1. Introduction
Deep vein thrombosis (DVT) affects approximately 900,000 individuals annually in the United States, and pulmonary emboli, a severe complication of DVT, are often observed in these patients [
1]. Unlike in arterial thrombus, which is observed to initiate upon exposed extracellular matrix (ECM), venous thrombus initiates from dysfunctional endothelium, in part due to platelet binding to and activation on endothelium [
1]. The binding and activation of platelets not only supports thrombus formation, but also further contributes to the inflammatory state of the venous endothelium, thus exacerbating the diseased vessel state [
2].
Vascular endothelial cells (ECs) line the entire cardiovascular system in the human body. As the innermost layer of the blood vessel lumen, ECs are in constant contact with the blood and perform many critical functions. These include involvement in metabolism, regulating blood vessel tone, permeability and growth, vascular smooth muscle cell (SMC) proliferation, inflammation, platelet and leukocyte interactions, thrombosis and fibrinolysis [
3,
4,
5]. Given these overlapping and wide-ranging activities, it is not surprising that disruption of normal EC function is associated with many different disease states. Indeed, EC dysfunction has been shown to play a role in DVT, atherosclerosis, myocardial infarction, peripheral vascular disease, stroke, hypertension, diabetes, chronic kidney disease, infections including sepsis, and even cancer [
3,
4,
6,
7,
8,
9,
10,
11].
In general, characteristics of endothelial dysfunction include a reduction in ability of vessels to vasodilate, along with increases in inflammation and prothrombotic properties [
3,
12]. Additionally, loss of the glycocalyx, an anionic layer comprised primarily of glycosaminoglycans (GAGs) that lines the endothelium, is a key feature of EC dysfunction [
13,
14]. The loss of this delicate layer causes a significant increase in vascular wall permeability and subsequent migration of fluid, protein and other cell types into the vessel [
15]. Lack of a glycocalyx layer also results in exposure of EC adhesion molecules such as E-selectin, P-selectin, intracellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1) and platelet-endothelial cell adhesion molecule 1 (PECAM-1) [
13,
16], some of which are known to play a role in platelet binding to the endothelial cell surface. Once bound, platelets release cytokines, chemokines and platelet activation factors, such as platelet activating factor (PAF), neutrophil-activating peptide-2 (NAP-2) and platelet factor-4 (PF-4), which leads to thrombus formation and a further increase in inflammatory cell recruitment to the site [
17,
18,
19,
20].
Endothelial dysfunction is likely a reversible disorder as eliminating cardiovascular risk factors through such means as smoking cessation, hypertension control, cholesterol lowering and physical activity have been shown to improve endothelial health [
10]. Angiotensin converting enzyme (ACE) inhibitors,
N-acetylcysteine (NAC), ascorbic acid (Vitamin C), vitamin E, and erythropoietin (EPO) have all been studied as potential therapeutics for EC dysfunction [
21,
22,
23,
24]. While some positive outcomes have been observed, they have often displayed a minimal effect on overall endothelial health or resulted in unintended cardiovascular events [
4,
23]. Therefore, additional means of targeting and effectively treating EC dysfunction and reducing associated cardiovascular events are needed.
Targeting EC adhesion molecules, such as selectins, for therapeutic or diagnostic purposes has been relatively well studied. Examples include microparticle drug delivery [
25], therapeutic gene delivery [
26,
27], inflammation reduction [
28,
29,
30], cancer metastasis prevention [
31] and in vivo diagnostic imaging [
32,
33]. Specifically, antibodies and peptides have been utilized to target selectin receptors on EC surfaces to prevent platelet and leukocyte binding and the subsequent inflammatory response that occurs [
28,
29,
30,
34]. Therefore, our laboratory has recently developed several variants of a GAG derived, selectin targeting anti-adhesive coating (termed EC-SEAL) designed to utilize overexpressed selectins in EC dysfunction to prevent platelet binding and subsequent thrombus formation and restore a more quiescent endothelial state. Each variant consists of a dermatan sulfate (GAG component of the glycocalyx) [
13] backbone with multiple selectin-binding peptides attached. EC-SEAL variants differing in type and number of peptides attached were investigated to determine highest binding affinities to inflamed ECs and assess ability to prevent platelet binding along the EC surface. Results indicated effective molecule binding to inflamed ECs and subsequent prevention of platelet binding and activation, as well as suppression of thrombus formation in a murine model of DVT, highlighting the possibility of EC-SEAL as a therapeutic to treat EC dysfunction.
3. Discussion
Loss of the glycocalyx is a key characteristic of EC dysfunction [
13,
14]. Removal of this glycosaminoglycan (GAG)-rich layer results in the exposure and upregulation of EC adhesion molecules, including selectins [
13,
16]. Selectins and their ligands, located on both ECs and platelets, play a key role in the binding of platelets to inflamed endothelium. In the context of endothelial dysfunction, this process can lead to complications in a myriad of disease states including DVT, atherosclerosis, diabetes, chronic kidney disease and sepsis [
3,
4,
6,
8,
39]. Masking these selectins with antibodies or peptides to prevent platelet binding presents a possible avenue of treatment [
28,
29,
30]. We sought to develop and test multiple variants of a GAG derived, selectin-binding anti-adhesive coating (termed EC-SEAL) utilizing selectin-binding peptides to bind to an inflamed endothelial surface. We demonstrate the ability to successfully synthesize variants ranging from 2 to 30 peptides per dermatan sulfate molecule that bind to E-selectin and cells expressing selectins, and prevent platelet binding and activation in a dose-dependent manner. Additionally, we present evidence that the lead variant reduces thrombus size in vivo while exhibiting minimal effects on clotting time.
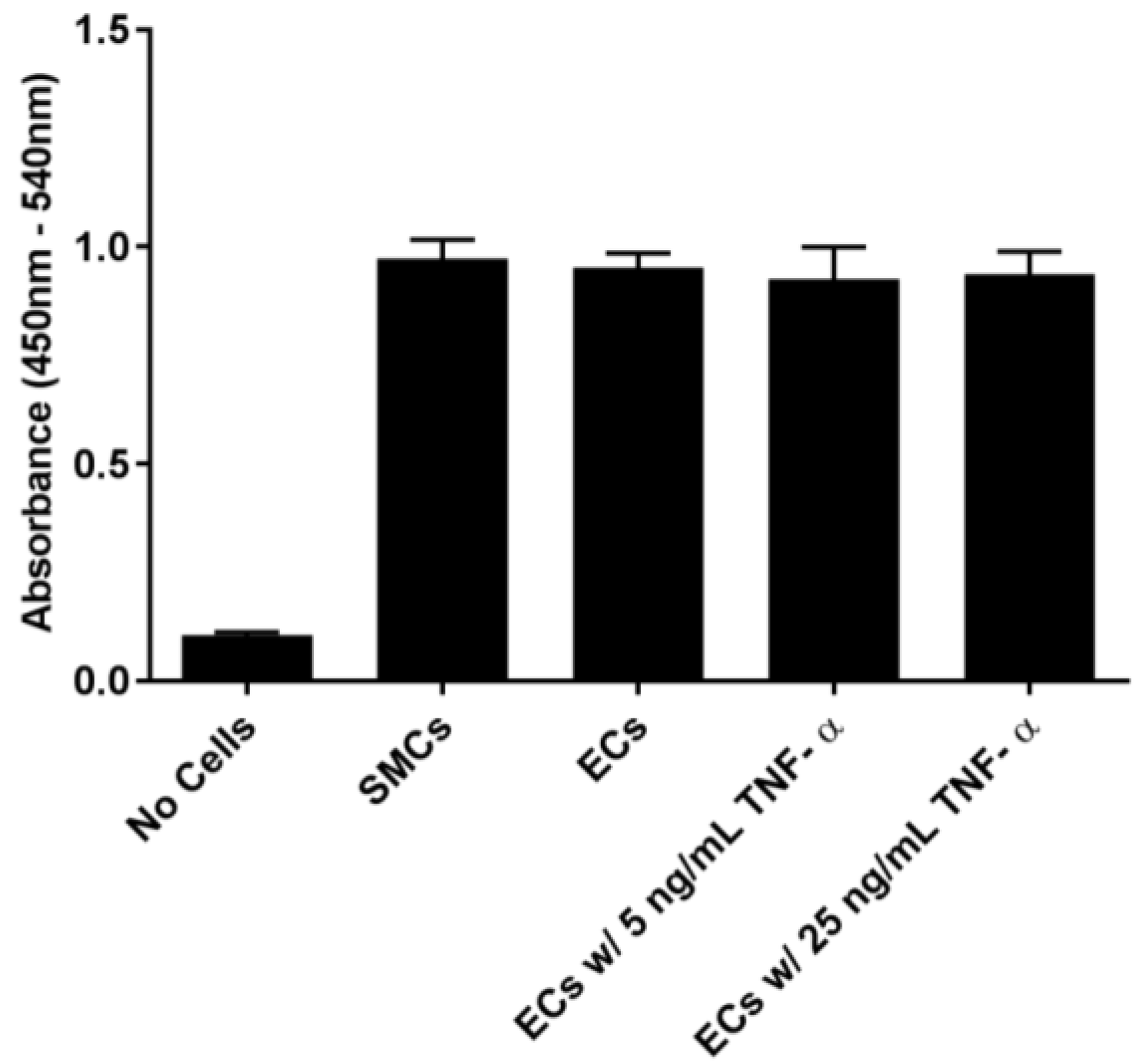
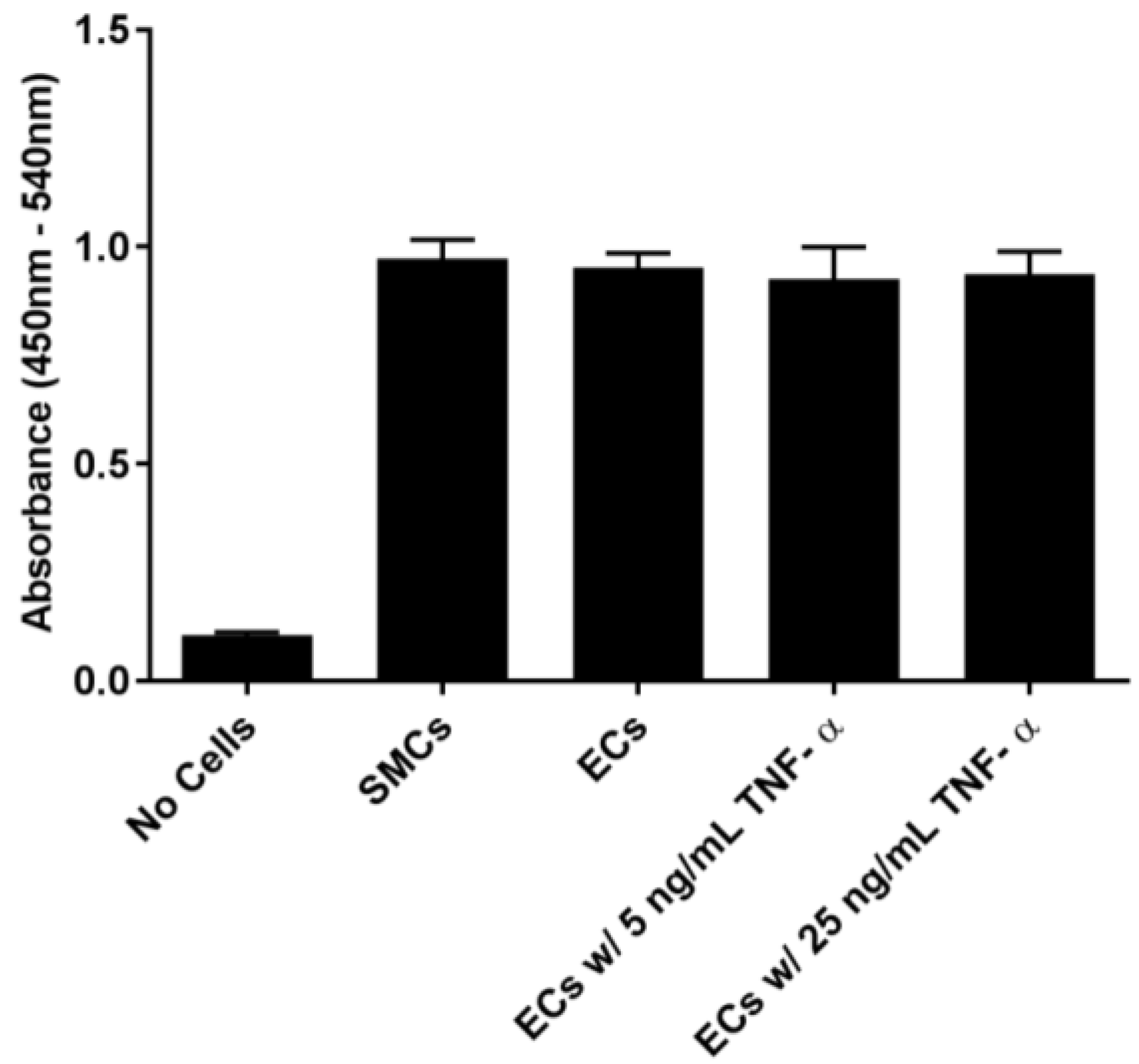
In order to examine the binding of EC-SEAL variants to vascular cells, we first established that selectin was present on the cell surfaces. As shown in
Figure 1 and
Supplementary Figures S1 and S2, ECs (which express both E and P-selectin) did not increase selectin levels following TNF-α stimulation or any other proinflammatory stimulants used. Although not anticipated because it has been shown previously that adding TNF-α to human aortic ECs leads to an increase in E-selectin expression 3–6 h after stimulation [
40,
41,
42], there is also evidence that many different factors including number of cell passages, seeding density, certain growth factors present in media, and even the culture substrate can lead to increased basal levels of adhesion molecules on cultured ECs [
43,
44,
45,
46]. It is also possible that the rinsing and manipulation of the live ECs throughout the experiments contributed to the observed basal selectin expression. The presence of selectins on the EC surface in the unstimulated cultures, the lack of response to multiple proinflammatory cytokines (TNF-α, LPS, IL-1β and IL-6), and the inability to form intact monolayers (
Supplementary Figure S3), all suggest that the EC cultures were in a constitutively activated state. SMCs, known to express P-selectin [
35,
36], but not E-selectin [
37], demonstrated P-selectin cross-reactivity with the anti-E-selectin antibody.
