
Structural Hypervariability of the Two Human Protein Kinase CK2 Catalytic Subunit Paralogs Revealed by Complex Structures with a Flavonol- and a Thieno[2,3-d]pyrimidine-Based Inhibitor †

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Overview of the CK2α/CK2α′ Co-Crystal Structures
2.2. Complex Structures with FLC21
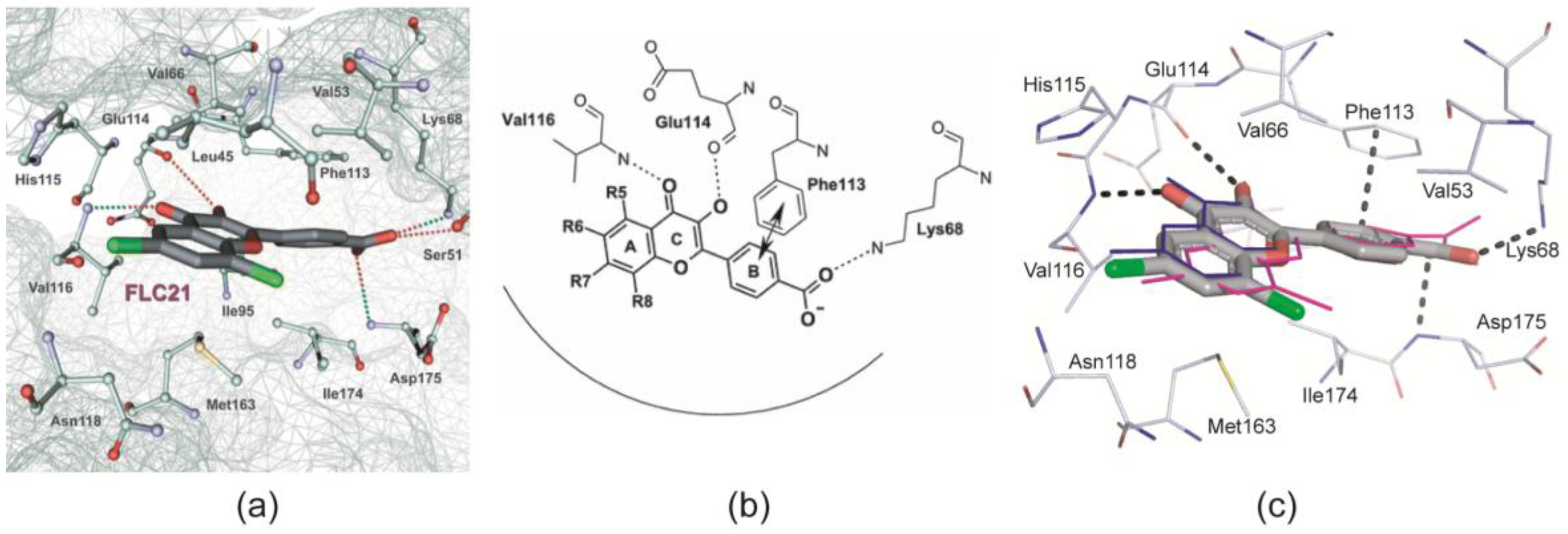
2.2.1. General Binding Mode of FLC21 to CK2α/CK2α′
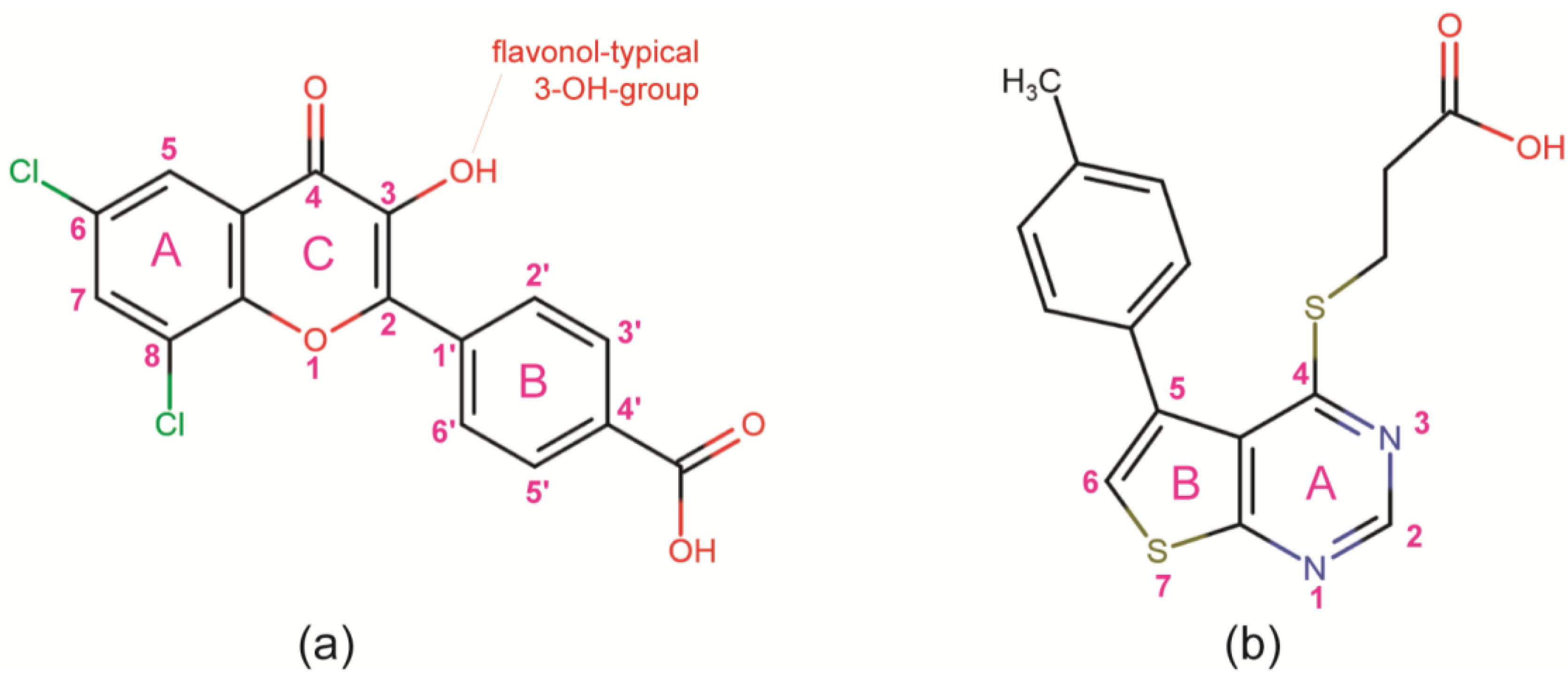
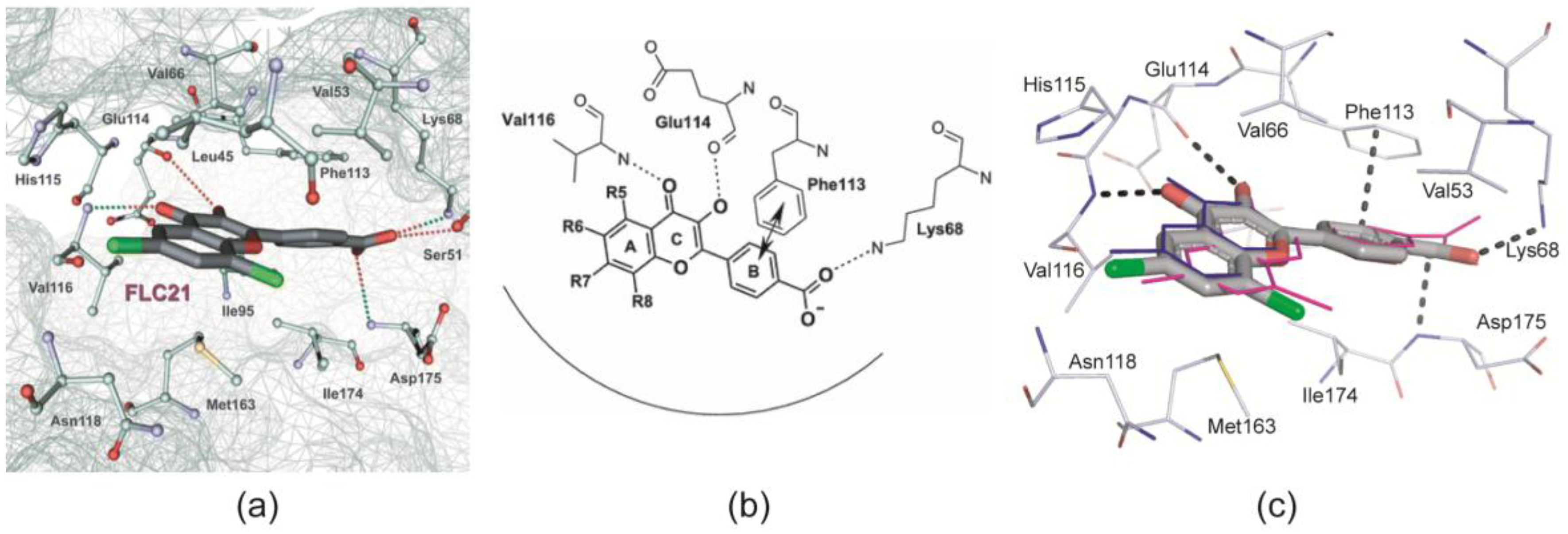
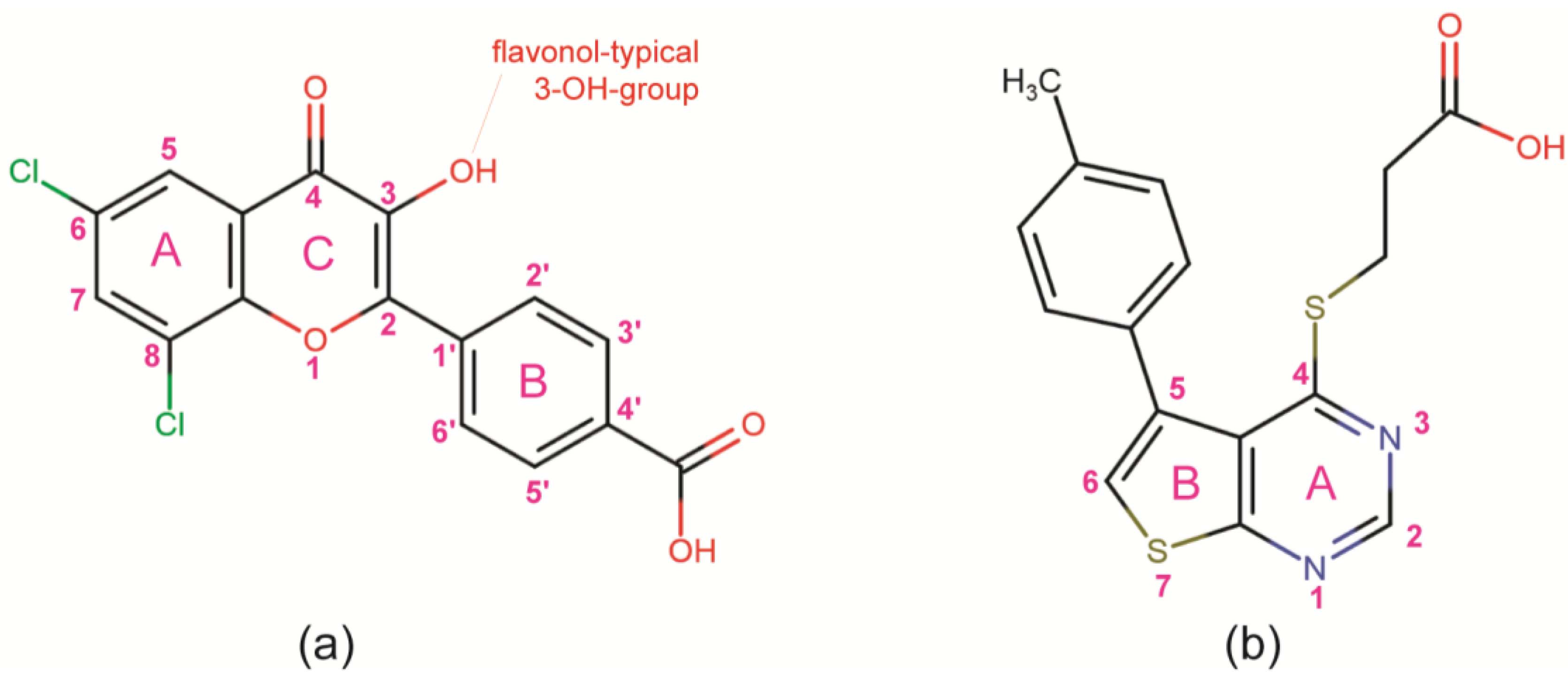
- Golub et al. [28] modelled FLC21 bound to CK2α as shown in Figure 2a. This CK2α/FLC21 complex model is based on a set of four predicted ionic, hydrogen bond and π/π interactions (Figure 2b). These interactions were assumed to be formed by the B and the C-ring of the flavone framework and its substituents whereas the A-ring with the two chloro atoms were supposed to be not involved.
