kruse et al. [42] | |
| Atopic asthma | 150 | 23.4 | N/S | ||
| German | Nonatopic | 33 | 24.1 | N/S | |
| Atopic | 72 | 25.4 | (IgE levels) | ||
| British | Controls | 146 | 24.7 | Bell et al. [12] | |
| Schizophrenia | 298 | 25.2 | N/S | ||
| Caucasian | Controls | 693 | 25.8 | Hoffmann et al. [43] | |
| CAD | 2,541 | 26.0 | N/S | ||
| German | Controls | 484 | 22.4 | Ninio et al. [45] | |
| CAD | 1,303 | 27.1 | 0.015 | ||
| Caucasian + African American + American Indian | Controls | 267 | 34.0 | Sutton et al. [8] | |
| CAD (< 56y/o) | 599 | 28.0 | 0.01-0.04 | ||
| CAD (> 56 y/o) | 207 | 21.0 | 0.0001-0.0002 | ||
| Myocardial infarction | 425 | 28.0 | 0.0008-0.002 | ||
| Chinese | Controls | 896 | 17.2 | Hou et al. [5] | |
| CHD | 806 | 18.2 | N/S | ||
| Myocardial infarction | 499 | 20.1 | N/S | ||
| Caucasian | ARDS | 41 | 20.7 | N/A | Li et al. [44] |
| African American | ARDS | 17 | 29.4 | N/A | |
| Caucasian | Controls | 355 | 27.5 | Limou et al. [48] | |
| AIDS (slow progressors) | 168 | 26.8 | N/S | ||
| AIDS (rapid progressors) | 54 | 29.6 | N/S | ||
| Japanese | Randomly selected | 1,878 | 21.1 | N/A | Kokubo et al. [49] |
| Caucasian | Randomly selected | 1,183 | 26.8 | N/A | Schnabel et al. [50] |
4.2. I198T (rs 1805018)
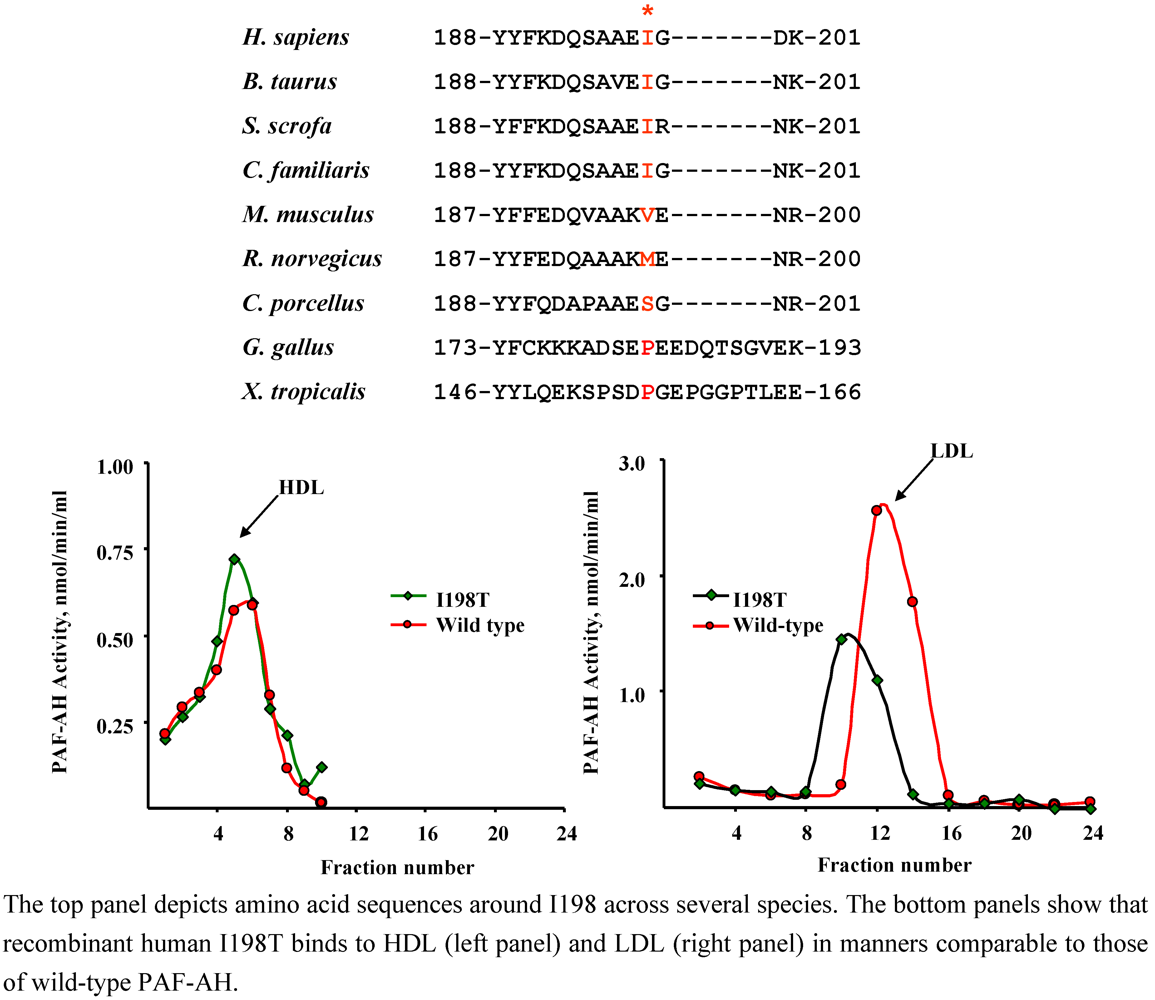
| Population | Clinical Status | n | Allele frequency,% | p | Authors [Ref.] |
|---|---|---|---|---|---|
| British | Controls | 150 | 7.6 | Kruse et al. [42] | |
| Atopic asthma | 150 | 11.7 | 0.008 | ||
| German | Nonatopic | 33 | 0.8 | ||
| Atopic | 72 | 3.7 | 0.0087 (IgE levels) | ||
| British | Controls | 99 | 2.5 | Bell et al. [12] | |
| Schizophrenia | 204 | 6.4 | 0.04 | ||
| Caucasian | Controls | 693 | 6.1 | Hoffmann et al. [43] | |
| CAD | 2,541 | 4.4 | 0.009 | ||
| German | Controls | 484 | 5.7 | Ninio et al. [45] | |
| CAD | 1,311 | 5.4 | N/S | ||
| Caucasian + African American + American Indian | Controls | 267 | 6.0 | Sutton et al. [8] | |
| CAD (< 56y/o) | 599 | 9.0 | N/S | ||
| CAD (> 56 y/o) | 207 | 7.0 | N/S | ||
| Myocardial infarction | 425 | 8.0 | N/S | ||
| Chinese | Controls | 909 | 8.7 | Hou et al. [5] | |
| CHD | 808 | 9.8 | N/S | ||
| Myocardial infarction | 502 | 9.9 | N/S | ||
| Caucasian | ARDS | 41 | 9.8 | N/A | Li et al. [44] |
| African American | ARDS | 17 | 11.8 | N/A | |
| Caucasian | Randomly selected | 1,202 | 5.2 | N/A | Schnabel et al. [13] |
| Japanese | Randomly selected | 1,878 | 20.7 | N/A | Kokubo et al. [49] |
| Japanese | Controls | 96 | 28.1 | N/A | Jinnai et al. [51] |
| Caucasian | Randomly selected | 1,202 | 5.2 | N/A | Schnabel et al. [13] |
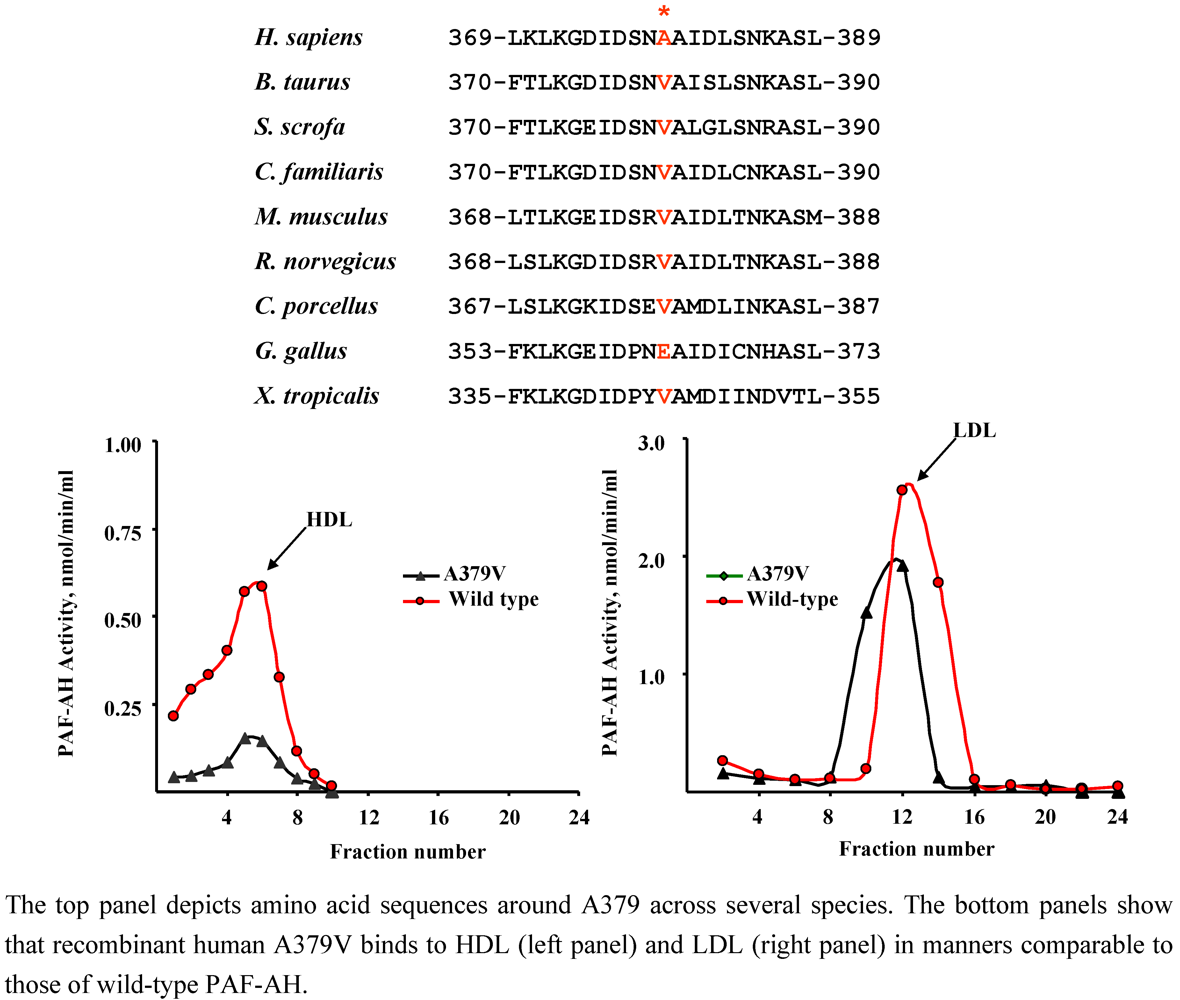
4.3. A379V (rs 1051931)
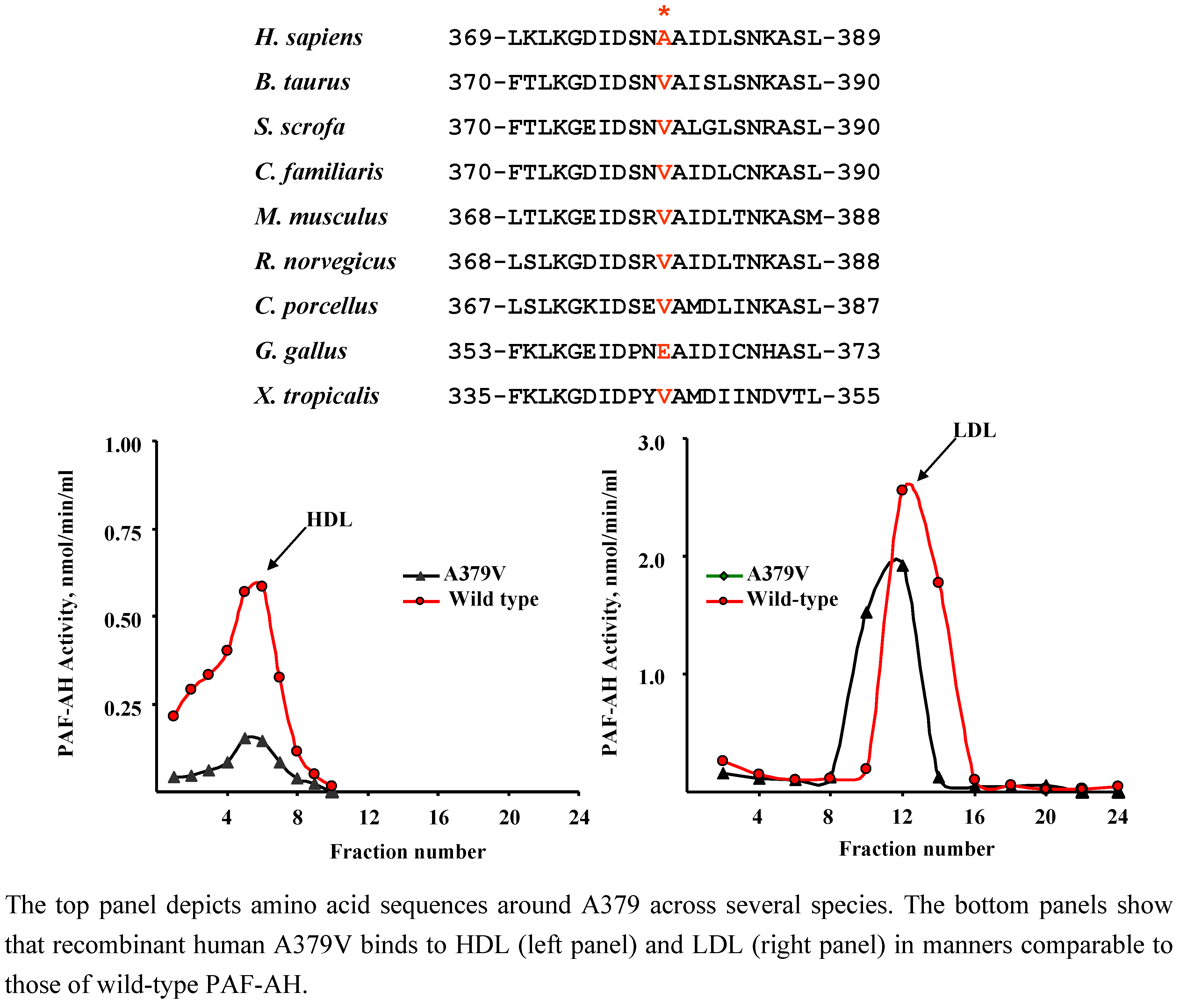
| Population | Clinical Status | n | Allele frequency | p | Authors [Ref.] |
|---|---|---|---|---|---|