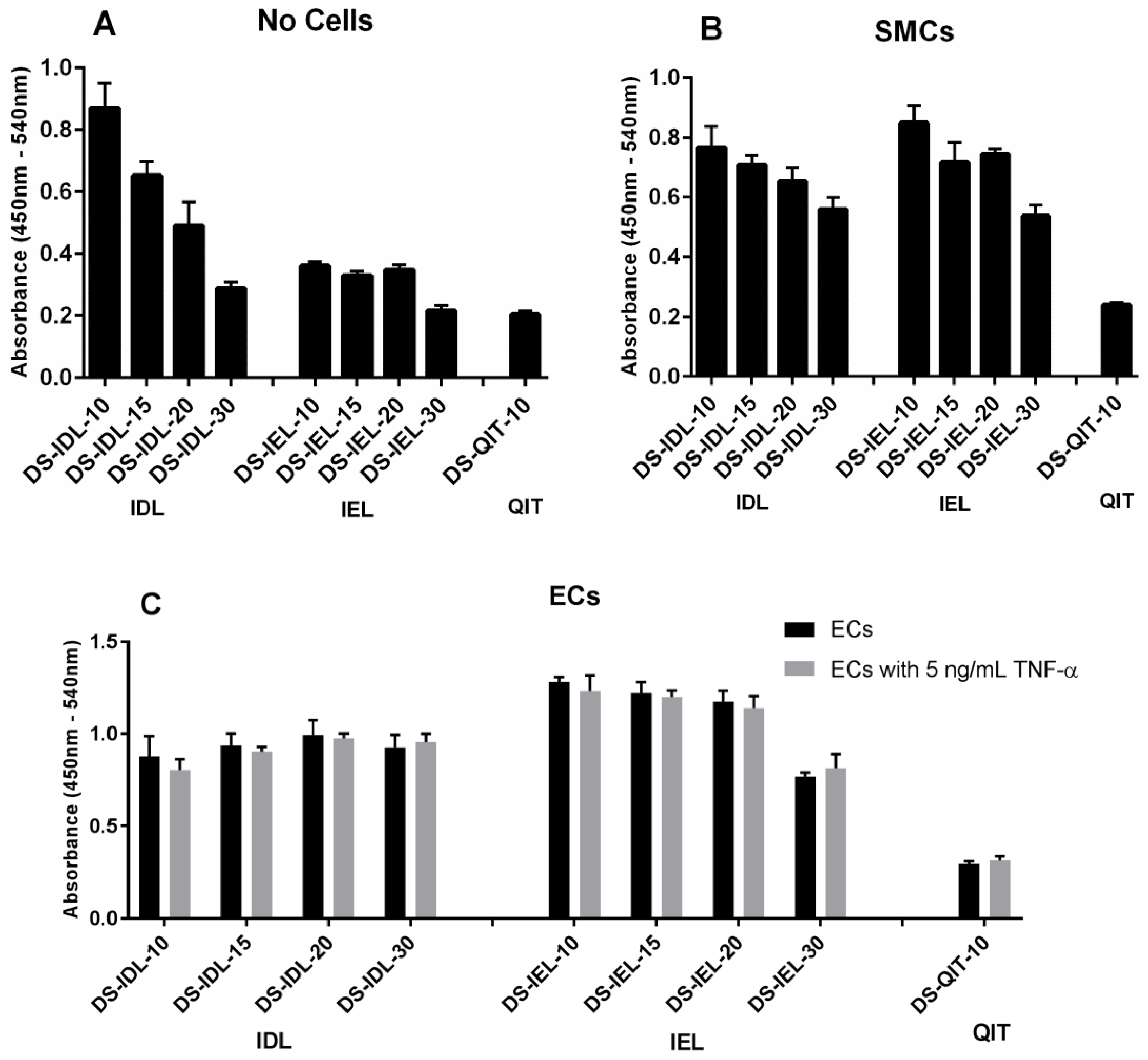
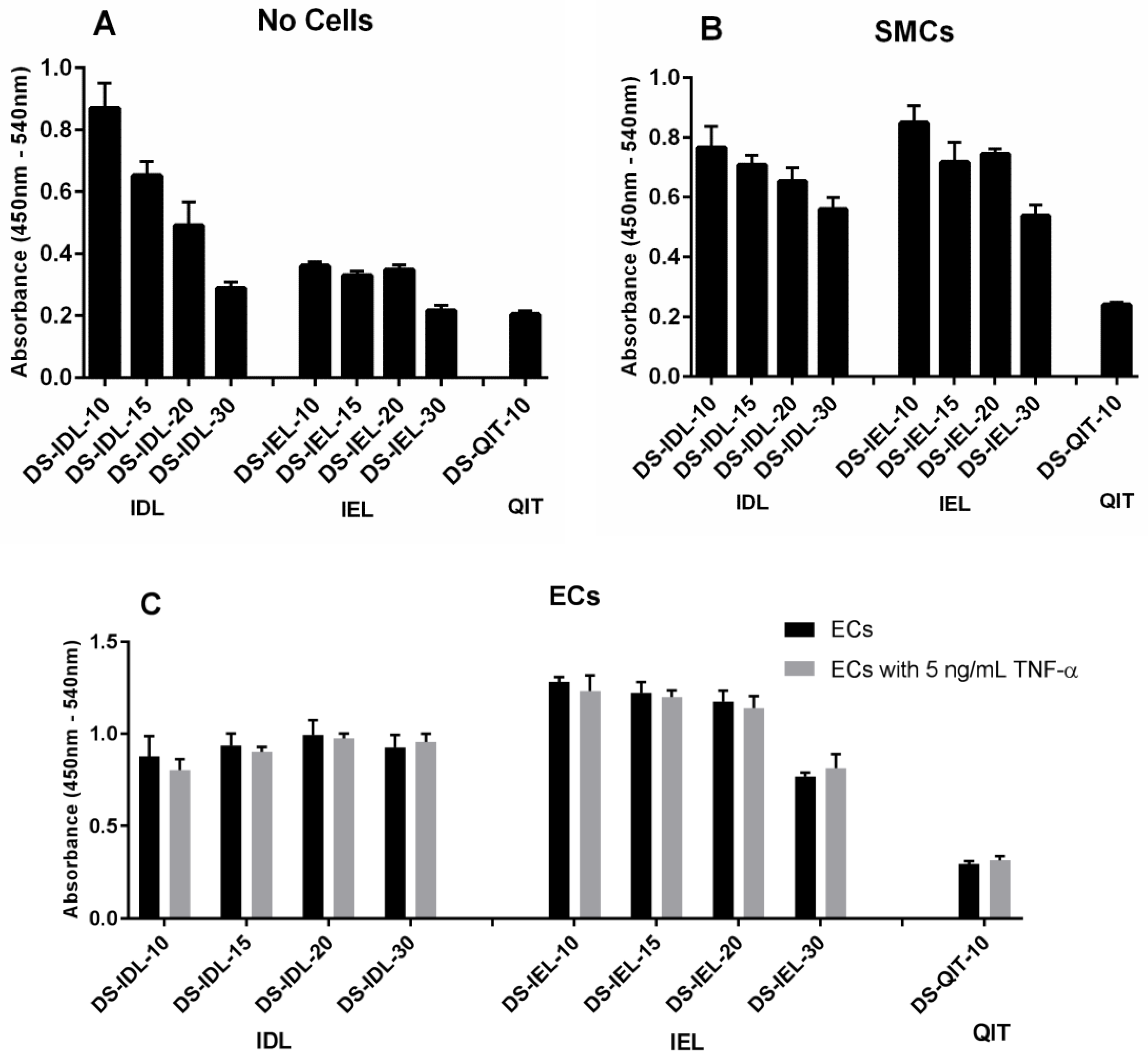
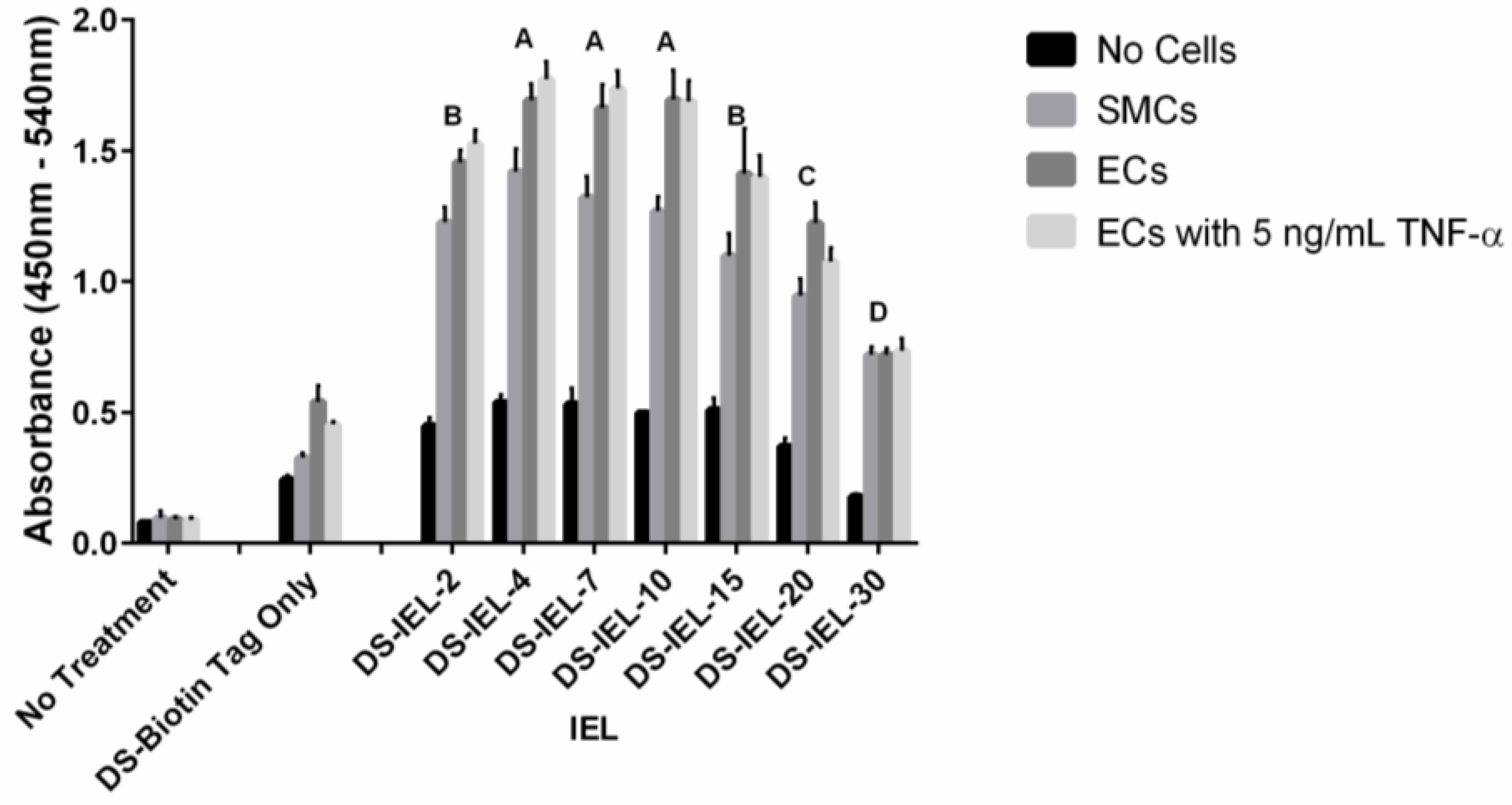
The selectin-expressing cells were then used to test and compare EC-SEAL variant binding. Both the type and number of peptides attached to the DS backbone had a significant impact on binding affinity (
Figure 2 and
Figure 3). There was no difference in binding to unstimulated ECs or ECs stimulated with TNF-α for any EC-SEAL variant, which is consistent with the selectin expression data discussed previously. Although nearly all variants had a higher binding affinity to ECs, they also adhered to SMCs, presumably through P-selectin. Prior studies on IDL and IEL peptides indeed indicated binding to both E and P-selectin [
31]. QIT was previously shown to have a significantly higher preference for E-selectin over P-selectin [
47], but in our hands the binding observed in the DS-QIT
x variants was primarily due to nonspecific interactions as indicated by the (relatively) high level of binding with no cells present compared to both SMCs and ECs (
Figure 2). IEL was selected as the peptide to examine further due to its increased binding to ECs and reduced nonspecific interactions when no cells were present. This is consistent with a previous study that showed IEL had a higher binding affinity to E-selectin expressing bacteria than IDL [
31]. Importantly, all DS-IEL
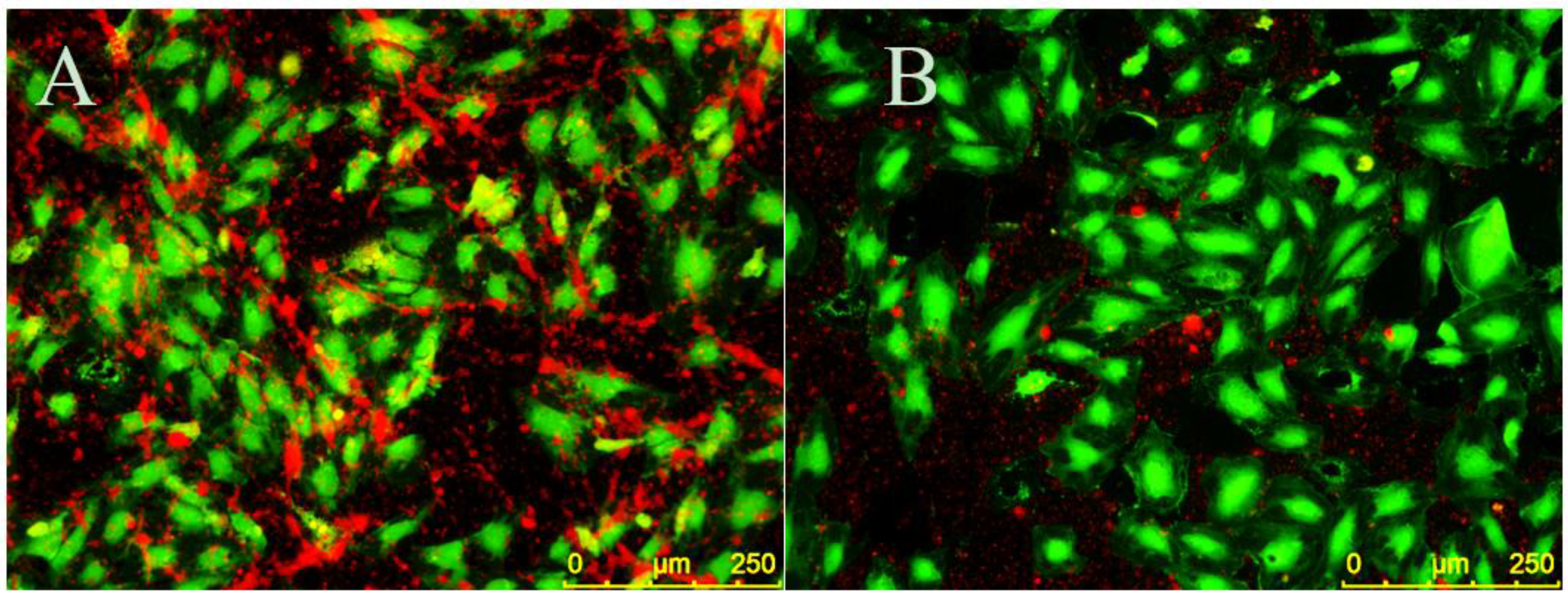