- Guerra et al. [22] published two complex structures (PDB 4UBA and 4UB7) of CK2α1–335 with FLC26 [28] which is the sister compound of FLC21 containing bromo rather than chloro substituents attached to the C-atoms 6 and 8 (Figure 1a). These structures revealed that the inhibitor was in fact bound to the enzyme in the predicted orientation and with exactly the set of non-covalent interactions suggested in Figure 2b.
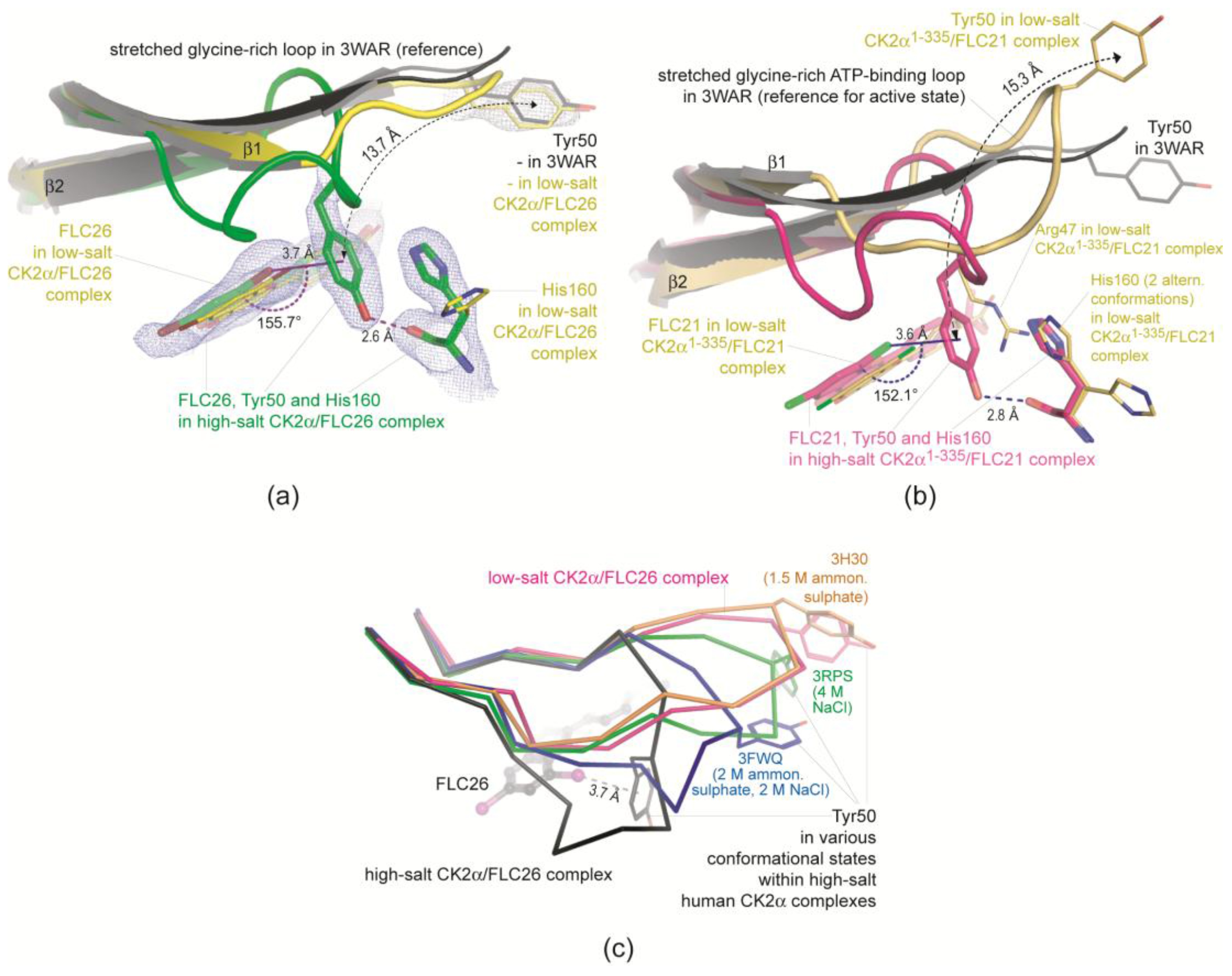
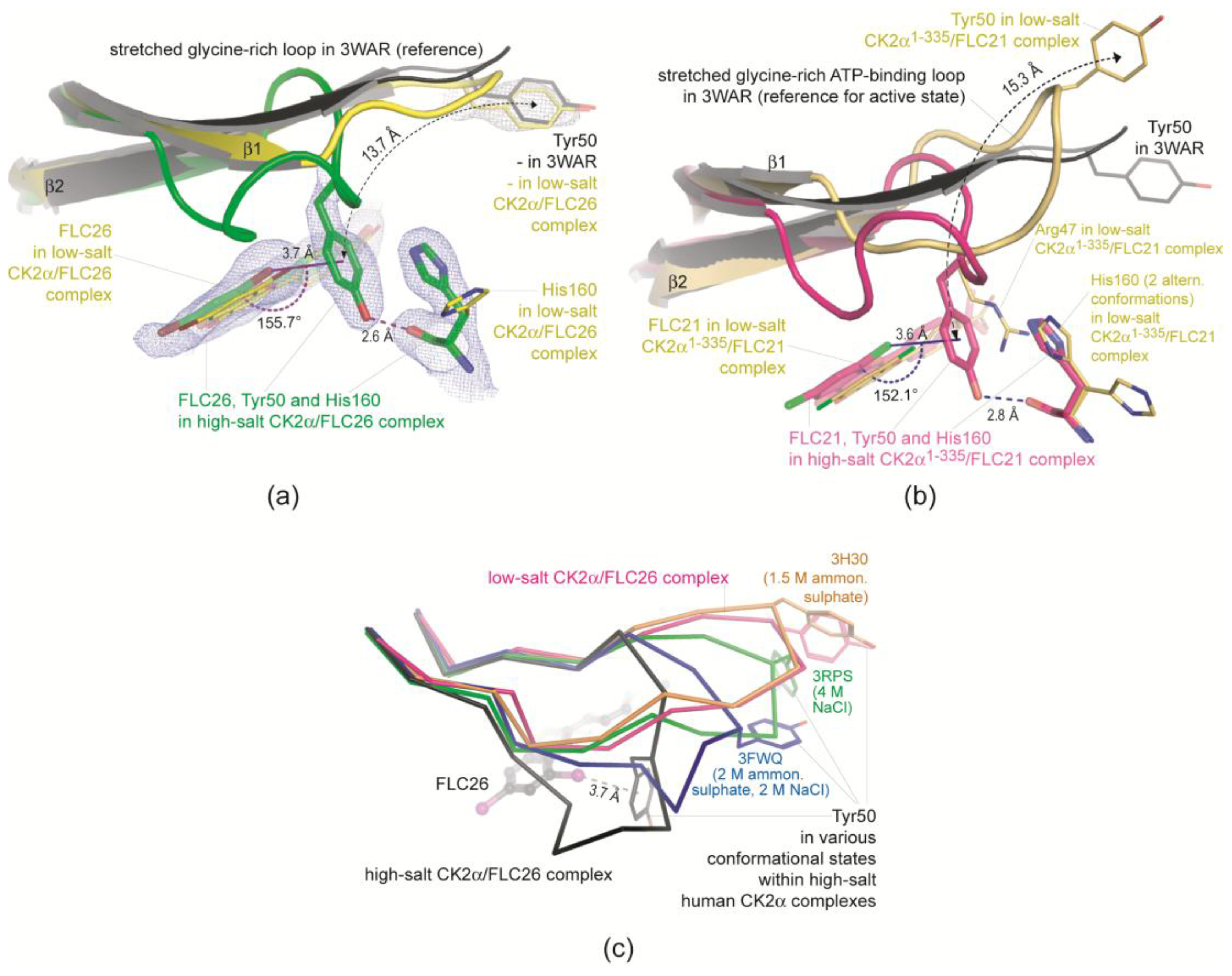
2.2.2. A π-Halogen Bond Enabled by an Extremely Distorted ATP-Binding Loop
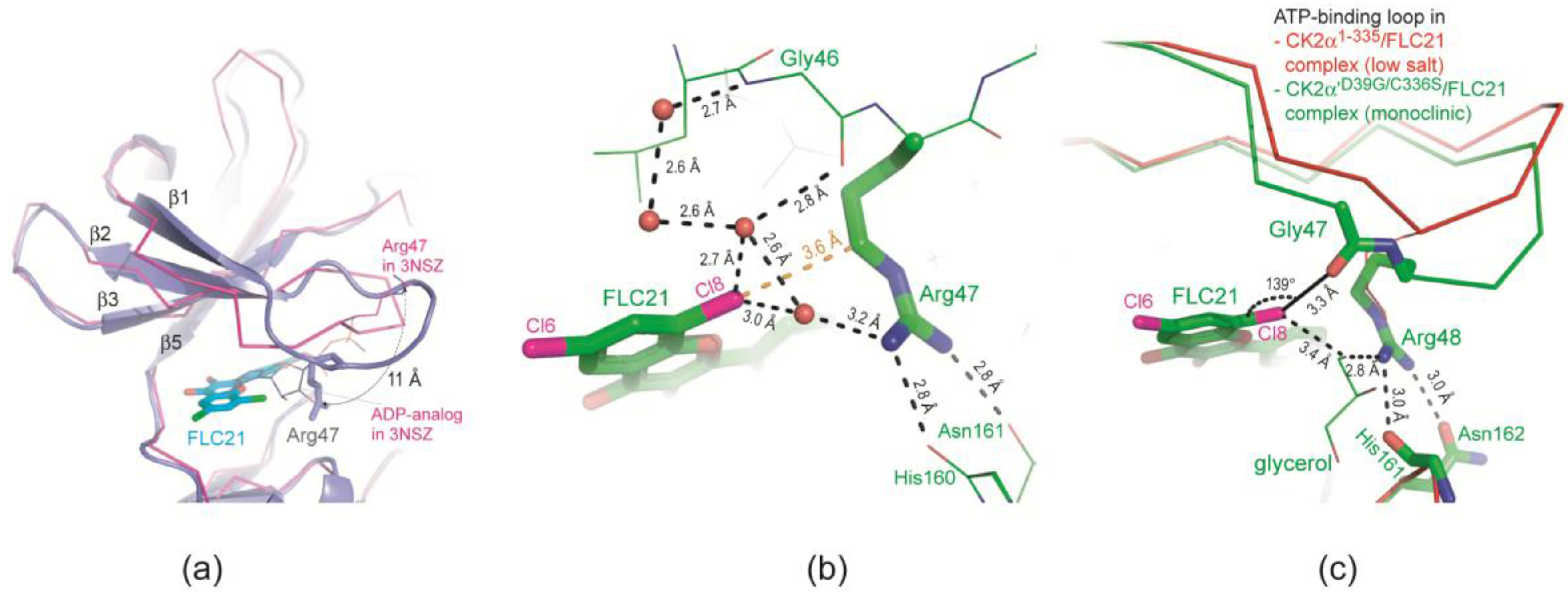
2.2.3. FLC21 Traps the Gly-Rich Loop Arginine of CK2α and CK2α′ in a Non-Functional Conformation
- In the low-salt CK2α1–335/FLC21 structure a network of hydrogen bonds around the Cl8 atom stabilizes the conformation; the propensity to operate as hydrogen bond acceptor is for chloro in fact higher than for bromo substituents (Figure 4b).
- Figure 4c was drawn from one of the two protomers of the monoclinic CK2α′Asp39Gly/Cys336Ser complex structure with FLC21. Here, the Arg48 side chain is hydrogen-bonded to His161 and Asn162 (the CK2α′ equivalents of His160 and Asn161 in CK2α) similar to what is seen in Figure 4b. However, in addition the Cl8 atom forms a geometrically well-established halogen bond with a peptide oxygen of the ATP-binding loop, namely the peptide group connecting Gly47 and Arg48 (Figure 4c). There is no reason to believe that FLC26 binding cannot support such a particular state via a halogen bond as well; co-crystallization efforts of FLC26 with CK2α′Asp39Gly/Cys336Ser might clarify this.