| British | Controls | 150 | 15.2 | 0.038 | Kruse et al. [42] |
| Atopic asthma | 150 | 21.6 | |||
| German | Nonatopic | 33 | 10.3 | 0.0017 | |
| Atopic | 72 | 21.9 | |||
| German | Controls | 484 | 24.3 | 0.0007 | Ninio et al. [45] |
| CAD | 1298 | 19.5 | |||
| Caucasian + African American + American Indian | Controls | 267 | 15.0 | Sutton et al. [8] | |
| CAD (< 56y/o) | 599 | 20.0 | 0.05 | ||
| CAD (> 56 y/o) | 207 | 26.0 | 0.002 | ||
| Myocardial infarction | 425 | 19.0 | 0.01 | ||
| Taiwanese | Controls | 200 | 21.0 | 0.01 | Liu et al. [3] |
| Myocardial infarction | 200 | 33.0 | |||
| Caucasian | Controls | 693 | 20.9 | N/S | Hoffmann et al. [43] |
| CAD | 2541 | 21.4 | |||
| Chinese | Controls | 904 | 15.9 | Hou et al. [5] | |
| CHD | 808 | 16.6 | N/S | ||
| Myocardial infarction | 503 | 15.5 | N/S | ||
| Korean | Controls | 670 | 14.6 | N/S | Jang et al. [4] |
| CVD | 532 | 15.5 | |||
| European | Controls | 556 | 24.0 | -- | Abuzeid et al. [53] |
| Myocardial infarction | 527 | 22.0 | |||
| Caucasian | Male controls | 359 | 20.2 | Wootton et al. [52] | |
| Male CHD | 104 | 21.2 | N/S | ||
| Female controls | 244 | 20.9 | |||
| Female CHD | 50 | 23.0 | N/S | ||
| Caucasian | Early ARDS | 41 | 13.4 | N/A | Li et al. [44] |
| African American | Early ARDS | 17 | 11.8 | N/A | |
| British | Controls | 93 | 25.8 | Bell et al. [12] | |
| Schizophrenia | 191 | 18.8 | 0.06 | ||
| British | Controls | 123 | 21.5 | N/A | Wootton et al. [54] |
| British Caucasian | Controls | 2695 | 19.6 | N/A | Rudd et. al. [55] |
| Mixed | Randomly selected | 8105 | 19.4 | N/A | Schnabel et al. [50] |
| Dutch | Randomly selected | 3575 | 19.0 | N/A | Van den Berg et al. [56] |
| Japanese | Randomly selected | 1,878 | 10.8 | N/A | Kokubo et al. [49] |
| Japanese | Controls | 96 | 4.2 | N/A | Jinnai et al. [51] |
4.4. L12L (rs 35142331), L45P (rs 45521937), K191N (rs 45454695), and L389S (rs 34159425)
5. Additional Measurements May Be Necessary to Elucidate Functional Consequences of PAF-AH Polymorphisms
5.1. Rate of hydrolysis at sub-saturating substrate levels and effect of lipoprotein environment

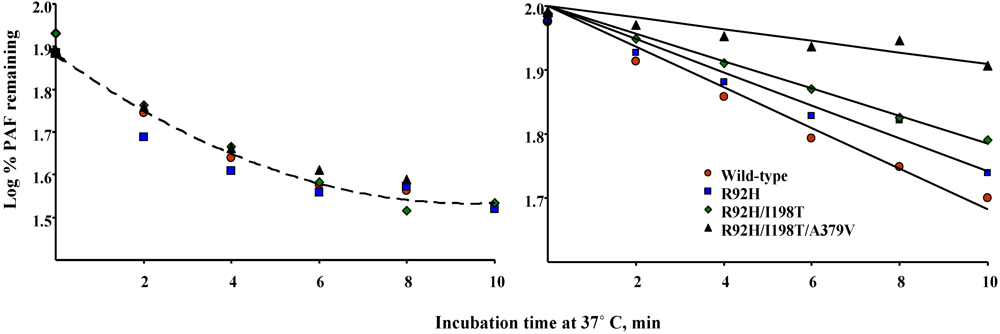
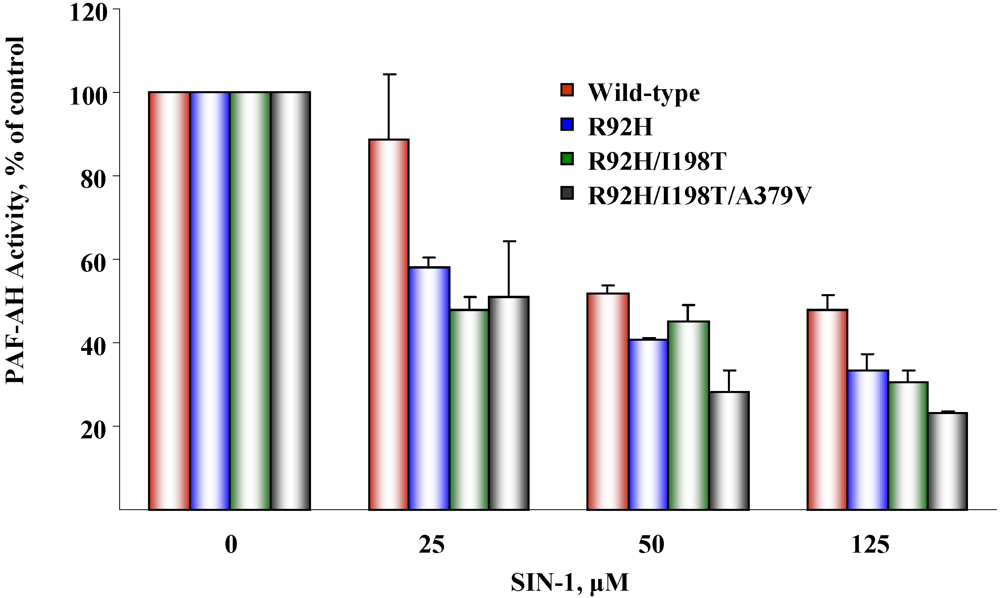
5.2. Susceptibility to oxidative inactivation

6. Conclusions
Acknowledgements
References and Notes
- Vadas, P.; Gold, M.; Perelman, B.; Liss, G.M.; Lack, G.; Blyth, T.; Simons, F.E.; Simons, K.J.; Cass, D.; Yeung, J. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl. J. Med. 2008, 358, 28–35. [Google Scholar] [PubMed]