x variants showed increased levels of binding compared to only DS, which as a native glycocalyx component will interact with the EC surface, indicating the benefit of the selectin-binding peptides. The apparent ability to bind to both E and P-selectin is also viewed as advantageous as it may allow for more complete coverage of: (1) ECs expressing both E and P-selectin; and (2) SMCs expressing P-selectin should they become exposed following EC injury or denudation.
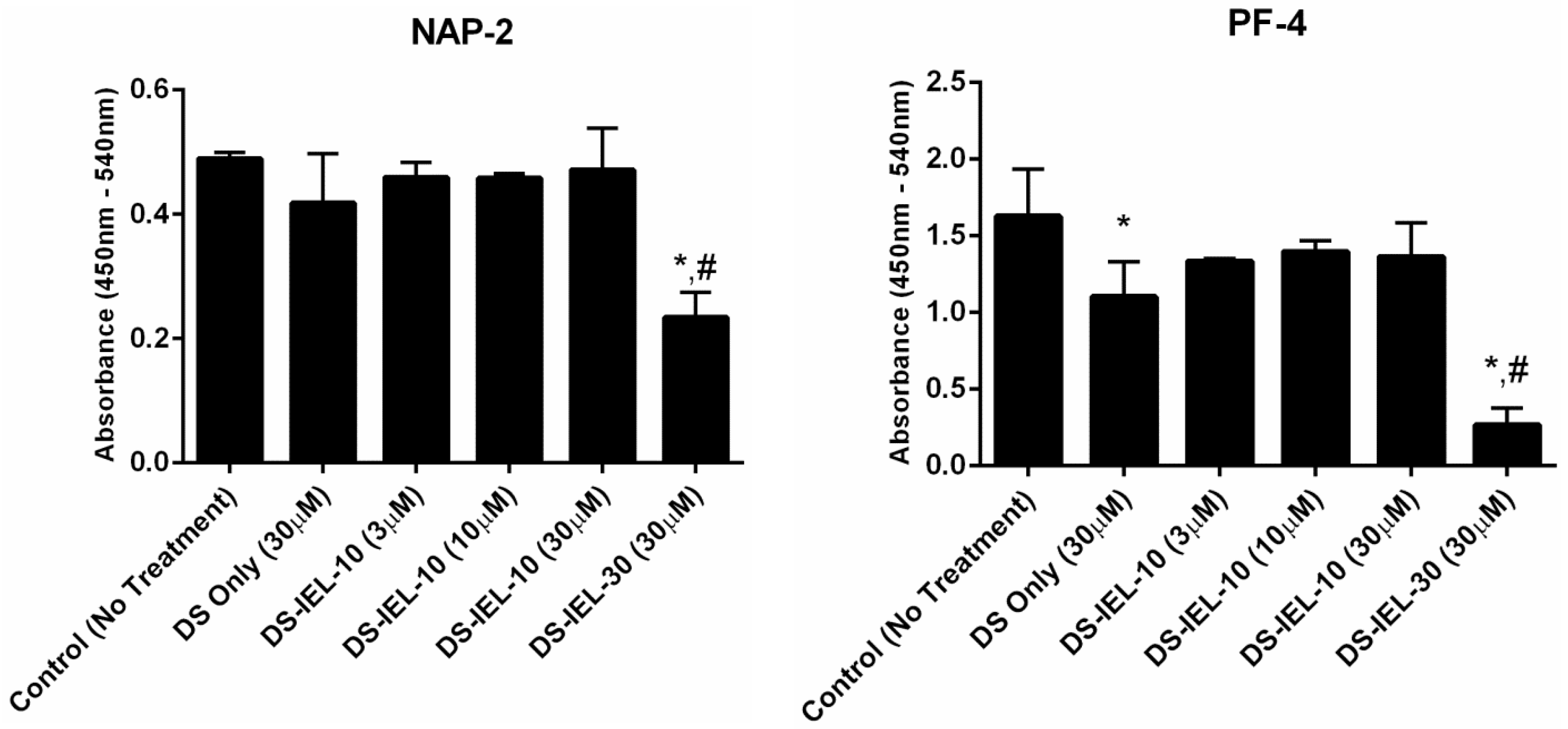
DS-IEL
10 was originally chosen for platelet binding and activation testing given that it possessed the greatest number of selectin-binding peptides among the highest binding group of DS-IEL
x variants (
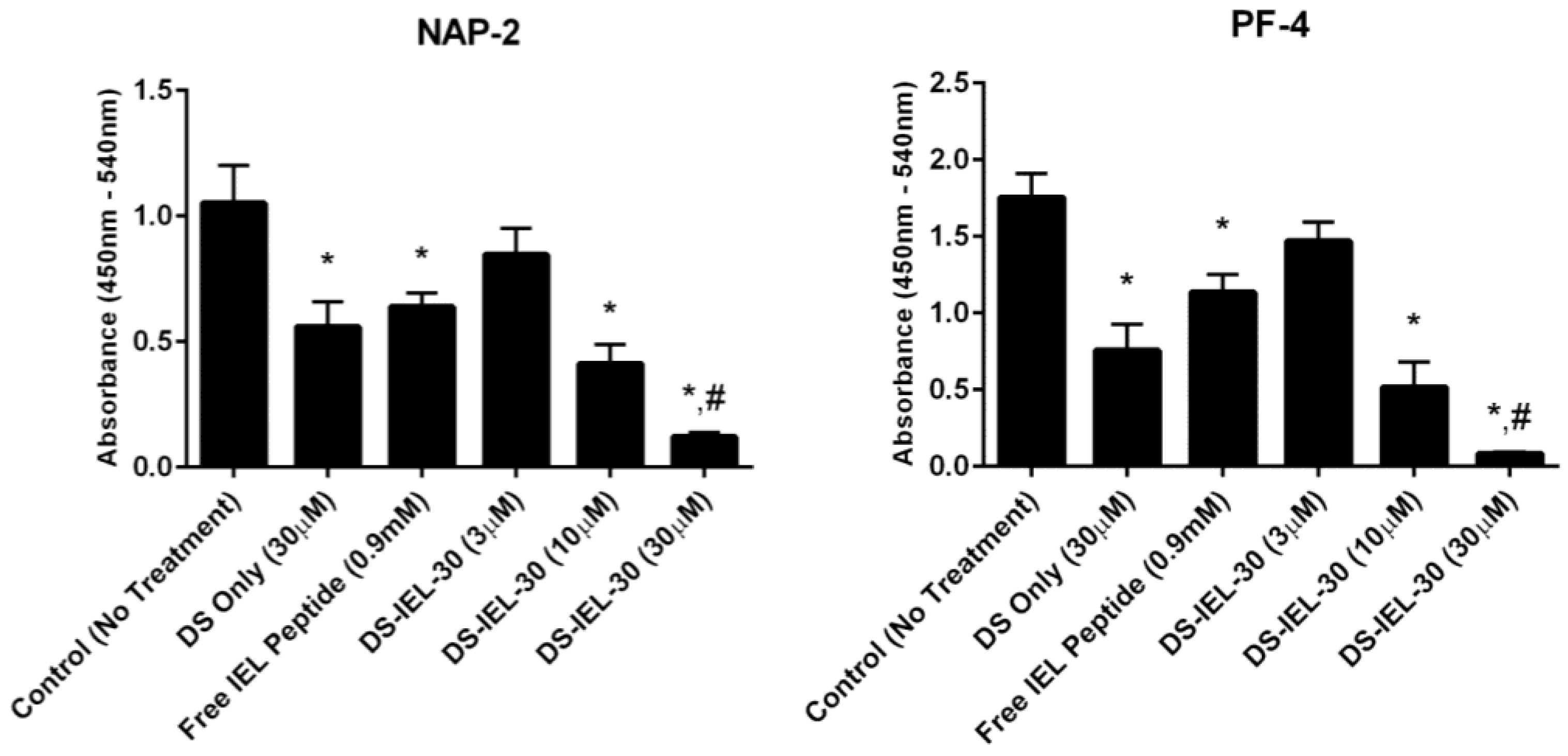
Figure 3.) However, using NAP-2 and PF-4 as platelet activation markers, DS-IEL
10 did not reduce platelet activation at any treatment concentration compared to untreated controls (
Figure 4). DS-IEL
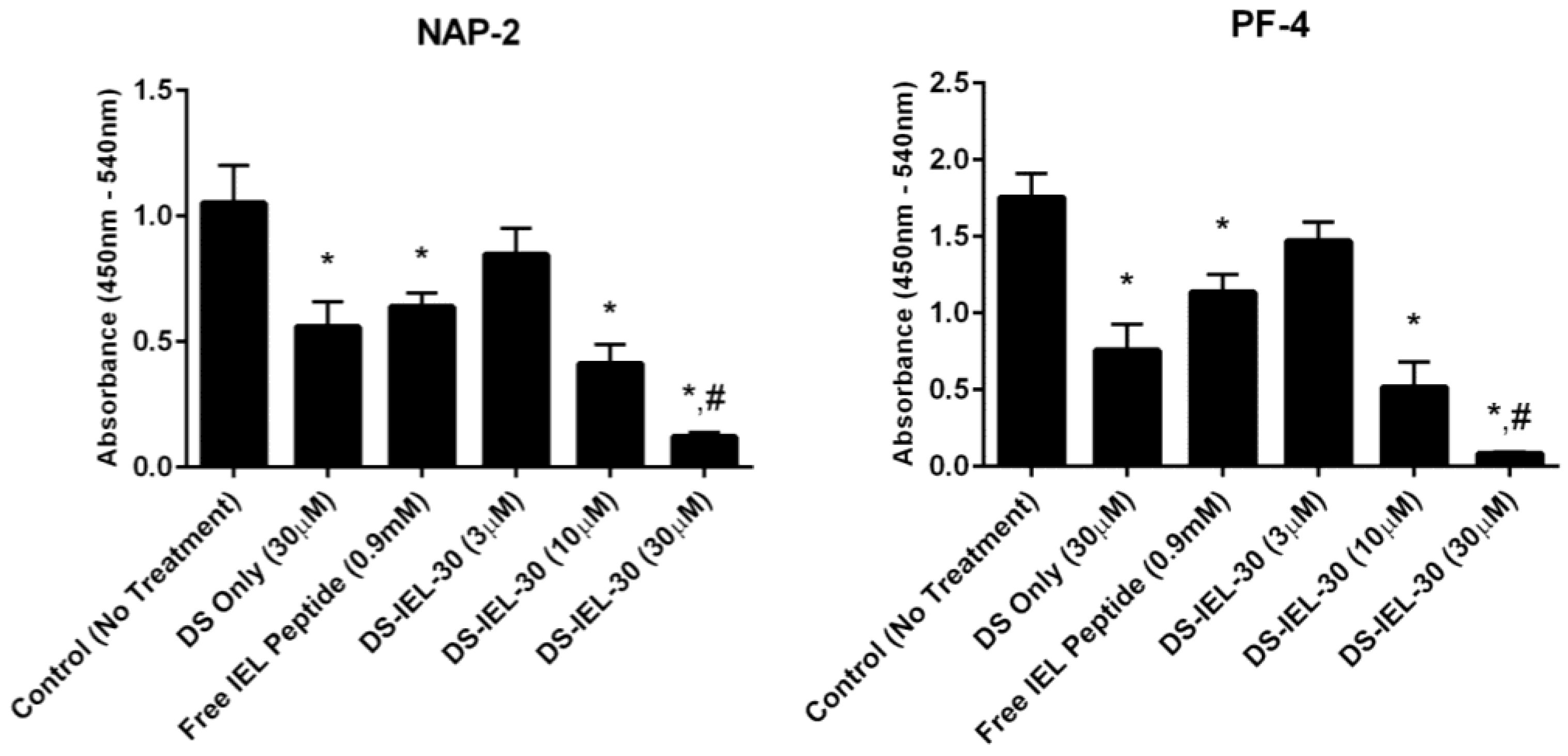
30 was also studied because the high density of selectin-binding peptide per backbone presented the possibility that some peptide would not be bound to the endothelial cell surface and might in fact bind to P-selectin on platelets and increase platelet binding. Importantly, there was no evidence for platelet-peptide interactions in these studies; in fact, DS-IEL
30 showed a dose-dependent response in reducing platelet activation compared to untreated controls (
Figure 5). Additionally, 30 µM DS-IEL
30 proved to be an extremely effective dose even when compared to equivalent amounts of its individual components (30 µM DS Only and 0.9 mM Free IEL Peptide). We believe that the apparent discrepancy between the binding studies (where DS-IEL