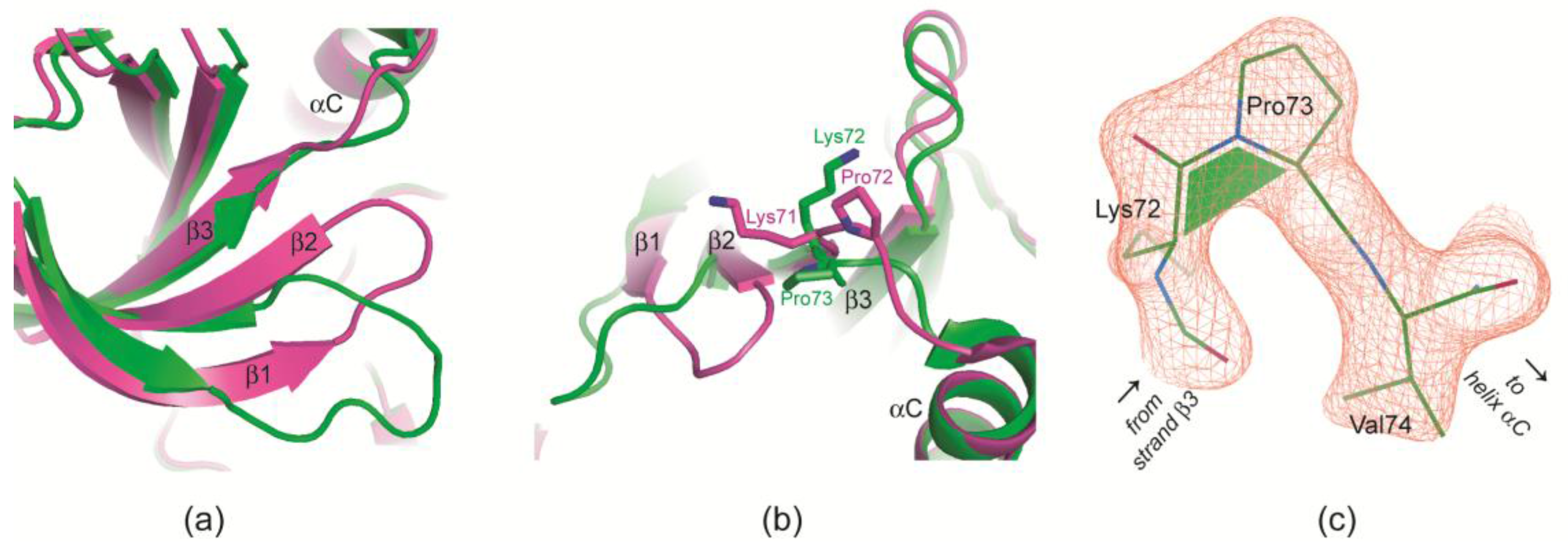
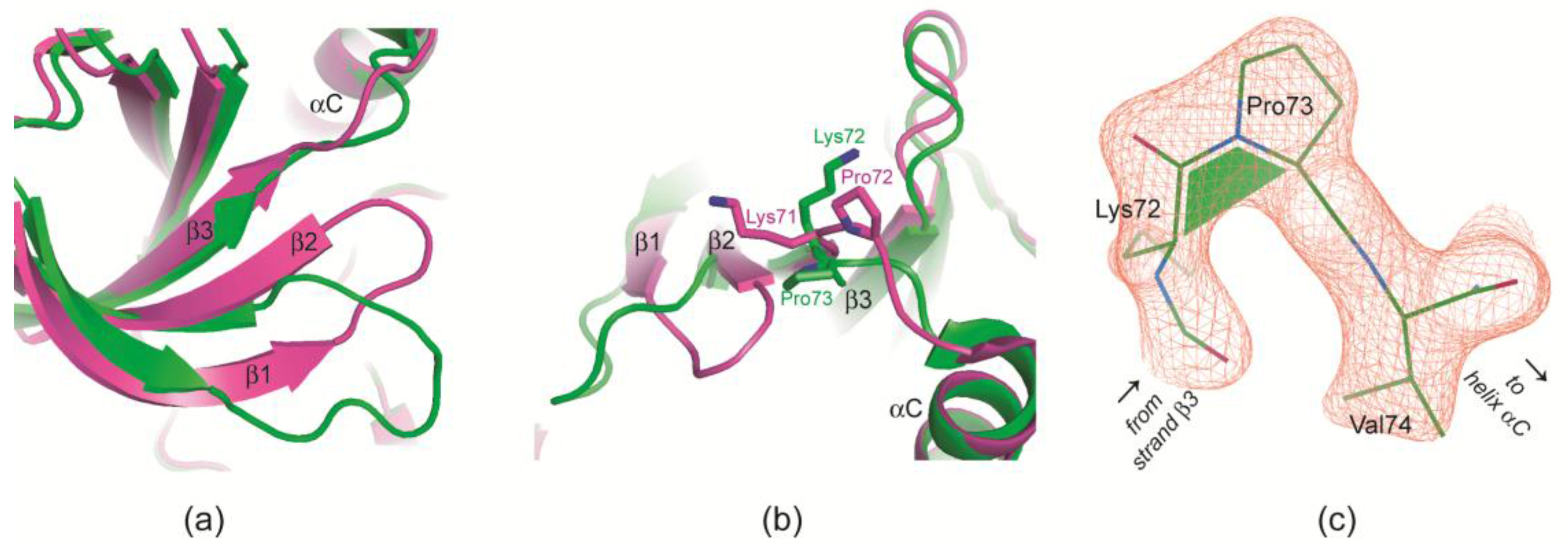
2.2.4. Prolyl cis/trans-Isomerization at the β3/αC Loop
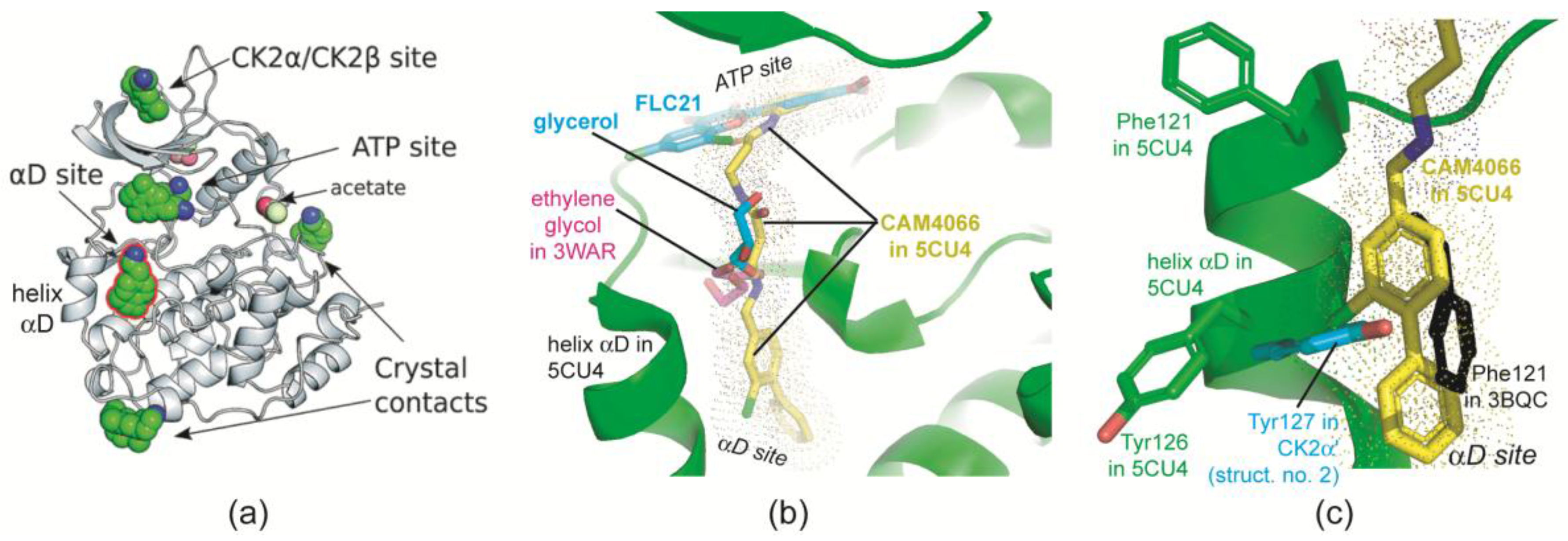
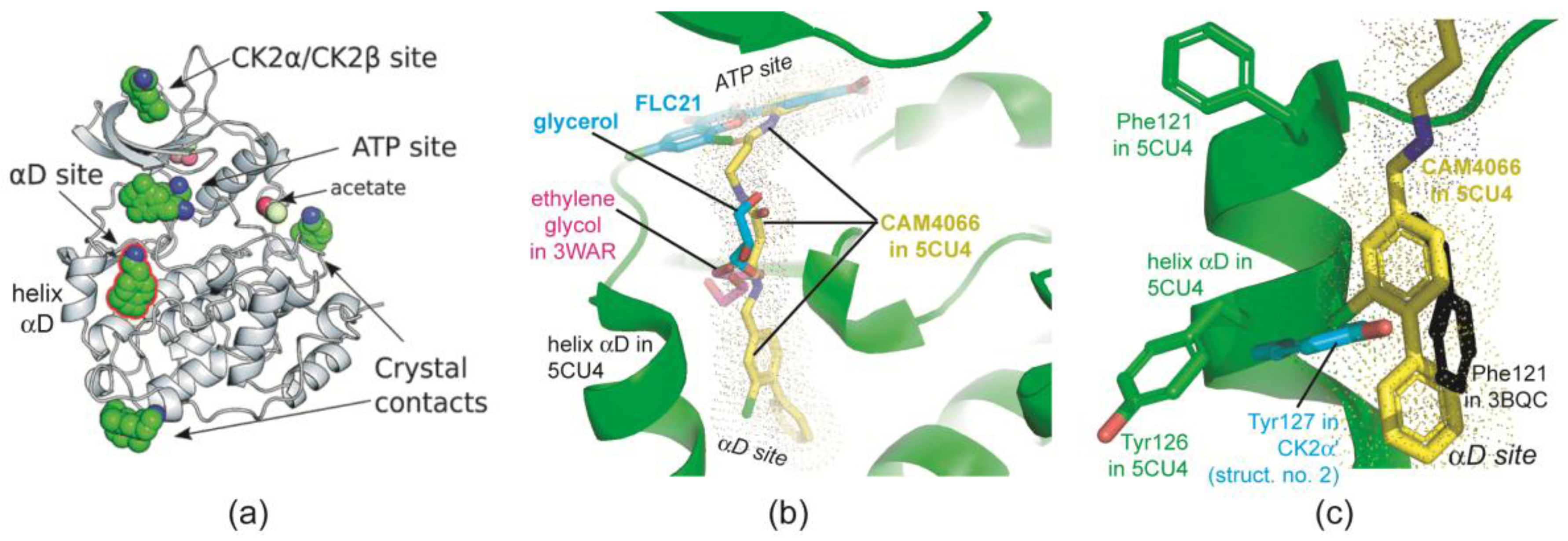
2.2.5. The Hinge/Helix αD Region Harbours a Novel αD Site
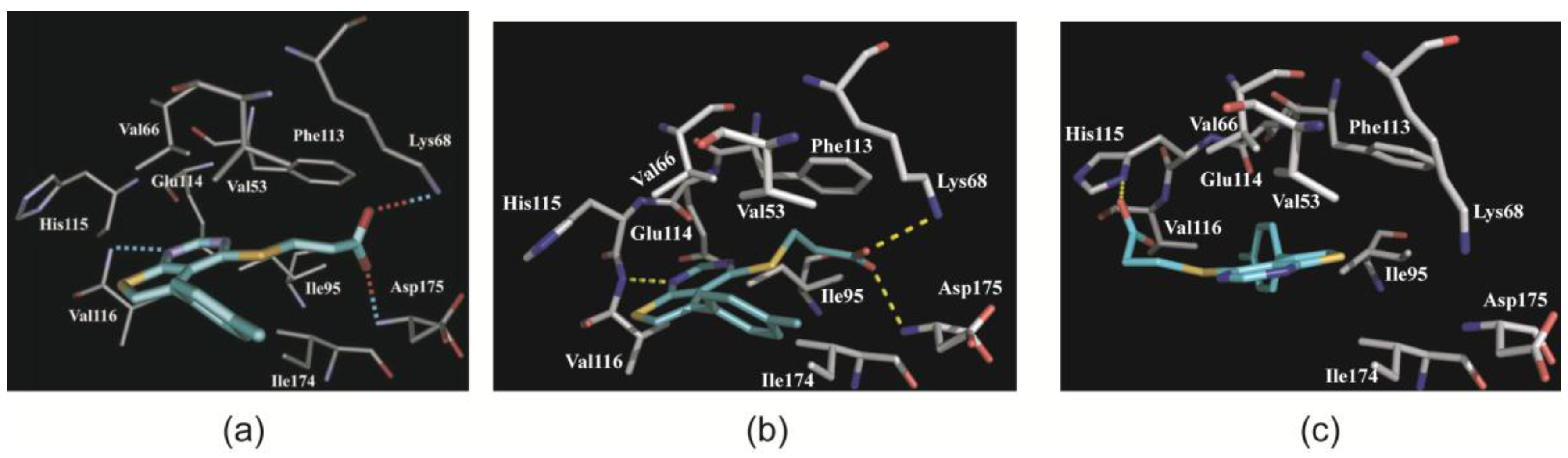
2.3. Complex Structures with TTP22
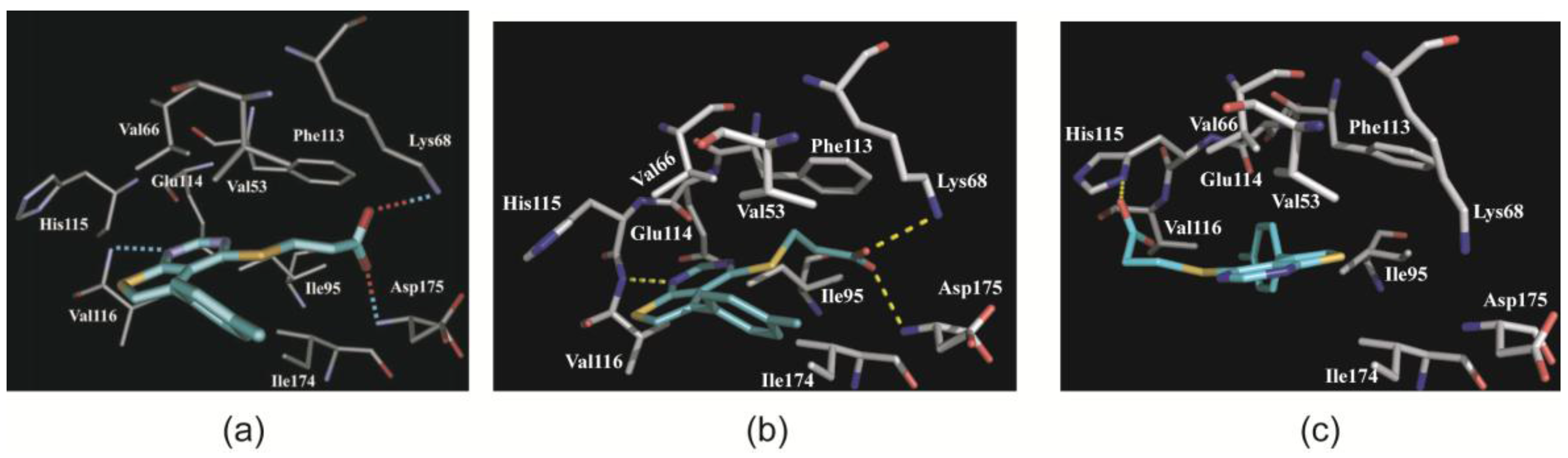
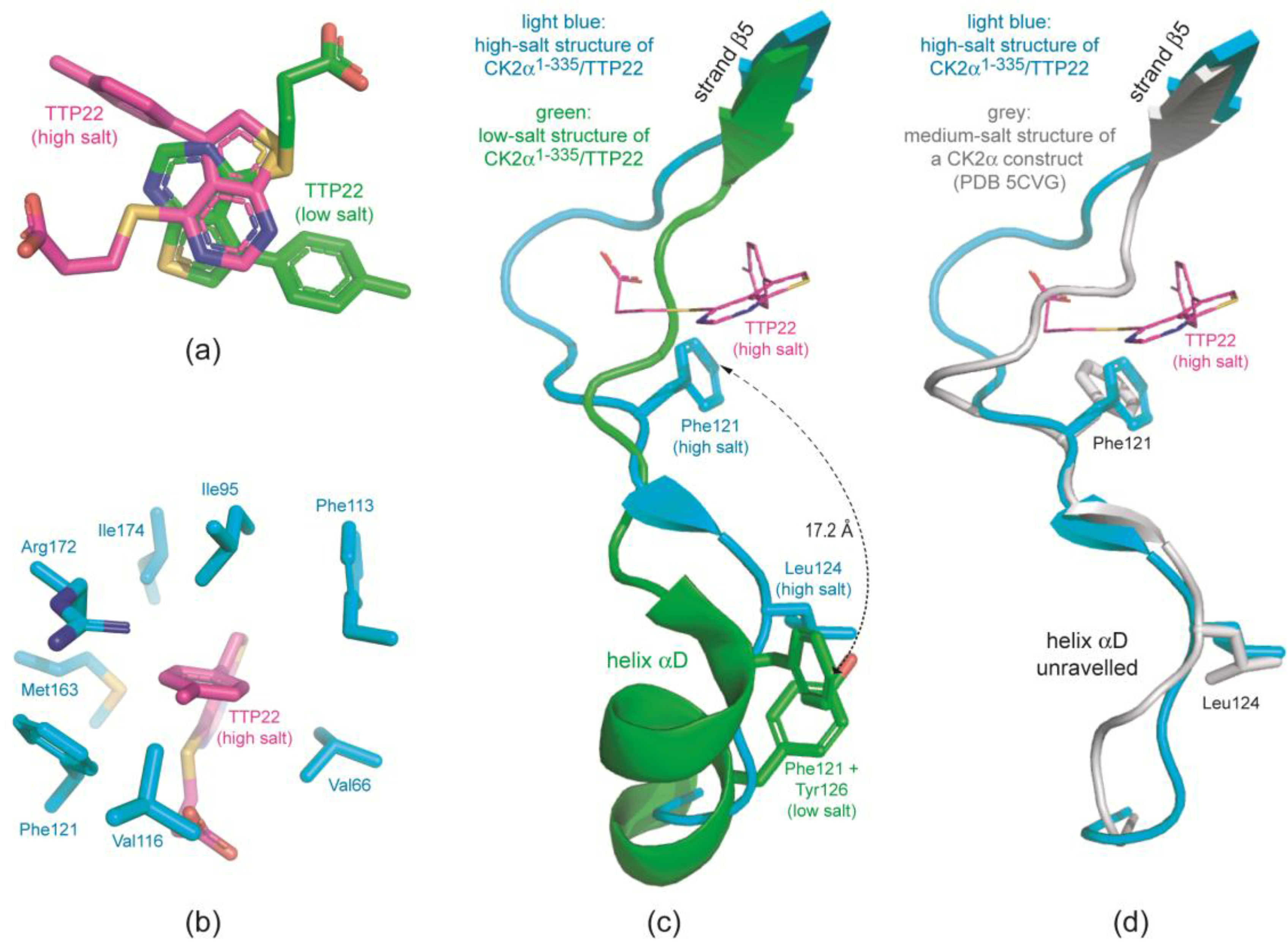
2.3.1. CK2α Binds TTP22 as Predicted under Low-Salt, but Differently under High-Salt Conditions
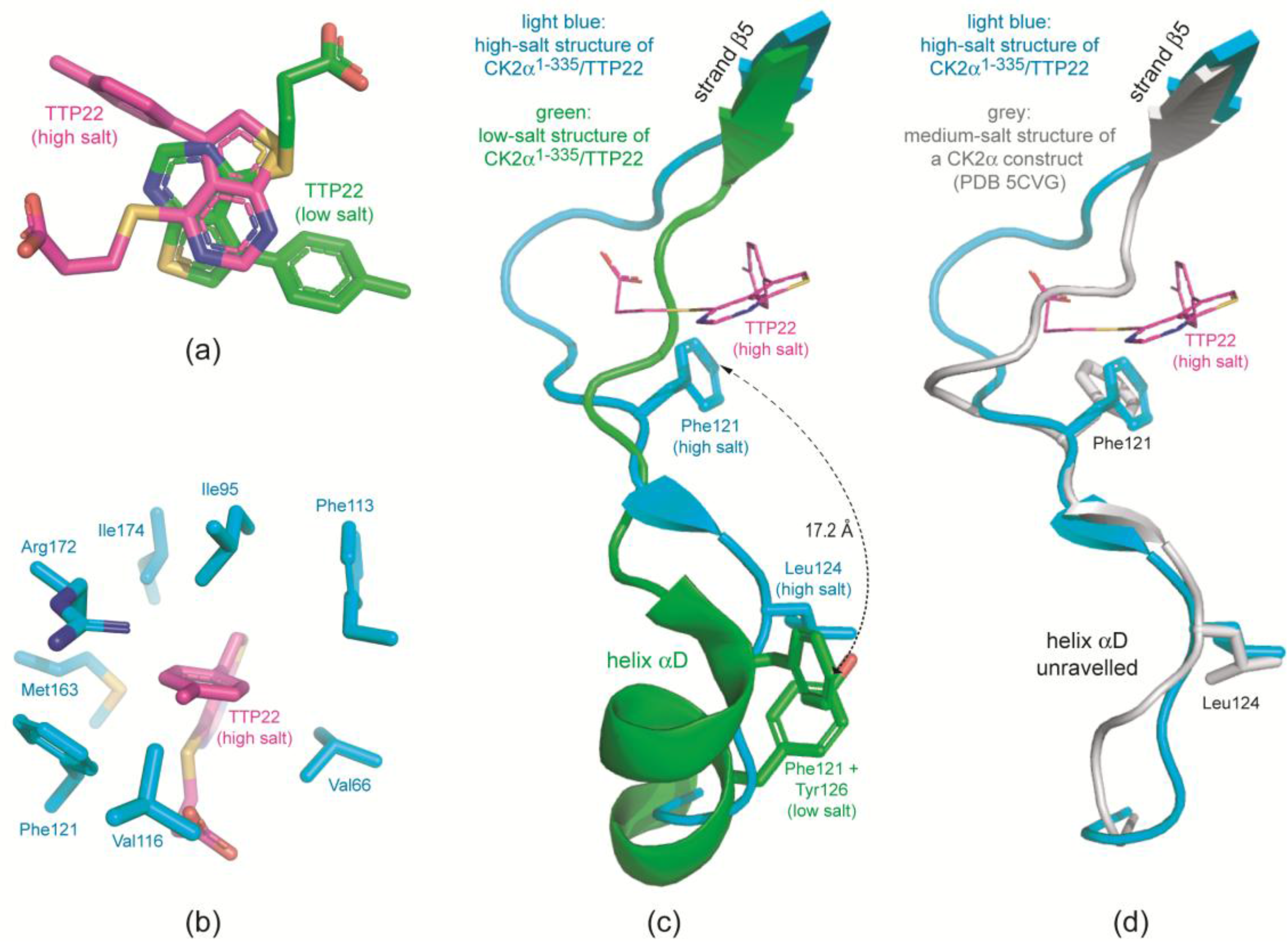
2.3.2. TTP22 Traps CK2α with an Unraveled Helix αD under High-Salt Conditions
3. Materials and Methods
3.1. CK2 Inhibitors
3.2. Proteins
3.3. Crystallization
3.4. X-Ray Diffractometry
3.5. Structure Solution, Refinement, Validation and Deposition
3.6. Illustration
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Nagar, B.; Bornmann, W.G.; Pellicena, P.; Schindler, T.; Veach, D.R.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [Google Scholar] [PubMed]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Bukhtiyarova, M.; Karpusas, M.; Northrop, K.; Namboodiri, H.V.; Springman, E.B. Mutagenesis of p38α MAP kinase establishes key roles of Phe169 in function and structural dynamics and reveals a novel DFG-OUT state. Biochemistry 2007, 46, 5687–5696. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Seeliger, M.A.; Eastwood, M.P.; Frank, F.; Xu, H.; Jensen, M.; Dror, R.O.; Kuriyan, J.; Shaw, D.E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl Acad Sci. USA 2009, 106, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Gerber, D.A.; Cochet, C. Joining the cell survival squad: An emerging role for protein kinase CK2. Trends Cell Biol. 2002, 12, 226–230. [Google Scholar] [CrossRef]
- Ruzzene, M.; Tosoni, K.; Zanin, S.; Cesaro, L.; Pinna, L.A. Protein kinase CK2 accumulation in “oncophilic” cells: Causes and effects. Mol. Cell. Biochem. 2011, 356, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Rabalski, A.J.; Gyenis, L.; Litchfield, D.W. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin. Cancer Res. 2016, 22, 2840–2847. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Raaf, J.; Issinger, O.G. Protein kinase CK2 in health and disease: Protein kinase CK2: From structures to insights. Cell. Mol. Life Sci. 2009, 66, 1800–1816. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Battistutta, R. Structural bases of protein kinase CK2 function and inhibition. In Protein Kinase CK2; Pinna, L.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 1–75. [Google Scholar]
- Niefind, K.; Yde, C.; Ermakova, I.; Issinger, O. Evolved to be active: Sulfate ions define substrate recognition sites of CK2α and emphasise its exceptional role within the CMGC family of eukaryotic protein kinases. J. Mol. Biol. 2007, 370, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Issinger, O.G. Conformational plasticity of the catalytic subunit of protein kinase CK2 and its consequences for regulation and drug design. Biochim. Biophys. Acta 2010, 1804, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Hochscherf, J.; Schnitzler, A.; Issinger, O.-G.; Niefind, K. Impressions from the Conformational and Configurational Space Captured by Protein Kinase CK2. In Protein Kinase CK2 Cellular Function in Normal and Disease States; Ahmed, K., Issinger, O.-G., Szyszka, R., Eds.; Springer International Publishing: New York, NY, USA, 2015; pp. 17–33. [Google Scholar]