- Miwa, M.; Miyake, T.; Yamanaka, T.; Sugatani, J.; Suzuki, Y.; Sakata, S.; Araki, Y.; Matsumoto, M. Characterization of serum platelet-activating factor (PAF) acetylhydrolase. Correlation between deficiency of serum PAF acetylhydrolase and respiratory symptoms in asthmatic children. J. Clin. Invest. 1988, 82, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Li, Y.H.; Wu, H.L.; Chao, T.H.; Tsai, L.M.; Lin, L.J.; Shi, G.Y.; Chen, J.H. Platelet-activating factor-acetylhydrolase A379V (exon 11) gene polymorphism is an independent and functional risk factor for premature myocardial infarction. J. Thromb. Haemost. 2006, 4, 1023–1028. [Google Scholar]
- Jang, Y.; Kim, O.Y.; Koh, S.J.; Chae, J.S.; Ko, Y.G.; Kim, J.Y.; Cho, H.; Jeong, T.S.; Lee, W.S.; Ordovas, J.M.; Lee, J.H. The Val279Phe Variant of the Lipoprotein-Associated Phospholipase A2 Gene Is Associated with Catalytic Activities and Cardiovascular Disease in Korean Men. J. Clin. Endocrinol. Metab. 2006, 91, 3521–3527. [Google Scholar]
- Hou, L.; Chen, S.; Yu, H.; Lu, X.; Chen, J.; Wang, L.; Huang, J.; Fan, Z.; Gu, D. Associations of PLA2G7 gene polymorphisms with plasma lipoprotein-associated phospholipase A2 activity and coronary heart disease in a Chinese Han population: the Beijing atherosclerosis study. Hum. Genet. 2009, 125, 11–20. [Google Scholar]
- Yamada, Y.; Yokota, M. Loss of activity of plasma platelet-activating factor acetylhydrolase due to a novel Gln281-->Arg mutation. Biochem. Biophys. Res. Commun. 1997, 236, 772–775. [Google Scholar]
- Ishihara, M.; Iwasaki, T.; Nagano, M.; Ishii, J.; Takano, M.; Kujiraoka, T.; Tsuji, M.; Hattori, H.; Emi, M. Functional impairment of two novel mutations detected in lipoprotein-associated phospholipase A2 (Lp-PLA2) deficiency patients. J. Hum. Genet. 2004, 49, 302–307. [Google Scholar]
- Sutton, B.S.; Crosslin, D.R.; Shah, S.H.; Nelson, S.C.; Bassil, A.; Hale, A.B.; Haynes, C.; Goldschmidt-Clermont, P.J.; Vance, J.M.; Seo, D.; Kraus, W.E.; Gregory, S.G.; Hauser, E.R. Comprehensive genetic analysis of the platelet activating factor acetylhydrolase (PLA2G7) gene and cardiovascular disease in case-control and family datasets. Hum. Mol. Genet. 2008, 17, 1318–1328. [Google Scholar]
- Stafforini, D.M.; Satoh, K.; Atkinson, D.L.; Tjoelker, L.W.; Eberhardt, C.; Yoshida, H.; Imaizumi, T.; Takamatsu, S.; Zimmerman, G.A.; McIntyre, T.M.; Gray, P.W.; Prescott, S.M. Platelet-activating factor acetylhydrolase deficiency. A missense mutation near the active site of an anti-inflammatory phospholipase. J. Clin. Invest. 1996, 97, 2784–2791. [Google Scholar]
- Stafforini, D.M.; Numao, T.; Tsodikov, A.; Vaitkus, D.; Fukuda, T.; Watanabe, N.; Fueki, N.; McIntyre, T.M.; Zimmerman, G.A.; Makino, S.; Prescott, S.M. Deficiency of platelet-activating factor acetylhydrolase is a severity factor for asthma. J. Clin. Invest. 1999, 103, 989–997. [Google Scholar]