10 was believed to be best) and platelet activation results (where DS-IEL
30 was most effective) lies in how the binding was quantified. Since each DS backbone has one biotinylated peptide, the streptavidin-HRP assay will quantify each DS molecule attached regardless of the number of selectin-binding peptides bound to the surface. Therefore, despite DS-IEL
10 showing a slightly more than two-fold increase in number of molecules bound compared to DS-IEL
30, there are three times the number of selectin-binding peptides on each molecule of DS-IEL
30, resulting in a higher number of peptides available to bind to the cell surface and block interaction with platelets. Furthermore, increasing the number of peptides per molecule appeared to decrease nonspecific interactions (
Figure 2 and
Figure 3) and therefore, more of the molecules likely reached and bound to their targeted site. This suggests that despite fewer molecules present, the higher local peptide density of DS-IEL
30 supports more efficient binding to the EC surface to prevent platelet binding and activation.
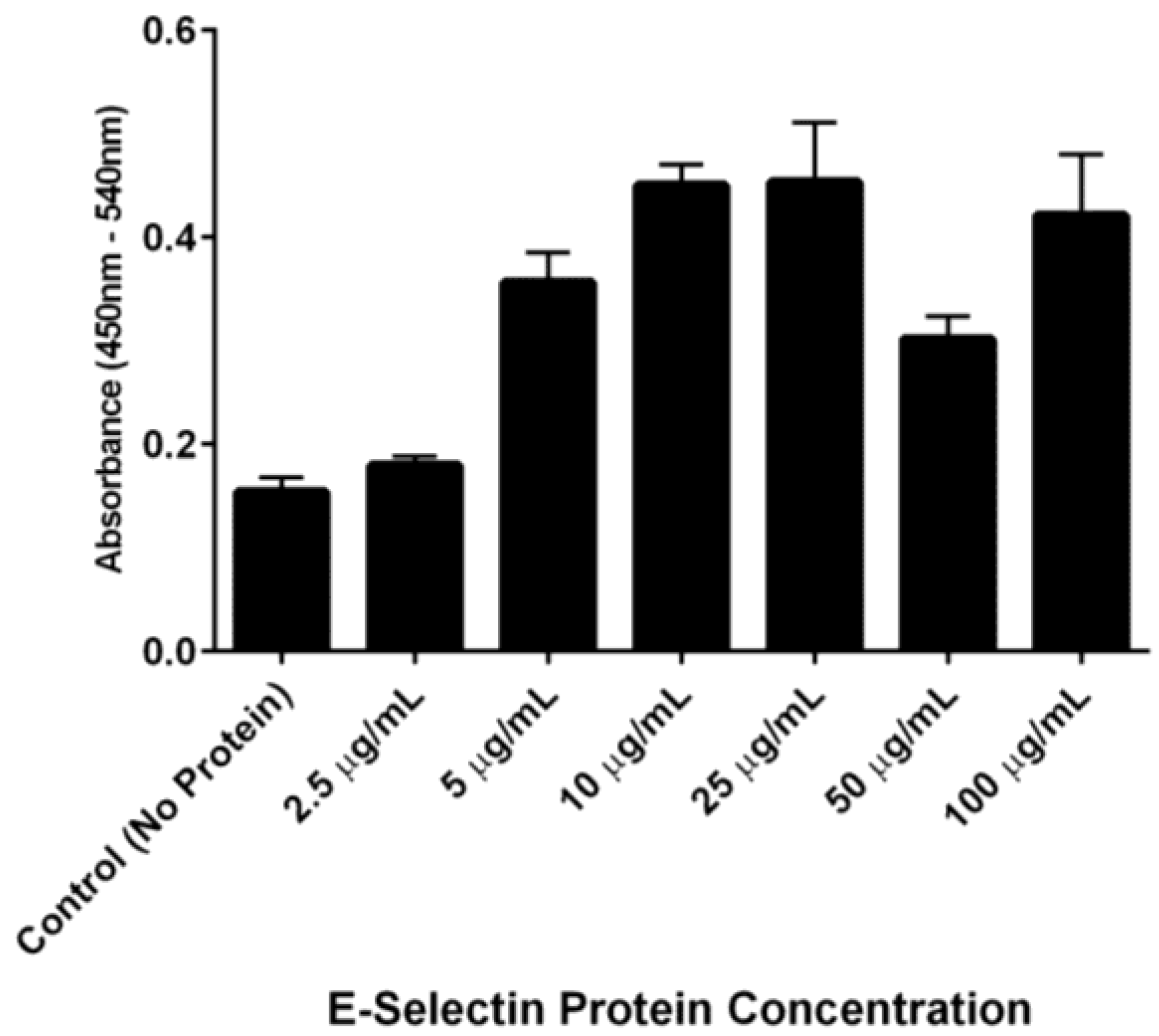
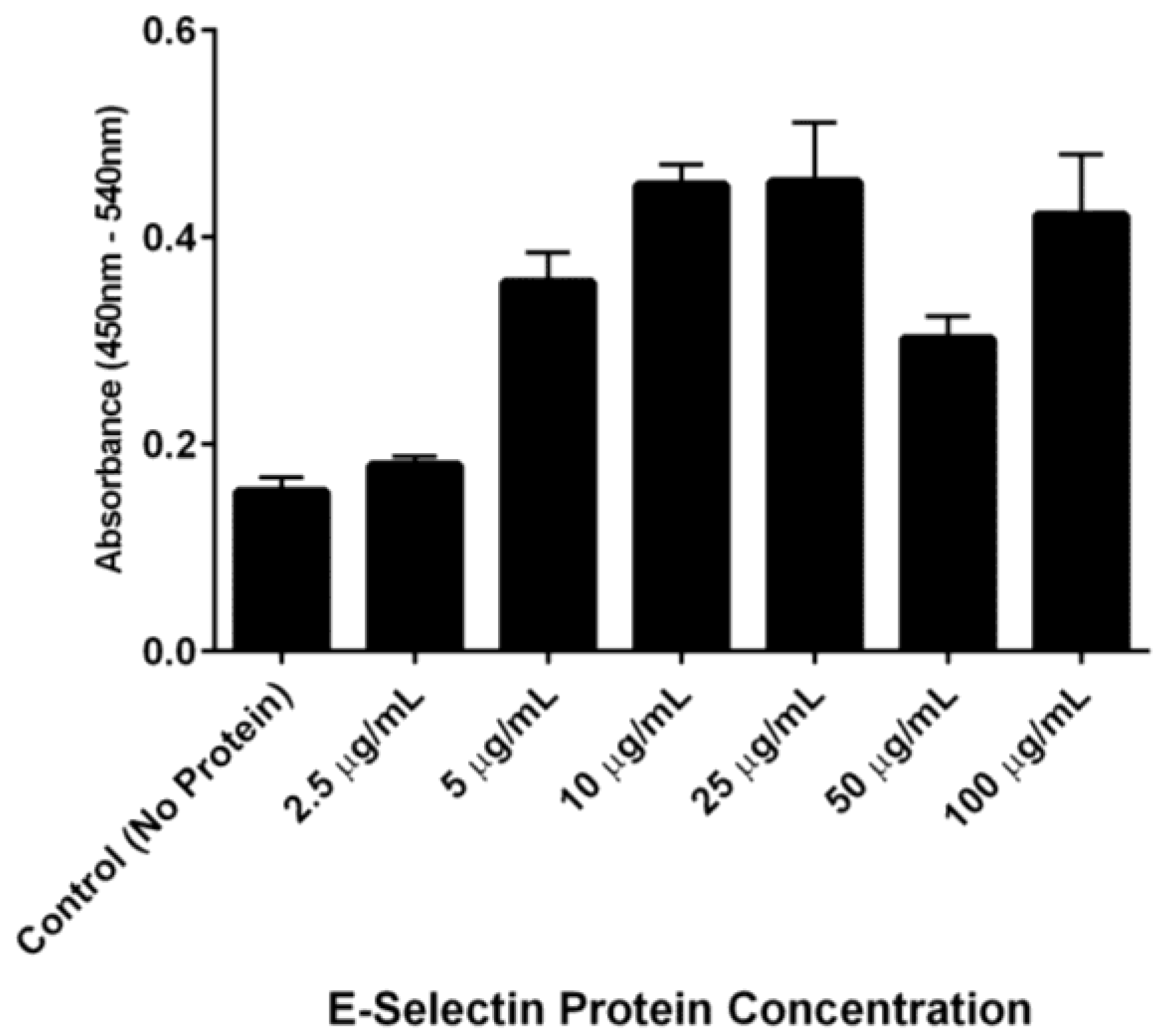
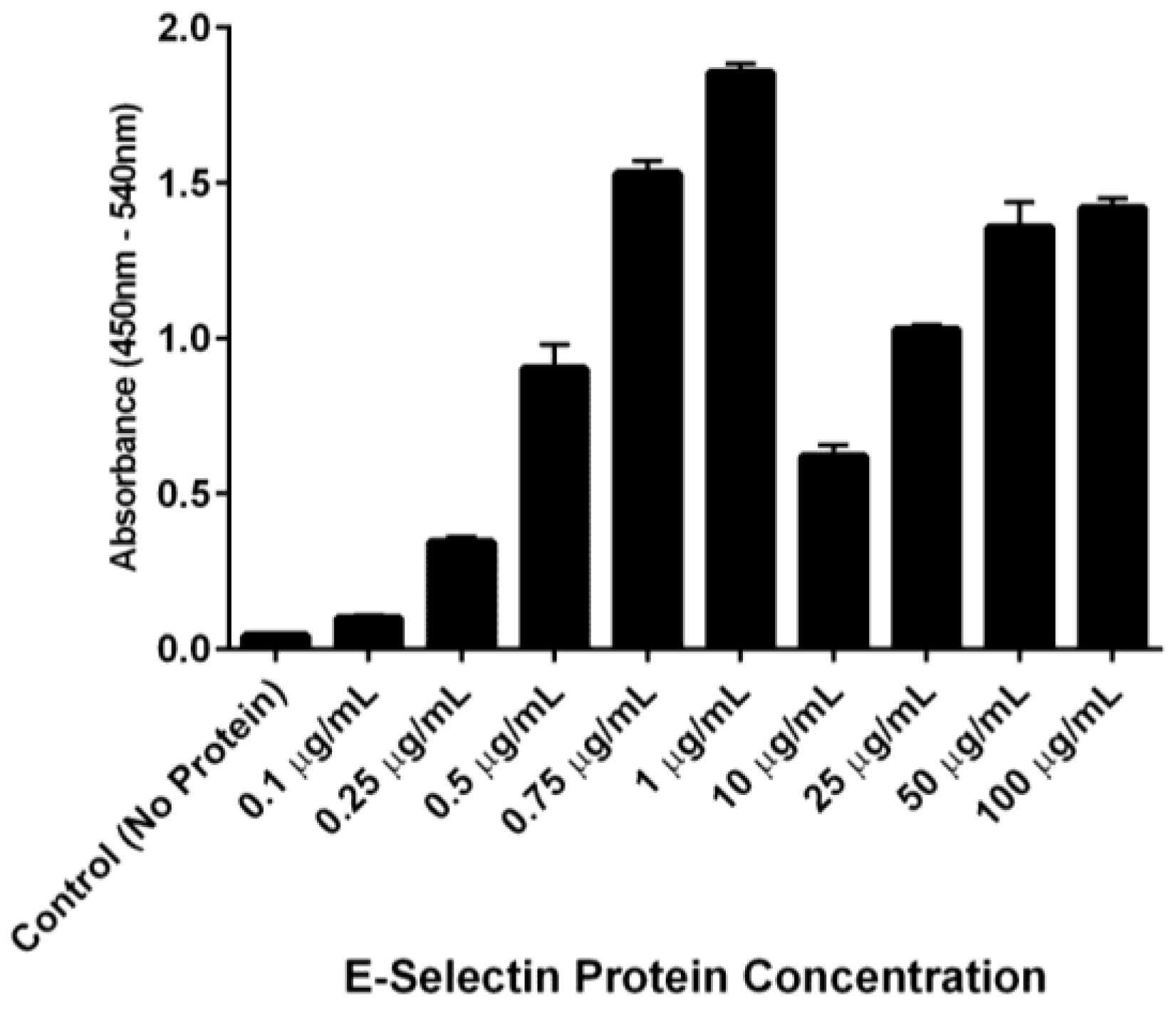
The ability of DS-IEL
30 to preferentially bind to recombinant human E-selectin over BSA was observed by incubating a single concentration of DS-IEL
30 on plates coated with increasing concentrations of E-selectin. DS-IEL