- Yde, C.W.; Ermakova, I.; Issinger, O.G.; Niefind, K. Inclining the purine base binding plane in protein kinase CK2 by exchanging the flanking side-chains generates a preference for ATP as a cosubstrate. J. Mol. Biol. 2005, 347, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J.; Klopffleisch, K.; Issinger, O.G.; Niefind, K. The catalytic subunit of human protein kinase CK2 structurally deviates from its maize homologue in complex with the nucleotide competitive inhibitor emodin. J. Mol. Biol. 2008, 377, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gouron, A.; Milet, A.; Jamet, H. Conformational flexibility of human casein kinase catalytic subunit explored by metadynamics. Biophys. J. 2014, 106, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Papinutto, E.; Ranchio, A.; Lolli, G.; Pinna, L.A.; Battistutta, R. Structural and functional analysis of the flexible regions of the catalytic α-subunit of protein kinase CK2. J. Struct. Biol. 2012, 177, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Lolli, G. Structural and functional determinants of protein kinase CK2α: Facts and open questions. Mol. Cell. Biochem. 2011, 356, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Klopffleisch, K.; Issinger, O.G.; Niefind, K. Low-density crystal packing of human protein kinase CK2 catalytic subunit in complex with resorufin or other ligands: A tool to study the unique hinge-region plasticity of the enzyme without packing bias. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Sarno, S.; De Moliner, E.; Papinutto, E.; Zanotti, G.; Pinna, L.A. The replacement of ATP by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J. Biol. Chem. 2000, 275, 29618–29622. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J.; Issinger, O.G.; Niefind, K. First inactive conformation of CK2α, the catalytic subunit of protein kinase CK2. J. Mol. Biol. 2009, 386, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Bischoff, N.; Bdzhola, V.G.; Yarmoluk, S.M.; Issinger, O.G.; Golub, A.G.; Niefind, K. A Note of Caution on the Role of Halogen Bonds for Protein Kinase/Inhibitor Recognition Suggested by High- and Low-Salt CK2α Complex Structures. ACS Chem. Biol. 2015, 10, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J.; Guerra, B.; Neundorf, I.; Bopp, B.; Issinger, O.G.; Jose, J.; Pietsch, M.; Niefind, K. First structure of protein kinase CK2 catalytic subunit with an effective CK2β-competitive ligand. ACS Chem. Biol. 2013, 8, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Gowda, C.; Sachdev, M.; Muthisami, S.; Kapadia, M.; Petrovic-Dovat, L.; Hartman, M.; Ding, Y.; Song, C.; Payne, J.L.; Tan, B.H.; et al. Casein kinase II (CK2) as a therapeutic target for hematological malignancies. Curr. Pharm. Des. 2016, in press. [Google Scholar]