- Balta, G.; Gurgey, A.; Kudayarov, D.K.; Tunc, B.; Altay, C. Evidence for the existence of the PAF acetylhydrolase mutation (Val279Phe) in non-Japanese populations: a preliminary study in Turkey, Azerbaijan, and Kyrgyzstan. Thromb. Res. 2001, 101, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.; Collier, D.A.; Rice, S.Q.; Roberts, G.W.; MacPhee, C.H.; Kerwin, R.W.; Price, J.; Gloger, I.S. Systematic screening of the LDL-PLA2 gene for polymorphic variants and case-control analysis in schizophrenia. Biochem. Biophys. Res. Commun. 1997, 241, 630–635. [Google Scholar]
- Schnabel, R.; Larson, M.G.; Dupuis, J.; Lunetta, K.L.; Lipinska, I.; Meigs, J.B.; Yin, X.; Rong, J.; Vita, J.A.; Newton-Cheh, C.; Levy, D.; Keaney, J.F., Jr.; Vasan, R.S.; Mitchell, G.F.; Benjamin, E.J. Relations of inflammatory biomarkers and common genetic variants with arterial stiffness and wave reflection. Hypertension 2008, 51, 1651–1657. [Google Scholar]
- Smith, J.M.; Jones, F.; Ciol, M.A.; Jelacic, S.; Boster, D.R.; Watkins, S.L.; Williams, G.D.; Tarr, P.I.; Henderson, W.R., Jr. Platelet-activating factor and Escherichia coliO157:H7 infections. Pediatr. Nephrol. 2002, 17, 1047–1052. [Google Scholar]
- Ito, S.; Noguchi, E.; Shibasaki, M.; Yamakawa-Kobayashi, K.; Watanabe, H.; Arinami, T. Evidence for an association between plasma platelet-activating factor acetylhydrolase deficiency and increased risk of childhood atopic asthma. J. Hum. Genet. 2002, 47, 99–101. [Google Scholar]
- Satoh, N.; Asano, K.; Naoki, K.; Fukunaga, K.; Iwata, M.; Kanazawa, M.; Yamaguchi, K. Plasma platelet-activating factor acetylhydrolase deficiency in Japanese patients with asthma. Am. J. Respir. Crit. Care Med. 1999, 159, 974–979. [Google Scholar]
- Unno, N.; Nakamura, T.; Mitsuoka, H.; Uchiyama, T.; Yamamoto, N.; Saito, T.; Sugatani, J.; Miwa, M.; Nakamura, S. Association of a G994 -->T missense mutation in the plasma platelet-activating factor acetylhydrolase gene with risk of abdominal aortic aneurysm in Japanese. Ann. Surg. 2002, 235, 297–302. [Google Scholar]
- Unno, N.; Nakamura, T.; Mitsuoka, H.; Saito, T.; Miki, K.; Ishimaru, K.; Sugatani, J.; Miwa, M.; Nakamura, S. Single nucleotide polymorphism (G994-->T) in the plasma platelet-activating factor-acetylhydrolase gene is associated with graft patency of femoropopliteal bypass. Surgery 2002, 132, 66–71. [Google Scholar]
- Unno, N.; Sakaguchi, T.; Nakamura, T.; Yamamoto, N.; Sugatani, J.; Miwa, M.; Konno, H. A single nucleotide polymorphism in the plasma PAF acetylhydrolase gene and risk of atherosclerosis in Japanese patients with peripheral artery occlusive disease. J. Surg. Res. 2006, 134, 36–43. [Google Scholar]
- Unno, N.; Nakamura, T.; Kaneko, H.; Uchiyama, T.; Yamamoto, N.; Sugatani, J.; Miwa, M.; Nakamura, S. Plasma platelet-activating factor acetylhydrolase deficiency is associated with atherosclerotic occlusive disease in japan. J. Vasc. Surg. 2000, 32, 263–267. [Google Scholar]