30 binding increased with initial increases in E-selectin concentration, but dropped off at a certain point, only to increase again upon a further increase in E-selectin concentration (
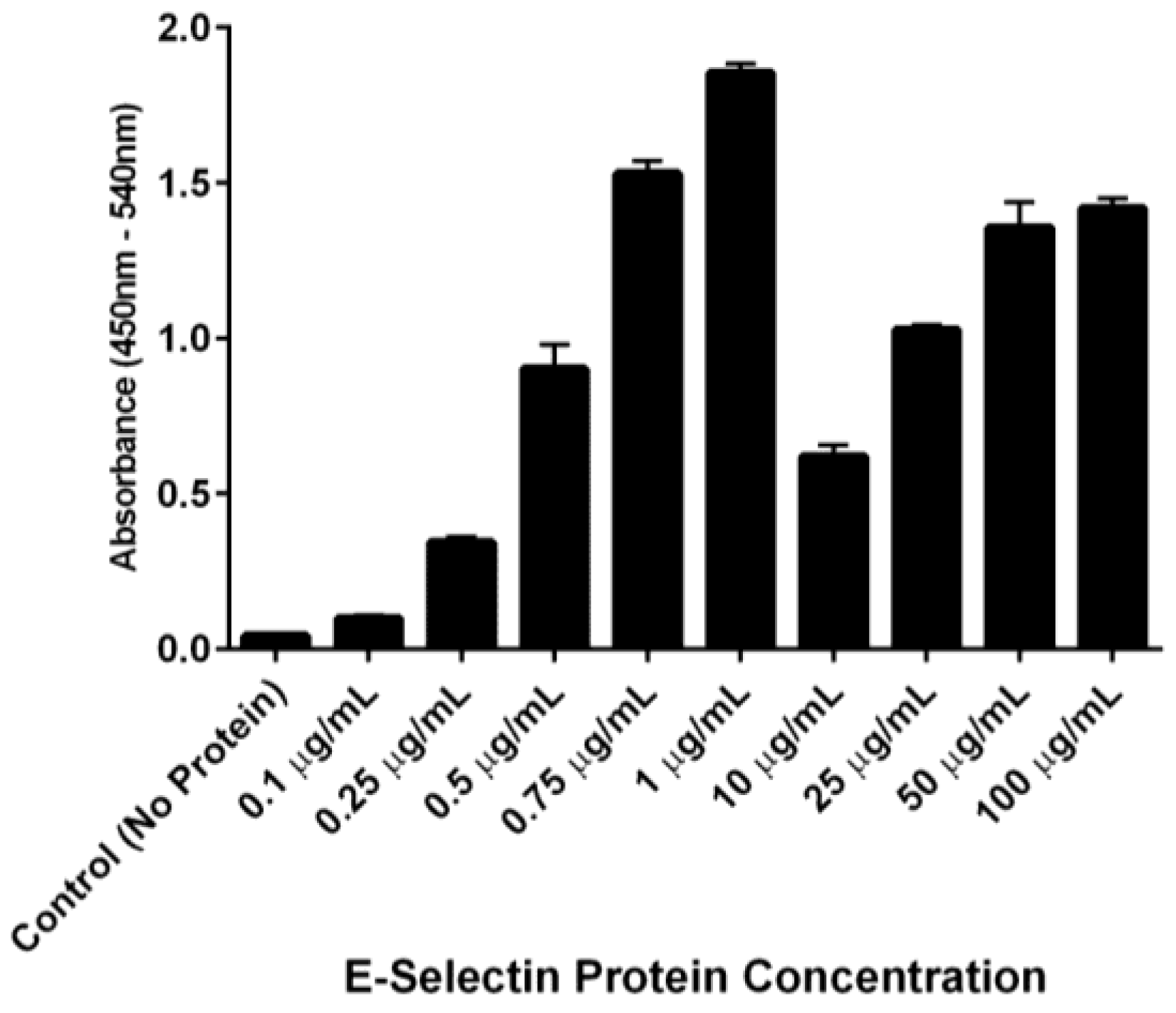
Figure 7). Interestingly, anti-E-selectin antibodies exhibited a similar binding trend, although at different concentrations of E-selectin (
Figure 8). We hypothesize that these binding characteristics are due to a potential selectin conformational change that is dependent upon concentration of both receptor (E-selectin) and ligand (DS-IEL
30 or antibody) and their respective binding affinities. Ligand binding itself is known to cause conformational changes at the binding site [
48,
49], including that of E-selectin [
50,
51]. It is also possible that the concentration of E-selectin protein coated on the plate’s surface influences the orientation in which the protein adsorbs and therefore, the exposure (or lack thereof) of ligand binding sites. Given that DS-IEL
30 and anti-E-selectin antibody may have different binding sites and affinities to E-selectin, it is not surprising that the drop off and subsequent rise in binding occur at different concentrations of plated E-selectin protein.
Given the vast number of vascular disease states associated with endothelial dysfunction, animal models in this area are wide-ranging. The utilization of mouse models has proven to be an extremely useful tool allowing for the study of underlying mechanisms and initial screenings of therapeutics [