- Martins, L.R.; Lúcio, P.; Melão, A.; Antunes, I.; Cardoso, B.A.; Stansfield, R.; Bertilaccio, M.T.; Ghia, P.; Drygin, D.; Silva, M.G.; et al. Activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia 2014, 28, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Rasmussen, T.D.; Schnitzler, A.; Jensen, H.H.; Boldyreff, B.S.; Miyata, Y.; Marcussen, N.; Niefind, K.; Issinger, O.G. Protein kinase CK2 inhibition is associated with the destabilization of HIF-1α in human cancer cells. Cancer Lett. 2015, 356, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Golub, A.G.; Bdzhola, V.G.; Kyshenia, Y.V.; Sapelkin, V.M.; Prykhod’ko, A.O.; Kukharenko, O.P.; Ostrynska, O.V.; Yarmoluk, S.M. Structure-based discovery of novel flavonol inhibitors of human protein kinase CK2. Mol. Cell. Biochem. 2011, 356, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Golub, A.G.; Bdzhola, V.G.; Briukhovetska, N.V.; Balanda, A.O.; Kukharenko, O.P.; Kotey, I.M.; Ostrynska, O.V.; Yarmoluk, S.M. Synthesis and biological evaluation of substituted (thieno[2,3-d]pyrimidin-4-ylthio)carboxylic acids as inhibitors of human protein kinase CK2. Eur. J. Med. Chem. 2011, 46, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Lolli, G.; Cozza, G.; Mazzorana, M.; Tibaldi, E.; Cesaro, L.; Donella-Deana, A.; Meggio, F.; Venerando, A.; Franchin, C.; Sarno, S.; et al. Inhibition of protein kinase CK2 by flavonoids and tyrphostins. A structural insight. Biochemistry 2012, 51, 6097–6107. [Google Scholar] [PubMed]
- Ermakova, I.; Boldyreff, B.; Issinger, O.G.; Niefind, K. Crystal structure of a C-terminal deletion mutant of human protein kinase CK2 catalytic subunit. J. Mol. Biol. 2003, 330, 925–934. [Google Scholar] [CrossRef]
- Bischoff, N.; Olsen, B.; Raaf, J.; Bretner, M.; Issinger, O.G.; Niefind, K. Structure of the human protein kinase CK2 catalytic subunit CK2α′ and interaction thermodynamics with the regulatory subunit CK2β. J. Mol. Biol. 2011, 407, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tuazon, P.T.; Traugh, J.A. Casein kinase I and II—Multipotential serine protein kinases: Structure, function, and regulation. In Advances in Second Messenger and Phosphoprotein Research; Greengard, P., Robinson, G.A., Eds.; Raven Press, Ltd.: New York, NY, USA, 1991; Volume 23, pp. 123–164. [Google Scholar]
- Gerlach, M.; Mueller, U.; Weiss, M.S. The MX beamlines BL14.1-3 at BESSY II. J. Large Scale Res. Facil. 2016, 2, A47. [Google Scholar] [CrossRef]