- Ichihara, S.; Yamada, Y.; Yokota, M. Association of a G994-->T missense mutation in the plasma platelet-activating factor acetylhydrolase gene with genetic susceptibility to nonfamilial dilated cardiomyopathy in Japanese. Circulation 1998, 98, 1881–1885. [Google Scholar]
- Yamada, Y.; Yoshida, H.; Ichihara, S.; Imaizumi, T.; Satoh, K.; Yokota, M. Correlations between plasma platelet-activating factor acetylhydrolase (PAF-AH) activity and PAF-AH genotype, age, and atherosclerosis in a Japanese population. Atherosclerosis 2000, 150, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Ichihara, S.; Fujimura, T.; Yokota, M. Identification of the G994--> T missense in exon 9 of the plasma platelet-activating factor acetylhydrolase gene as an independent risk factor for coronary artery disease in Japanese men. Metabolism 1998, 47, 177–181. [Google Scholar]
- Yamada, Y.; Ichihara, S.; Izawa, H.; Tanaka, M.; Yokota, M. Association of a G994 --> T (Val279 --> Phe) polymorphism of the plasma platelet-activating factor acetylhydrolase gene with myocardial damage in Japanese patients with nonfamilial hypertrophic cardiomyopathy. J. Hum. Genet. 2001, 46, 436–441. [Google Scholar]
- Sekuri, C.; Cam, F.S.; Tengiz, I.; Ercan, E.; Bayturan, O.; Berdeli, A. Association of platelet-activating factor acetylhydrolase gene polymorphism with premature coronary artery disease in Turkish patients. Anadolu Kardiyol Derg 2006, 6, 132–134. [Google Scholar]
- Hiramoto, M.; Yoshida, H.; Imaizumi, T.; Yoshimizu, N.; Satoh, K. A mutation in plasma platelet-activating factor acetylhydrolase (Val279-->Phe) is a genetic risk factor for stroke. Stroke 1997, 28, 2417–2420. [Google Scholar]
- Satoh, K. Plasma platelet-activating factor acetylhydrolase (PAF-AH) deficiency as a risk factor for stroke. Brain Nerve 2008, 60, 1319–1324. [Google Scholar]
- Yoshida, H.; Imaizumi, T.; Fujimoto, K.; Itaya, H.; Hiramoto, M.; Yoshimizu, N.; Fukushi, K.; Satoh, K. A mutation in plasma platelet-activating factor acetylhydrolase (Val279Phe) is a genetic risk factor for cerebral hemorrhage but not for hypertension. Thromb. Haemost. 1998, 80, 372–375. [Google Scholar]
- Zhang, X.; Yuan, C.L.; Zhang, H.Z.; Xu, J.; Wu, J.; Chen, B.L. Analysis of 994(G--> T) mutation in the plasma platelet-activating factor acetylhydrolase gene in the patients with cerebral infarction. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2005, 22, 450–452. [Google Scholar]
- Yamamoto, I.; Fujitsu, J.; Nohnen, S.; Igarashi, T.; Motomura, T.; Inaba, M.; Tsubakimori, S.; Azuma, J. Association of plasma PAF acetylhydrolase gene polymorphism with IMT of carotid arteries in Japanese type 2 diabetic patients. Diabetes. Res. Clin. Pract. 2003, 59, 219–224. [Google Scholar]