52]. Thus, we employed a mouse model of deep vein thrombosis mediated endothelial dysfunction to observe the anti-thrombotic effects of EC-SEAL in vivo. This inferior vena cava (IVC) partial ligation model provides valuable information regarding vessel wall-blood interactions during thrombus formation and has been used previously to evaluate therapeutic agents [
52,
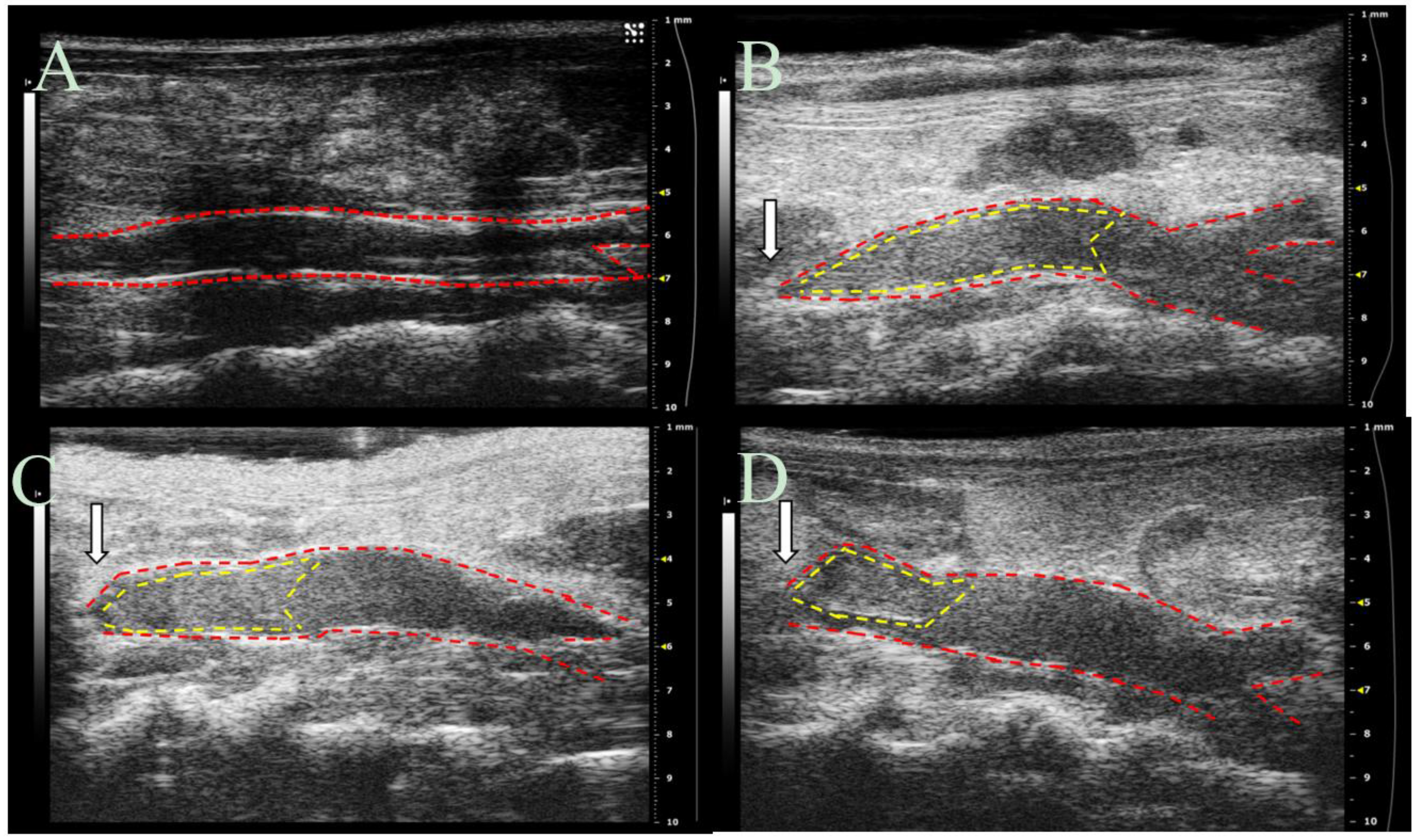
53]. Study results, obtained via ultrasound imaging, indicate that DS-IEL
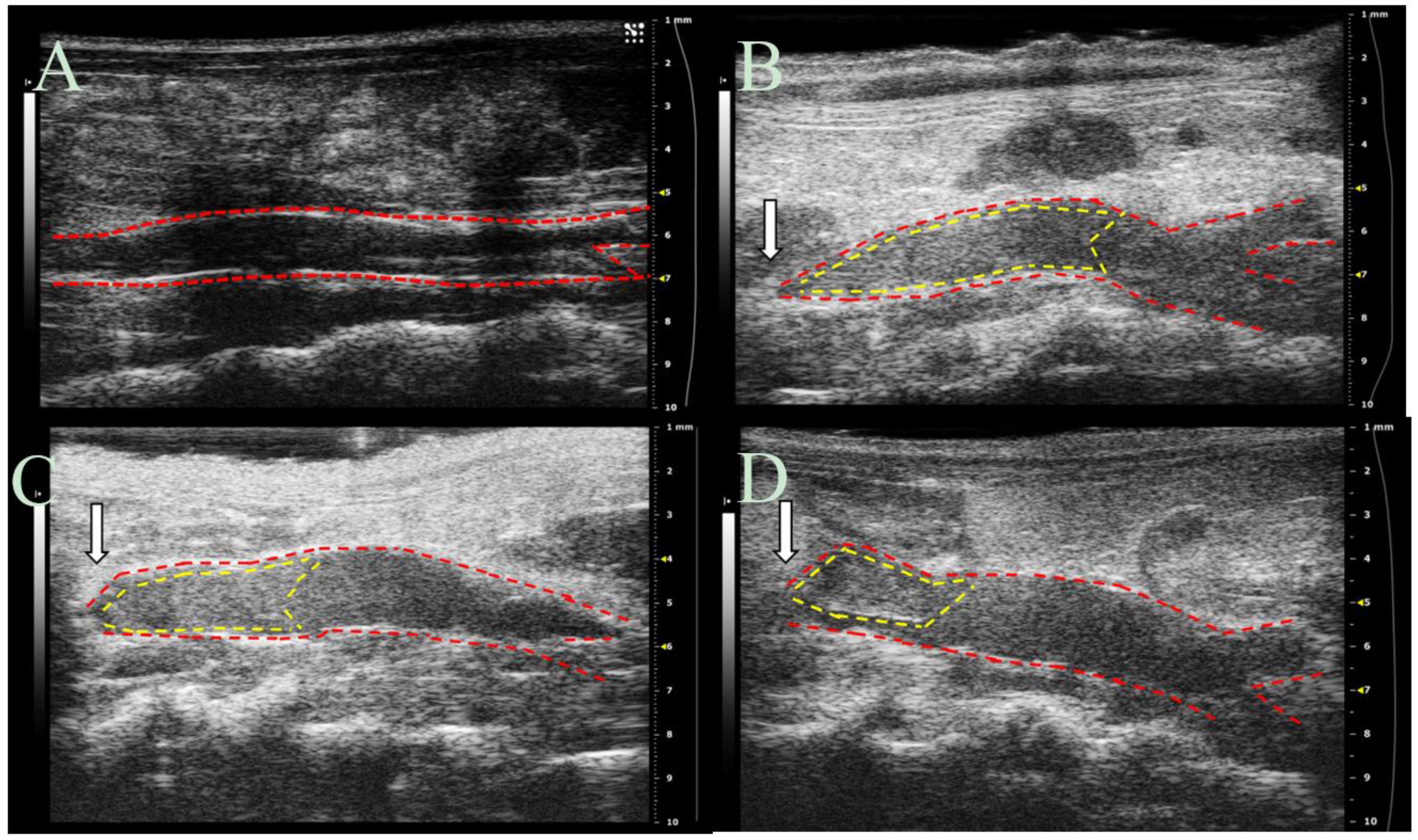
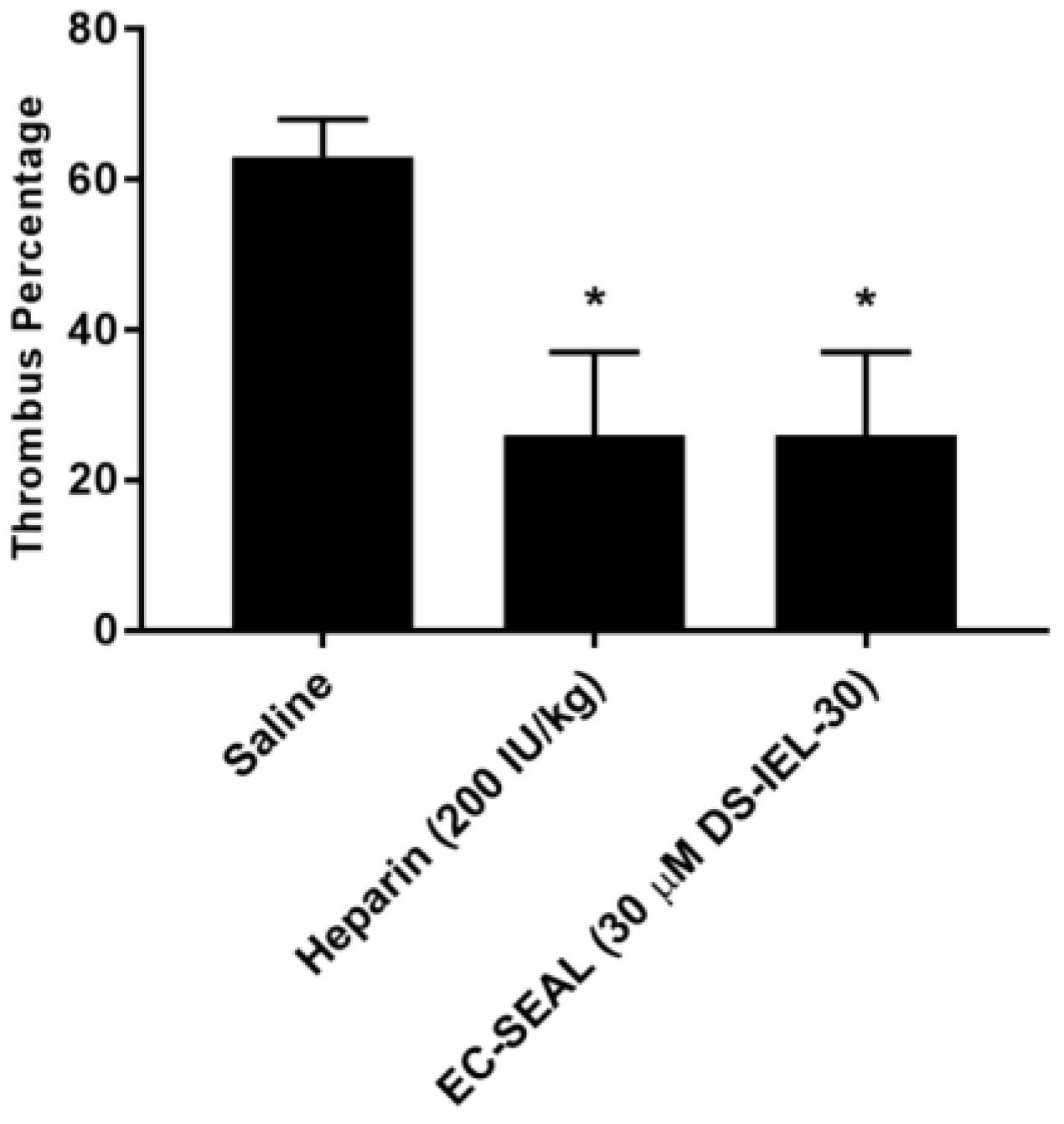
30 significantly decreases thrombus formation at six hours following IVC partial ligation (
Figure 10) and highlight the potential for DS-IEL
30 to act locally following systemic administration. It is believed that these therapeutic effects are due to DS-IEL
30 binding to the vessel wall and subsequently preventing platelet binding and not through direct anticoagulant properties. Dermatan sulfate from porcine origin (which is the type used for EC-SEAL synthesis) has been shown to have very minimal anticoagulant properties depending on the concentration [
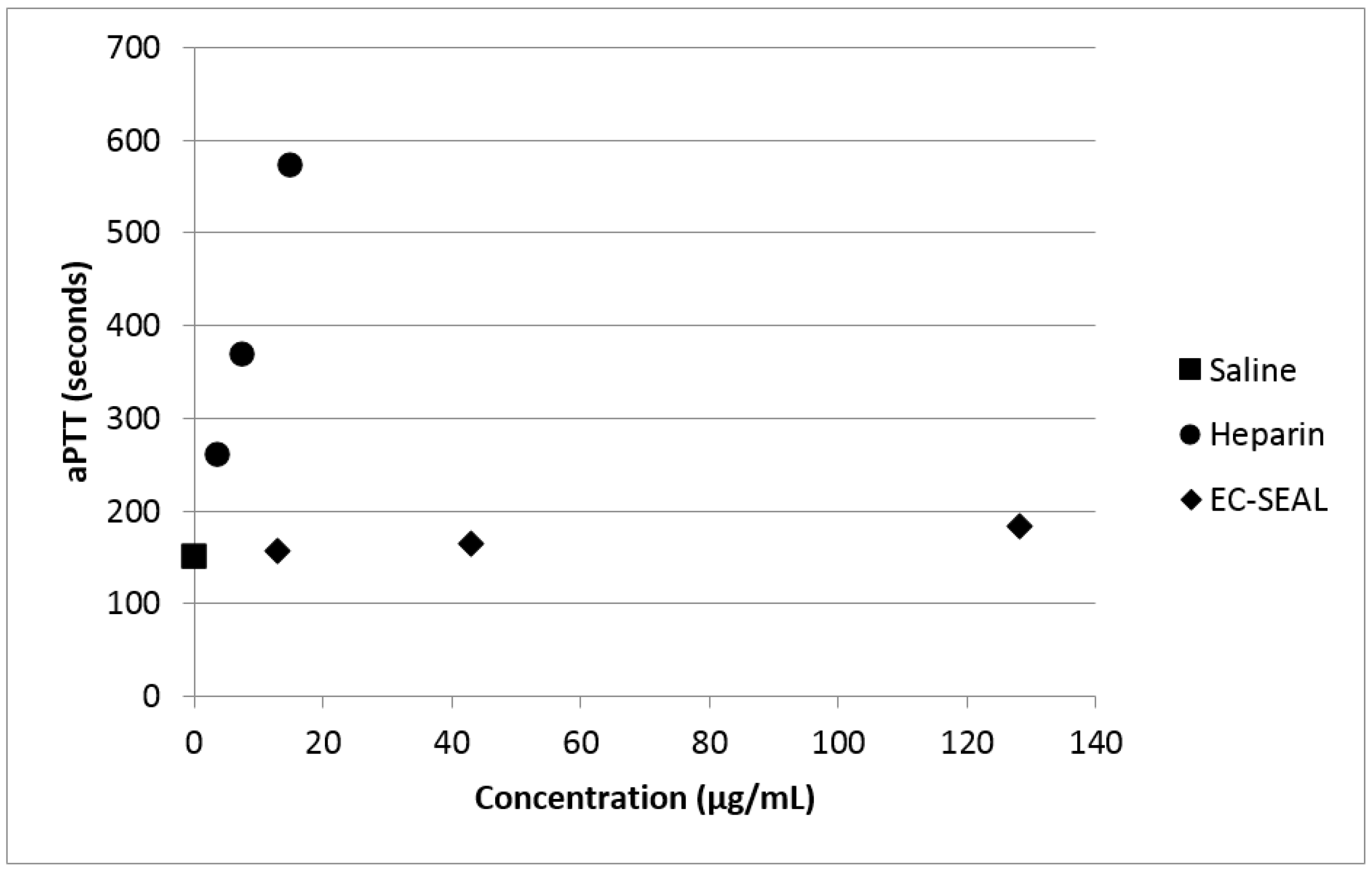
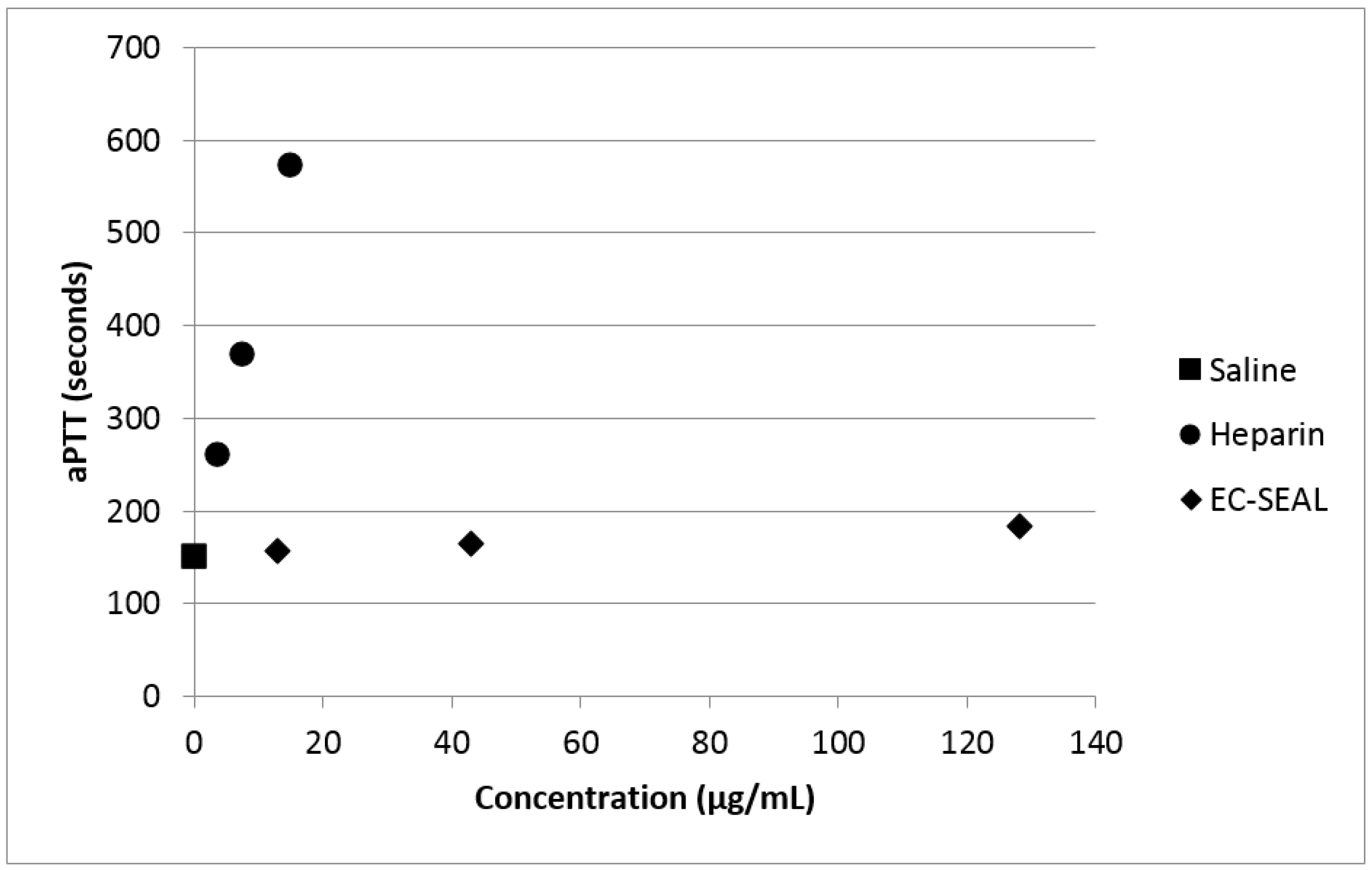
54]. By measuring aPTT of whole blood that had been incubated with varying concentrations of both heparin and DS-IEL
30, we have confirmed the minimal effect of DS-IEL
30 on clotting time (
Figure 11). Therefore, EC-SEAL has the potential to be utilized as a treatment for DVT without the negative side effects of current standard of care anticoagulant therapies such as heparin.