- Grant, S.K.; Lunney, E.A. Kinase inhibition that hinges on halogen bonds. Chem. Biol. 2011, 18, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Nakaniwa, T.; Sekiguchi, Y.; Sogabe, Y.; Sakurai, A.; Nakamura, S.; Nakanishi, I. Crystal structure of human CK2α at 1.06 Å resolution. J. Synchrotron Radiat. 2013, 20, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Sheth, P.R.; Basso, A.D.; Paliwal, S.; Gray, K.; Fischmann, T.O.; Le, H.V. Structural basis of CX-4945 binding to human protein kinase CK2. FEBS Lett. 2011, 585, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Brear, P.; De Fusco, C.; Georgiou, K.H.; Francis-Newton, N.J.; Stubbs, C.J.; Sore, H.F.; Venkitaraman, A.R.; Abell, C.; Spring, D.R.; Hyvönen, M. Specific inhibition of CK2α from an anchor outside the active site. Chem. Sci. 2016, 7, 6839–6845. [Google Scholar] [CrossRef]
- Bischoff, N.; Raaf, J.; Olsen, B.; Bretner, M.; Issinger, O.; Niefind, K. Enzymatic activity with an incomplete catalytic spine: Insights from a comparative structural analysis of human CK2α and its paralogous isoform CK2α′. Mol. Cell. Biochem. 2011, 356, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Pinna, L.A.; Issinger, O.G.; Schomburg, D. Crystal structure of the catalytic subunit of protein kinase CK2 from Zea mays at 2.1 A resolution. EMBO J. 1998, 17, 2451–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- The PyMol Molecular Graphics System, Version 1.7; Schrödinger, LLC: New York, NY, USA, 2013.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure No. | 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|---|
| PDB Code | 5M4U | 5M56 | 5M4F | 5M4I | 5M4C | 5M44 | |
| Crystallization | |||||||
| Crystallized complex | CK2α′Asp39Gly/Cys336Ser + FLC21 | CK2α1–335 + FLC21 | CK2α1–335 + TTP22 | ||||
| Vapour diffusion reservoir composition | 25% PEG5000 MME, 0.2 M ammonium sulphate, 0.1 M MES, pH 6.5 | 25% PEG4000, 15% glycerol, 0.17 M sodium acetate, 0.08 M Tris/HCl, pH 8.5 | 24% PEG3350, 0.2 M KCl | 4.3 M NaCl, 0.1 M sodium citrate, pH 5.2 | 24% PEG8000, 0.2 M KCl | 4.2 M NaCl, 0.1 M sodium citrate, pH 5.0 | |
| Sitting drop composition before equilibration | 1 µL reservoir + 1 µL enzyme/FLC21 mixture (90 µL 5.5 mg/mL enzyme, 0.5 M NaCl, 25 mM Tris/HCl, pH 8.5, mixed and pre-equilibrated with 10 µL 10 mM FLC21 in DMSO) | 1 µL reservoir + 1 µL enzyme/inhibitor mixture (90 µL 6 mg/mL enzyme, 0.5 M NaCl, 25 mM Tris/HCl, pH 8.5, mixed and pre-equilibrated with 10 µL 10 mM inhibitor in DMSO) | |||||
| X-ray Diffraction Data Collection | |||||||
| Wavelength (Å) | 1.0000 | 1.0000 | 0.91841 | 0.91841 | 1.0000 | 1.54179 | |
| Synchrotron (beamline) | SLS (X06DA) | SLS (X06DA) | HZB BESSY II (MX-14.1 [34]) | HZB BESSY II (MX-14.1 [34]) | SLS (X06DA) | Home source (rot. Cu anode) | |
| Space group | P212121 | P21 | P212121 | P43212 | P212121 | P43212 | |
| Unit cell | a, b, c (Å) | 46.85, 83.78, 142.34 | 69.34, 87.62, 72.98 | 48.03, 79.57, 82.14 | 72.59, 72.59, 133.25 | 48.10, 79.42, 82.34 | 72.06, 72.06, 131.58 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90, 109.69, 90 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | |
| Protomers per asymmetric unit | 1 | 2 | 1 | 1 | 1 | 1 | |
| Resolution (Å) (highest res. shell) | 44.50–2.195 (2.274–2.195) 1 | 40.94–2.237 (2.317–2.237) 1 | 41.12–1.519 (1.574–1.519) 1 | 37.89–2.218 (2.297–2.218) 1 | 41.14–1.935 (2.004–1.935) 1 | 27.84–2.710 (2.807–2.710) 1 | |