- Tanaka, R.; Iijima, K.; Xu, H.; Inoue, Y.; Murakami, R.; Shirakawa, T.; Nishiyama, K.; Miwa, M.; Shiozawa, S.; Nakamura, H.; Yoshikawa, N. Role of platelet-activating factor acetylhydrolase gene mutation in Japanese childhood IgA nephropathy. Am. J. Kidney. Dis. 1999, 34, 289–295. [Google Scholar]
- Xu, H.; Iijima, K.; Shiozawa, S.; Tanaka, S.S.; Inoue, Y.; Shirakawa, T.; Nishiyama, K.; Miwa, M.; Nakamura, H.; Yoshikawa, N. Platelet-activating factor acetylhydrolase gene mutation in Japanese nephrotic children. Kidney. Int. 1998, 54, 1867–1871. [Google Scholar]
- Xu, H.; Iijima, K.; Shirakawa, T.; Shiozawa, S.; Miwa, M.; Yamaoka, K.; Kawamura, N.; Nakamura, H.; Yoshikawa, N. Platelet-activating factor acetylhydrolase gene mutation in Japanese children with Escherichia coli O157-associated hemolytic uremic syndrome. Am. J. Kidney. Dis. 2000, 36, 42–46. [Google Scholar]
- Nakamura, T.; Sakaguchi, T.; Unno, N.; Sugatani, J.; Miwa, M.; Nakamura, S. Relationship between the platelet activating factor acetylhydrolase gene and intractability of ulcerative colitis. Dis. Colon. Rectum. 2002, 45, 389–393. [Google Scholar]
- Ohtsuki, T.; Watanabe, H.; Toru, M.; Arinami, T. Lack of evidence for associations between plasma platelet-activating factor acetylhydrolase deficiency and schizophrenia. Psychiatry Res 2002, 109, 93–96. [Google Scholar]
- Osoegawa, M.; Niino, M.; Ochi, H.; Kikuchi, S.; Murai, H.; Fukazawa, T.; Minohara, M.; Tashiro, K.; Kira, J. Platelet-activating factor acetylhydrolase gene polymorphism and its activity in Japanese patients with multiple sclerosis. J. Neuroimmunol. 2004, 150, 150–156. [Google Scholar]
- Minami, T.; Suzuki, H.; Takeuchi, T.; Uemura, S.; Sugatani, J.; Yoshikawa, N. A polymorphism in plasma platelet-activating factor acetylhydrolase is involved in resistance to immunoglobulin treatment in Kawasaki disease. J. Pediatr. 2005, 147, 78–83. [Google Scholar]
- Wang, T.; Karino, K.; Yamasaki, M.; Zhang, Y.; Masuda, J.; Yamaguchi, S.; Shiwaku, K.; Nabika, T. Effects of G994T in the Lp-PLA2 gene on the plasma oxidized LDL level and carotid intima-media thickness in Japanese: the Shimane study. Am. J. Hypertens. 2009, 22, 742–747. [Google Scholar]
- Stafforini, D.M.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. The platelet-activating factor acetylhydrolase from human plasma prevents oxidative modification of low-density lipoprotein. Trans. Assoc. Am. Physicians 1992, 105, 44–63. [Google Scholar]
- Kujiraoka, T.; Iwasaki, T.; Ishihara, M.; Ito, M.; Nagano, M.; Kawaguchi, A.; Takahashi, S.; Ishi, J.; Tsuji, M.; Egashira, T.; Stepanova, I.P.; Miller, N.E.; Hattori, H. Altered distribution of plasma PAF-AH between HDLs and other lipoproteins in hyperlipidemia and diabetes mellitus. J. Lipid. Res. 2003, 44, 2006–2014. [Google Scholar]
- Stafforini, D.M.; Carter, M.E.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Lipoproteins alter the catalytic behavior of the platelet-activating factor acetylhydrolase in human plasma. Proc. Natl. Acad. Sci. USA 1989, 86, 2393–2397. [Google Scholar]