Since DS-IEL
30 proved to be the most effective EC-SEAL variant and no variants were synthesized with greater than 30 peptides per DS molecule, it is possible that further increases to the number of peptides per molecule may be beneficial. Previous work has shown, however, that excessive oxidation of GAGs can lead to chain scission [
55,
56], so a balance between number of selectin-binding peptides and DS backbone integrity will need to be considered. It is also possible that targeting additional EC adhesion molecules, such as ICAM-1 or VCAM-1, may further increase binding and therapeutic effect. Future studies on DS-IEL
30 and any additional EC-SEAL variants can focus on prevention of leukocyte binding and subsequent diapedesis. Additional experiments further assessing the specificity of EC-SEAL binding as well as observing the binding characteristics of EC-SEAL over time and under flow conditions to account for shear forces would be valuable.
4. Materials and Methods
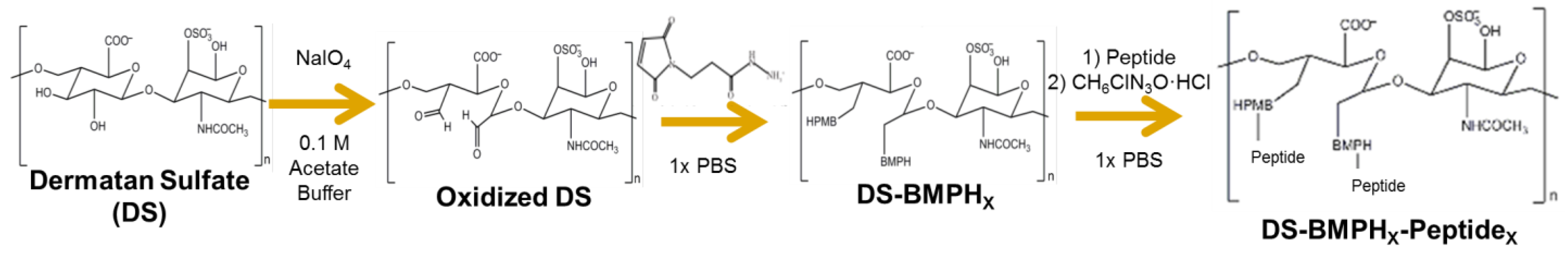
4.1. GAG Dervied, Selectin Targeting Anti-Adhesive Coating (EC-SEAL) Synthesis
The synthesis of the GAG derived, selectin targeting anti-adhesive coating variants (
Scheme 1) was performed at room temperature as follows: vicinal diol groups on dermatan sulfate (DS, MW
avg 46,275 Da, Celsus Laboratories, Cincinnati, OH, USA) were oxidized to aldehydes using sodium meta-periodate (Thermo Fisher Scientific, Waltham, MA, USA) in 0.1 M acetate buffer. The ratio of DS to sodium meta-periodate varied depending on the desired degree of oxidation. Following oxidation, the modified DS was isolated via size exclusion chromatography (SEC) (AKTA Purifier FPLC, GE Healthcare). An excess of
N-[β-maleimidopropionic acid] hydrazide, trifluoroacetic acid salt (BMPH, Thermo Fisher Scientific), was then conjugated via the hydrazide to the DS aldehyde groups in 1× phosphate buffered saline (PBS, 1.05 mM KH
2PO
4, 155.17 mM NaCl, 2.97 Na
2HPO
4-7H
2O; pH 7.4, Thermo Fisher Scientific) to form DS-BMPH
x. Unbound BMPH was removed via SEC and quantified by calculating the area under the peak to determine the average number (
x) of BMPH molecules bound to each DS backbone. Next, biotinylated peptide was added at a 1:1 molar ratio of DS:biotinylated peptide in 1× PBS to conjugate an average of one biotinylated peptide per molecule of DS, and then an excess of one of three selectin-binding peptides (Genscript, Piscataway, NJ, USA) was conjugated via the cysteine thiol to the maleimide groups on DS-BMPH
x in 1× PBS. The three peptide sequences used were as follows: IDLMQARGC (IDL) [
31], IELLQARGC (IEL) [
31,
32,
34,
57], and QITWAQLWNMMKGC (QIT) [
47]. After the peptide reaction was complete, semicarbazide hydrochloride (Sigma-Aldrich, St. Louis, MO, USA) was added directly to the solution to reduce any unreacted aldehyde groups. Subsequent SEC isolation and lyophilization resulted in the purified sixteen variants containing from one to thirty peptides per DS backbone of DS-BMPH
x-Peptide
x, biotin (hereby simply referred to in the form of DS-Peptide
x, ex: DS-IEL
20).
4.2. Cell Cultures
Human aortic endothelial cells (ECs, Thermo Fisher Scientific) were cultured in Medium 200 (M 200, Thermo Fisher Scientific) with low serum growth supplement (LSGS, Thermo Fisher Scientific). Human coronary artery smooth muscle cells (SMCs, Thermo Fisher Scientific) were cultured in Medium 231 (M 231, Thermo Fisher Scientific) with smooth muscle growth supplement (SMGS, Thermo Fisher Scientific). Cells were maintained at 37 °C and 5% CO2. Unless otherwise noted, passage 3–7 cells were seeded at a density of 1 × 105 cells/cm2 on tissue culture polystyrene and allowed to adhere for 24 h prior to any stimulation or treatment.
4.3. Selectin Expression
Monolayers of SMCs and ECs were seeded separately on Biocoat Collagen I Cellware 96-well plates (Corning, Corning, NY, USA). ECs were stimulated for four hours with 0.05–50 ng/mL tumor necrosis factor-α (TNF-α, Thermo Fisher Scientific), 10–100 ng/mL interleukin-1β (IL-1β, PeproTech, Rocky Hill, NJ, USA), 10–100 ng/mL interleukin-6 (IL-6, PeproTech), 10–100 ng/mL lipopolysaccharide (LPS, Sigma-Aldrich) or control (unstimulated) media. SMCs were cultured with only control (unstimulated) media. To block nonspecific binding, cells were treated with 1% bovine serum albumin (BSA, Sigma-Aldrich) in 0.05 M tris-buffered saline (TBS, 0.138 M NaCl, 0.0027 M KCl; pH 8.0, Sigma-Aldrich) for 30 min at room temperature with shaking. Primary rabbit polyclonal anti-E-selectin IgG (Santa Cruz Biotechnology, Dallas, TX, USA) or mouse monoclonal anti-E-selectin IgG2a (Santa Cruz Biotechnology) antibody was added (1:50 dilution of initial 200 μg/mL in 1% BSA in TBS) for one hour at 4 °C with shaking. Cells were rinsed three times with 1% BSA in TBS and secondary donkey anti-rabbit IgG HRP-conjugated (Thermo Fisher Scientific) or goat anti-mouse IgG HRP-conjugated (Thermo Fisher Scientific) antibody was added (1:1000 dilution of initial 0.5 mg/mL in 1% BSA in TBS) for 30 min at room temperature with shaking. Cells were rinsed three times with 1% BSA in TBS and colorimetric change was induced with 1:1 hydrogen peroxide: tetramethylbenzidine (R&D Systems, Minneapolis, MN, USA). Following 20 min of incubation with shaking, 2N sulfuric acid was added to cease the reaction and absorbance was measured at 450 nm and 540 nm using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA, USA). For each individual well, 540 nm was subtracted from 450 nm to obtain final absorbance reading.
4.4. EC Monolayer Permeability
ECs were seeded in control (unstimulated) media on tissue culture treated Transwell polyester membrane inserts with 3.0 μm pores on a 24-well plate (Corning) and cultured for 48 h to form monolayers. Cell culture media in both the upper and lower chambers was then changed with fresh, unstimulated media, and Rhodamine B isothiocyanate (RITC)-dextran (Sigma-Aldrich) was added to obtain a final concentration of 3 mg/mL in the upper chamber. The monolayers were incubated at 37 °C and 5% CO2 for four hours, then the media in the lower chamber was removed and fluorescence was read using an M5 plate reader (Ex: 520 nm; Em: 590 nm).
4.5. EC-SEAL Binding to Cells
Monolayers of SMCs and ECs were seeded separately on Biocoat Collagen I Cellware 96-well plates or CellBIND surface 96-well plates (Corning). ECs were stimulated for four hours with 5 ng/mL TNF-α or control (unstimulated) media, and SMCs were treated with only control (unstimulated) media. Nonspecific binding was blocked using 1% BSA in TBS with 150 mM CaCl2 for 20 min at room temperature with shaking. Cells were then treated with EC-SEAL variants (3 µM in 1% BSA in TBS with 150 mM CaCl2) for one hour at 37 °C and 5% CO2. After rinsing three times with 1% BSA in TBS with 150 mM CaCl2, streptavidin-HRP (R&D Systems) was diluted 1:200 in 1% BSA in TBS with 150 mM CaCl2 and added for 20 min at room temperature with shaking. After three more rinses in TBS with 150 mM CaCl2, a 1:1 hydrogen peroxide: tetramethylbenzidine solution was added to induce colorimetric change. After 20 min of incubation with shaking, 2N sulfuric acid was used to stop the reaction and an M5 plate reader was utilized to measure absorbance at 450 nm and 540 nm as described above.
4.6. Platelet Activation
EC monolayers were seeded on Biocoat Collagen I Cellware 96-well plates and stimulated with 5 ng/mL TNF-α for four hours. One-percent BSA in TBS with 150 mM CaCl2 was added for 15 min at room temperature to decrease nonspecific binding. ECs were then treated with 3–30 µM EC-SEAL variants for 1 h at 37 °C and 5% CO2. Whole blood was obtained from healthy volunteers in collection vials containing sodium citrate and centrifuged for 20 min at 200× g and 25 °C. Platelet rich plasma (PRP) was isolated and after EC-SEAL treatments, ECs were rinsed three times with TBS with 150 mM CaCl2 and 100 µL of PRP was added to each well. After 1 h of incubation at room temperature, 45 µL of PRP was then removed and added to tubes containing 5 µL of ETP (107 mM Ethylenediaminetetraacetic acid, disodium salt (EDTA, Promega, Madison, WI, USA), 12 mM Theophylline (Sigma-Aldrich) and 2.8 mM Prostaglandin E1 (Enzo Life Sciences, Farmingdale, NY, USA) in water). Samples were then centrifuged for 30 min at 2000× g and 4 °C and supernatant was collected and stored at −80 °C for use in subsequent assays.