| Rsym (%) | 19.1 (118.5) 1 | 9.3 (65.7) 1 | 5.9 (78.7) 1 | 11.1 (116.9) 1 | 9.8 (73.1) 1 | 13.1 (80.8) 1 | |
| CC1/2 | 0.993 (0.684) 1 | 0.996 (0.685) 1 | 0.999 (0.661) 1 | 0.999 (0.616) 1 | 0.998 (0.758) 1 | 0.996 (0.693) 1 | |
| Signal-to-noise ratio (I/σI) | 9.99 (1.72) 1 | 9.76 (1.78) 1 | 15.82 (1.89) 1 | 15.35 (1.84) 1 | 15.25 (2.26) 1 | 15.75 (2.32) 1 | |
| No. of unique refl. | 29246 (2680) 1 | 39,108 (3544) 1 | 49,151 (4808) 1 | 18,350 (1795) 1 | 23,280 (1476) 1 | 9935 (947) 1 | |
| Completeness (%) | 99.0 (93.0) 1 | 98.0 (90.0) 1 | 100.0 (99.0) 1 | 100.0 (100.0) 1 | 96.0 (62.0) 1 | 100.0 (98.0) 1 | |
| Multiplicity | 6.4 (5.6) 1 | 3.3 (2.9) 1 | 4.1 (4.0) 1 | 7.9 (7.9) 1 | 6.3 (5.2) 1 | 6.9 (5.9) 1 | |
| Wilson B-fact (Å2) | 21.44 | 29.76 | 15.41 | 36.83 | 21.84 | 40.32 | |
| Structure Refinement and Validation | |||||||
| No. of reflections for Rwork/Rfree | 1142 | 37,938/1161 | 48,116/1031 | 17,314/1034 | 22,177/1104 | 8979/956 | |
| Rwork/Rfree (%) | 21.41/17.04 | 16.35/20.64 | 16.28/18.26 | 18.80/22.83 | 15.64/19.68 | 21.86/25.94 | |
| Number of non-H-atoms | 3175 | 5987 | 3248 | 2936 | 3069 | 2820 | |
| Protein | 2789 | 5537 | 2821 | 2798 | 2806 | 2782 | |
| Ligand/ion | 71 | 72 | 43 | 28 | 35 | 27 | |
| Water | 315 | 378 | 384 | 110 | 228 | 11 | |
| Aver. B-factor (Å2) | 28.61 | 38.76 | 20.89 | 52.22 | 28.55 | 52.32 | |
| Protein | 27.33 | 38.57 | 19.30 | 52.64 | 28.01 | 52.52 | |
| Ligand/ion | 43.49 | 40.20 | 26.19 | 45.09 | 27.14 | 44.05 | |
| water | 36.62 | 41.20 | 31.95 | 43.45 | 35.37 | 22.12 | |
| RMS deviations | |||||||
| Bond lengths (Å) | 0.003 | 0.002 | 0.014 | 0.002 | 0.009 | 0.002 | |
| Bond angles (°) | 0.570 | 0.50 | 1.28 | 0.46 | 0.96 | 0.45 | |
| Ramachandran plot | |||||||
| favoured (%) | 97.0 | 95.9 | 97.9 | 96.4 | 97.6 | 95.4 | |
| allowed (%) | 2.7 | 3.8 | 1.8 | 3.6 | 2.1 | 4.0 | |
| outliers (%) | 0.3 | 0.3 | 0.3 | 0.0 | 0.3 | 0.6 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niefind, K.; Bischoff, N.; Golub, A.G.; Bdzhola, V.G.; Balanda, A.O.; Prykhod’ko, A.O.; Yarmoluk, S.M. Structural Hypervariability of the Two Human Protein Kinase CK2 Catalytic Subunit Paralogs Revealed by Complex Structures with a Flavonol- and a Thieno[2,3-d]pyrimidine-Based Inhibitor. Pharmaceuticals 2017, 10, 9. https://doi.org/10.3390/ph10010009
Niefind K, Bischoff N, Golub AG, Bdzhola VG, Balanda AO, Prykhod’ko AO, Yarmoluk SM. Structural Hypervariability of the Two Human Protein Kinase CK2 Catalytic Subunit Paralogs Revealed by Complex Structures with a Flavonol- and a Thieno[2,3-d]pyrimidine-Based Inhibitor. Pharmaceuticals. 2017; 10(1):9. https://doi.org/10.3390/ph10010009
Chicago/Turabian StyleNiefind, Karsten, Nils Bischoff, Andriy G. Golub, Volodymyr G. Bdzhola, Anatoliy O. Balanda, Andriy O. Prykhod’ko, and Sergiy M. Yarmoluk. 2017. "Structural Hypervariability of the Two Human Protein Kinase CK2 Catalytic Subunit Paralogs Revealed by Complex Structures with a Flavonol- and a Thieno[2,3-d]pyrimidine-Based Inhibitor" Pharmaceuticals 10, no. 1: 9. https://doi.org/10.3390/ph10010009