- Kruse, S.; Mao, X.Q.; Heinzmann, A.; Blattmann, S.; Roberts, M.H.; Braun, S.; Gao, P.S.; Forster, J.; Kuehr, J.; Hopkin, J.M.; Shirakawa, T.; Deichmann, K.A. The Ile198Thr and Ala379Val variants of plasmatic PAF-acetylhydrolase impair catalytical activities and are associated with atopy and asthma. Am. J. Hum. Genet. 2000, 66, 1522–1530. [Google Scholar]
- Hoffmann, M.M.; Winkler, K.; Renner, W.; Winkelmann, B.R.; Seelhorst, U.; Wellnitz, B.; Boehm, B.O.; Marz, W. Genetic variants and haplotypes of lipoprotein associated phospholipase A2 and their influence on cardiovascular disease (The Ludwigshafen Risk and Cardiovascular Health Study). J. Thromb. Haemost. 2009, 7, 41–48. [Google Scholar]
- Li, S.; Stuart, L.; Zhang, Y.; Meduri, G.U.; Umberger, R.; Yates, C.R. Inter-individual variability of plasma PAF-acetylhydrolase activity in ARDS patients and PAF-AH genotype. J. Clin. Pharm. Ther. 2009, 34, 447–455. [Google Scholar]
- Ninio, E.; Tregouet, D.; Carrier, J.L.; Stengel, D.; Bickel, C.; Perret, C.; Rupprecht, H.J.; Cambien, F.; Blankenberg, S.; Tiret, L. Platelet-activating factor-acetylhydrolase and PAF-receptor gene haplotypes in relation to future cardiovascular event in patients with coronary artery disease. Hum. Mol. Genet. 2004, 13, 1341–1351. [Google Scholar]
- Gardner, A.A.; Reichert, E.C.; Topham, M.K.; Stafforini, D.M. Identification of a domain that mediates association of platelet-activating factor acetylhydrolase with high density lipoprotein. J. Biol. Chem. 2008, 283, 17099–17106. [Google Scholar]
- Luo, G.; Zheng, L.; Zhang, X.; Zhang, J.; Nilsson-Ehle, P.; Xu, N. Genotyping of single nucleotide polymorphisms using base-quenched probe: a method does not invariably depend on the deoxyguanosine nucleotide. Anal. Biochem. 2009, 386, 161–166. [Google Scholar]
- Limou, S.; Coulonges, C.; Foglio, M.; Heath, S.; Diop, G.; Leclerc, S.; Hirtzig, T.; Spadoni, J.L.; Therwath, A.; Lambeau, G.; Gut, I.; Zagury, J.F. Exploration of associations between phospholipase A2 gene family polymorphisms and AIDS progression using the SNPlex method. Biomed. Pharmacother. 2008, 62, 31–40. [Google Scholar]
- Kokubo, Y.; Tomoike, H.; Tanaka, C.; Banno, M.; Okuda, T.; Inamoto, N.; Kamide, K.; Kawano, Y.; Miyata, T. Association of sixty-one non-synonymous polymorphisms in forty-one hypertension candidate genes with blood pressure variation and hypertension. Hypertens. Res. 2006, 29, 611–619. [Google Scholar]
- Schnabel, R.; Dupuis, J.; Larson, M.G.; Lunetta, K.L.; Robins, S.J.; Zhu, Y.; Rong, J.; Yin, X.; Stirnadel, H.A.; Nelson, J.J.; Wilson, P.W.; Keaney, J.F.; Vasan, R.S.; Benjamin, E.J. Clinical and genetic factors associated with lipoprotein-associated phospholipase A2 in the Framingham Heart Study. Atherosclerosis 2009, 204, 601–607. [Google Scholar]