4.7. NAP-2 and PF-4 ELISA
To quantify platelet activation, neutrophil activating peptide-2 (NAP-2) and platelet factor-4 (PF-4) levels in PRP collected from platelet activation experiments were measured utilizing sandwich ELISAs. Mouse monoclonal anti-hNAP-2 IgG and anti-hPF-4 IgG2B capture antibodies (R&D Systems) were coated on 96-well EIA/RIA high binding plates (Corning) at 2 µg/mL in 1× phosphate buffered saline (PBS) and incubated at 4 °C overnight. After rinsing three times with 1× PBS + 0.05% Tween and blocking with 1% BSA in 1× PBS for 1 h at room temperature with shaking, previously collected PRP samples were thawed, diluted 1:10,000 in 1% BSA in 1× PBS and added for 2 h at room temperature with shaking. Samples were removed, wells were triple rinsed with 1× PBS + 0.05% Tween and biotinylated polyclonal goat anti-hNAP-2 IgG and anti-hPF-4 IgG detection antibodies (R&D Systems) were added at 0.2 µg/mL in 1× PBS for 2 h at room temperature with shaking. After three rinses in 1× PBS + 0.05% Tween, streptavidin-HRP (diluted 1:200 in 1% BSA in 1× PBS) was added and detected as described above.
4.8. Platelet Binding
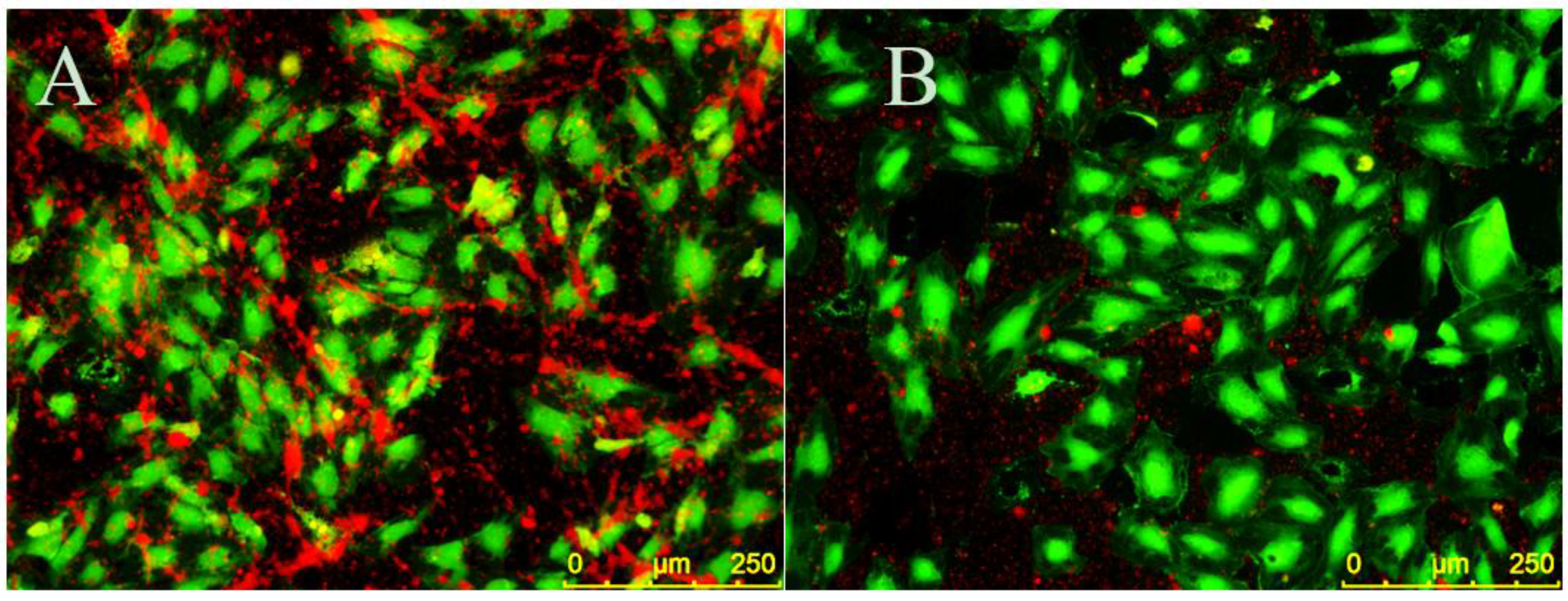
EC monolayers were seeded on CellBIND surface 96-well plates and stimulated with 5 ng/mL TNF-α for four hours to create a proinflammatory environment. CellTracker Green CMFDA (5-chloromethylfluorescein diacetate) (Thermo Fisher Scientific) was dissolved in dimethyl sulfoxide (DMSO), diluted in EC media and added to ECs for 20 min at 37 °C and 5% CO2. Cells were then rinsed three times in TBS with 150 mM CaCl2 and 1% BSA in TBS with 150 mM CaCl2 was added to ECs for 10 min at room temperature to decrease nonspecific binding. ECs were then treated with 30 µM EC-SEAL variants for 1 h at 37 °C and 5% CO2. PRP was isolated from human whole blood as described above and labeled with CellTracker Orange CMRA (Thermo Fisher Scientific) (which was previously dissolved in DMSO) for 25 min at 37 °C and 5% CO2. Platelets were then pelleted via centrifugation for 10 min at 900× g and 25 °C, supernatant was removed and remaining platelets were suspended in EC media. Following EC-SEAL treatments and rinsing three times in TBS with 150 mM CaCl2, labeled platelets in EC media were added to ECs for 1 h at 37 °C and 5% CO2. After incubation, media was removed and the cells were rinsed in TBS with 150 mM CaCl2 and then fixed with 3% paraformaldehyde for 20 min at room temperature. Three final rinses with TBS with 150 mM CaCl2 were performed and cells were imaged using a Leica DMI6000 B microscope with EL6000 external light source (Leica Microsystems, Wetzler, Germany) and CoolSNAP HQ2 camera (Photometrics, Tucson, AZ, USA). Leica application suite (LAS) AF6000 software (Leica Microsystems) was utilized for image acquisition. Excitation/emission spectra used were 460–500 nm/512–542 nm for ECs (green) and 540–552 nm/580–620 nm for platelets (orange). Contrast and brightness editing of acquired images was accomplished using the program ImageJ (National Institutes of Health, Bethesda, MD, USA) and identical settings were applied to all images.
4.9. EC-SEAL Binding (Selectin Protein)
Recombinant human E-selectin (PeproTech) was dissolved in water and added to a 96-well EIA/RIA high binding plate at 0.1–100 µg/mL. Following overnight incubation at 4 °C, the plate was rinsed three times with TBS with 150 mM CaCl2 and blocked with 1% BSA in TBS with 150 mM CaCl2 for 1 h at room temperature with shaking. The plate was then rinsed three more times with TBS with 150 mM CaCl2. In experiments observing EC-SEAL binding, 3 µM EC-SEAL (in 1%BSA in TBS with 150 mM CaCl2) was added for 1 h at room temperature with shaking. Following three rinses with TBS with 150 mM CaCl2, streptavidin-HRP (1:200 in 1% BSA in TBS with 150 mM CaCl2) was added for 20 min at room temperature with shaking. For experiments observing antibody binding, primary mouse monoclonal anti-E-selectin IgG2a antibody was added (1:50 dilution of initial 200 μg/mL in 1% BSA in TBS) for two hours at room temperature with shaking. Following three rinses with TBS with 150 mM CaCl2, secondary goat anti-mouse IgG HRP-conjugated antibody was added (1:1000 dilution of initial 0.5 mg/mL in 1% BSA in TBS) for 1 h at room temperature with shaking. HRP activity was detected as described above.
4.10. DVT Mouse Model
Ten-week-old C57BL/6 male mice (average weight: 26.4 g) were utilized in a model of surgically-induced deep vein thrombosis (DVT). All animals used in this study were obtained from Jackson Laboratory (Bar Harbor, ME) and fed standard chow diet. A well-established model of DVT caused by a significant inferior vena cava (IVC) flow restriction was utilized following a protocol (1505001247) approved by the institutional animal care and use committee at Purdue University. Mice were anesthetized using 1%–3% isoflurane and oxygen at a flow rate of 225 mL/min [
58]; toe pinch was be used to determine sufficient anesthetic induction. Buprenorphine (0.03 mg/mL) was subcutaneously injected near the incision site at 0.05 mg/kg for pain management. Using aseptic technique, a small incision was made in the abdomen and the entrails were carefully exteriorized onto a sterile saline soaked gauze pad to prevent desiccation. The skin and organs were further retracted to fully expose the IVC, its branching vessels, and the infrarenal aorta. The aorta was then carefully separated from the IVC directly below the left renal vein. In an attempt to ligate all side and back branching vessels of the IVC, 6-0 black silk-braided sutures and a low power cautery pen (Bovie Medical, Purchase, NY, USA) were used, respectively. To achieve flow restriction, the proximal region of the IVC below the left renal vein was partially ligated by placing a 30-guage needle adjacent to the IVC and then tying a 6-0 black silk-braided suture around both the IVC and needle. The needle was then removed, resulting in an approximate 90% reduction in flow through the vessel. The muscle and skin layer were closed using 5-0 polypropylene sutures. A syringe with a 30-gauge needle was utilized to inject 100 µL of 30 μM EC-SEAL (
n = 4), 200 IU/kg heparin (
n = 6), or saline (
n = 6) via tail vein. Tail vein injections were made approximately 10 min post-partial ligation of the IVC.
4.11. Ultrasound Imaging
Mice were anesthetized using 2%–3% isoflurane in 1.5 L/min oxygen. Cardiac and respiration rates were noninvasively monitored using gold-plated stage electrodes as part of the Vevo imaging station (FUJIFILM VisualSonics Inc., Toronto, ON, Canada). The mice were placed in a supine position on heated animal stage and the temperature was monitored using a rectal probe such that animals remained near 37°C. Depilatory cream was applied to remove hair on the abdomen prior to imaging, and warm ultrasound gel was applied on the skin to the entire abdomen just below the xiphoid process. IVC images were obtained using a Vevo2100 ultrasound imaging system with MS550D (40 MHz center frequency) and MS700 (50 MHz center frequency) transducers (FUJIFILM VisualSonics). The transducers were locked in the adjustable arm of the Vevo integrated rail system (FUJIFILM VisualSonics) for consistent image collection. In order to prevent distortion of the IVC, minimal pressure was applied to the abdomen to maintain original diameter of the vessel. The walls of the IVC were visualized with both long and short axes views. The angle of the stage was adjusted to optimize the view of the IVC and to minimize artifacts due to air and gas. Long and short axis B-mode images of the IVC were acquired at baseline (pre-operation) and at 6 h post-ligation of the IVC. Color Doppler images were used to assess blood flow through the IVC and branching vessels, and 3D scans of sequential slices were used to obtain vessel volumes as described previously [
59,
60] . Suture placement and the bifurcation of the IVC were used as landmarks to ensure consistent measurements between mice.
4.12. Ultrasound Image Analysis
All vessel measurements were made by the same ultrasonographer using Vevo LAB software (FUJIFILM VisualSonics). Thrombus length and volume and vessel volume were calculated using both the long axis and short axis ultrasound images. To determine the location of the thrombus, Color Doppler flow and B-mode images were utilized to visualize blood flow. The thrombus was also visualized as a hyperechoic region within the vessel lumen. Thrombus length was determined from the suture (point of IVC partial ligation) to the farthest point of thrombus and was calculated using an average of five measurements per image. The length of the thrombus in the 3D scans was compared to the length of thrombus in the long axis B-mode to ensure agreement. Thrombus volume was calculated by lofting together individual 2D segmentations from the first hyperechoic region in the lumen to the suture (where the lumen signal changed from hyperechoic to hypoechoic). Total IVC vessel volume was obtained using these same volumetric images and was measured from the suture to the IVC bifurcation. Percentage of IVC occlusion was obtained by determining the amount of thrombus volume within the total volume of the vessel.
4.13. Clotting Time (aPTT)
Activated partial thromboplastin time (aPTT) was measured using a Hemochron® Response (Accriva Diagnostics, San Diego, CA, USA) with associated reagents. The protocol was run according to manufacturer’s instructions. Briefly, whole blood was obtained from healthy volunteers in sodium citrate collection vials and 2 mL was added to the Hemochron® aPTT (citrated blood) test tube. One hundred microliters of saline, heparin or EC-SEAL was then added (final concentrations: 3.75–15 µg/mL heparin; 13–128 µg/mL EC-SEAL), and the test tube was shaken vigorously ten times from end-to-end. The test tube was then placed into the Hemochron® Response device and clotting time was recorded at completion.
4.14. Statistics
Unless otherwise noted, all experiments were performed in triplicate or quadruplicate (n ≥ 2) and results are presented as mean ± standard deviation. Statistical analysis was performed using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA). All results were analyzed using ANOVA with post-hoc Tukey test. Statistical significance threshold was set at p < 0.05.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}