- Jinnai, N.; Sakagami, T.; Sekigawa, T.; Kakihara, M.; Nakajima, T.; Yoshida, K.; Goto, S.; Hasegawa, T.; Koshino, T.; Hasegawa, Y.; Inoue, H.; Suzuki, N.; Sano, Y.; Inoue, I. Polymorphisms in the prostaglandin E2 receptor subtype 2 gene confer susceptibility to aspirin-intolerant asthma: a candidate gene approach. Hum. Mol. Genet. 2004, 13, 3203–3217. [Google Scholar]
- Wootton, P.T.; Stephens, J.W.; Hurel, S.J.; Durand, H.; Cooper, J.; Ninio, E.; Humphries, S.E.; Talmud, P.J. Lp-PLA2 activity and PLA2G7 A379V genotype in patients with diabetes mellitus. Atherosclerosis 2006, 189, 149–156. [Google Scholar]
- Abuzeid, A.M.; Hawe, E.; Humphries, S.E.; Talmud, P.J. Association between the Ala379Val variant of the lipoprotein associated phospholipase A2 and risk of myocardial infarction in the north and south of Europe. Atherosclerosis 2003, 168, 283–288. [Google Scholar]
- Wootton, P.T.; Flavell, D.M.; Montgomery, H.E.; World, M.; Humphries, S.E.; Talmud, P.J. Lipoprotein-associated phospholipase A2 A379V variant is associated with body composition changes in response to exercise training. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 24–31. [Google Scholar]
- Rudd, M.F.; Webb, E.L.; Matakidou, A.; Sellick, G.S.; Williams, R.D.; Bridle, H.; Eisen, T.; Houlston, R.S. Variants in the GH-IGF axis confer susceptibility to lung cancer. Genome Res. 2006, 16, 693–701. [Google Scholar]
- van den Berg, S.W.; Dolle, M.E.; Imholz, S.; van der, A.D.; van 't Slot, R.; Wijmenga, C.; Verschuren, W.M.; Strien, C.; Siezen, C.L.; Hoebee, B.; Feskens, E.J.; Boer, J.M. Genetic variations in regulatory pathways of fatty acid and glucose metabolism are associated with obesity phenotypes: a population-based cohort study. Int. J. Obes. (London) 2009, 33, 1143–1152. [Google Scholar] [CrossRef]
- MacRitchie, A.N.; Gardner, A.A.; Prescott, S.M.; Stafforini, D.M. Molecular basis for susceptibility of plasma platelet-activating factor acetylhydrolase to oxidative inactivation. Faseb. J. 2007, 21, 1164–1176. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stafforini, D.M. Functional Consequences of Mutations and Polymorphisms in the Coding Region of the PAF Acetylhydrolase (PAF-AH) Gene. Pharmaceuticals 2009, 2, 94-117. https://doi.org/10.3390/ph2030094
Stafforini DM. Functional Consequences of Mutations and Polymorphisms in the Coding Region of the PAF Acetylhydrolase (PAF-AH) Gene. Pharmaceuticals. 2009; 2(3):94-117. https://doi.org/10.3390/ph2030094
Chicago/Turabian StyleStafforini, Diana M. 2009. "Functional Consequences of Mutations and Polymorphisms in the Coding Region of the PAF Acetylhydrolase (PAF-AH) Gene" Pharmaceuticals 2, no. 3: 94-117. https://doi.org/10.3390/ph2030094