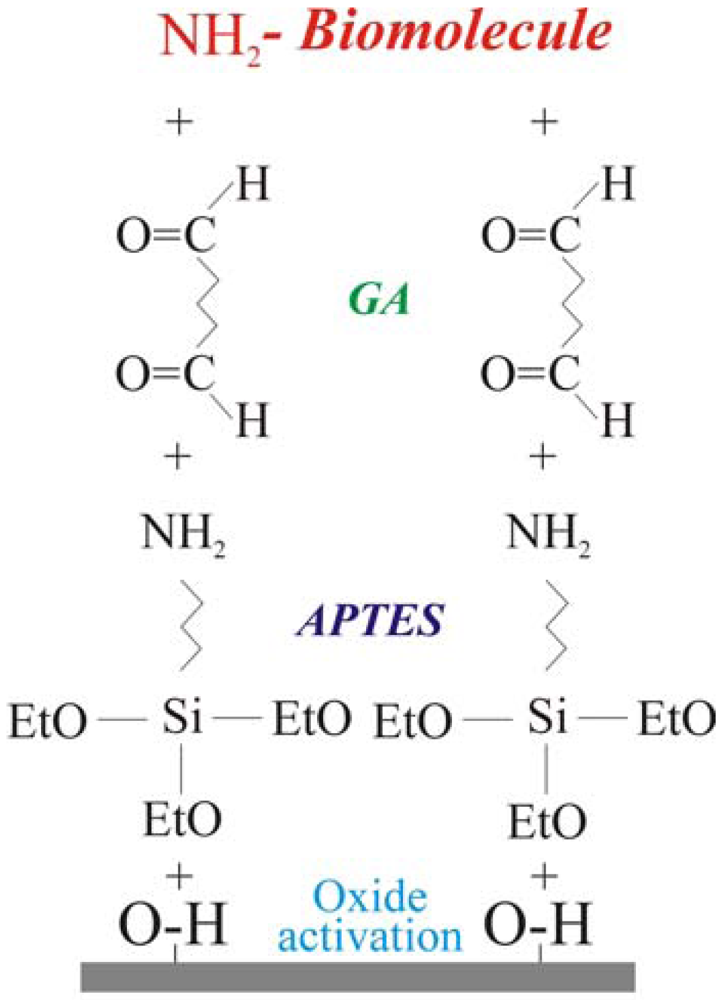
2.1. Immobilization protocol: definition and optimization
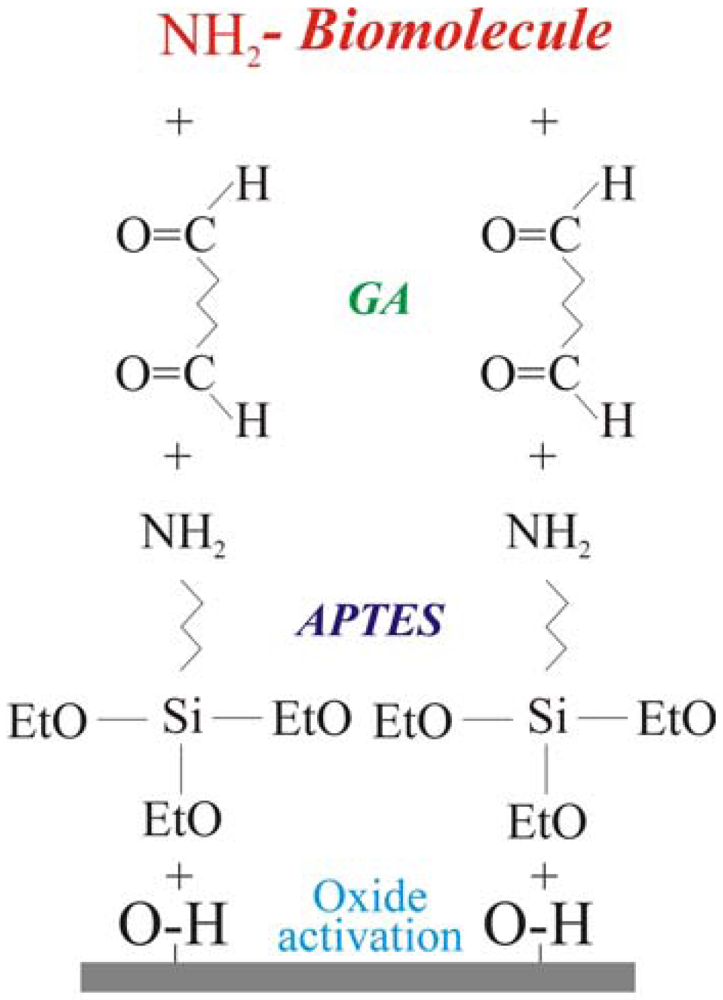
The biological molecule immobilization procedure, reported schematically in
Figure 1, requires four steps. The first one is oxide activation. It consists in the sample immersion in an ammonia and hydrogen peroxide water solution (SSC, ratio NH
2:H
2O
2:H
2O 1:1:10) at 70°C for 20 minutes. The process leaves OH terminated groups on the surface. This step is fundamental when the oxidation process is performed in dry ambient, in fact, the surface must be pre-treated before the second step (silanization) in order to maximize the -OH groups available to bond the silane groups [
23]. The second step consisted of a treatment in vapours of 3-aminopropyltriethoxysilane NH
2-(CH
2)
3-Si(OC
2H
5)
3 (APTES) for 1 h. After silanization, the samples were cured under vacuum at 80 °C for 40 min. This process terminated the surface with -NH
2 groups. The last two steps were linker molecule immobilization [surface terminated with –(C=O)H groups] and amino terminated biological molecule bonding. The linker molecule deposition was carried out using glutaraldehyde (GA), 2.5%, in phosphate buffer solution (PBS). GA is a linear molecule (CHO-(CH
2)
3-CHO) with one aldehydic group (CHO) at each end. Finally, the biological molecule immobilization was carried out for times ranging from 2 h up to 48 h at room temperature using water or PBS solutions depending on the molecule to immobilize (see experimental for details). The power of this method is that, once optimized, it may be used for any biological molecule having amino groups free for bonding. The details of the immobilization procedure for GOx [
30,
31,
40], DNA [
41] and MT [
42] probes are fully reported elsewhere.
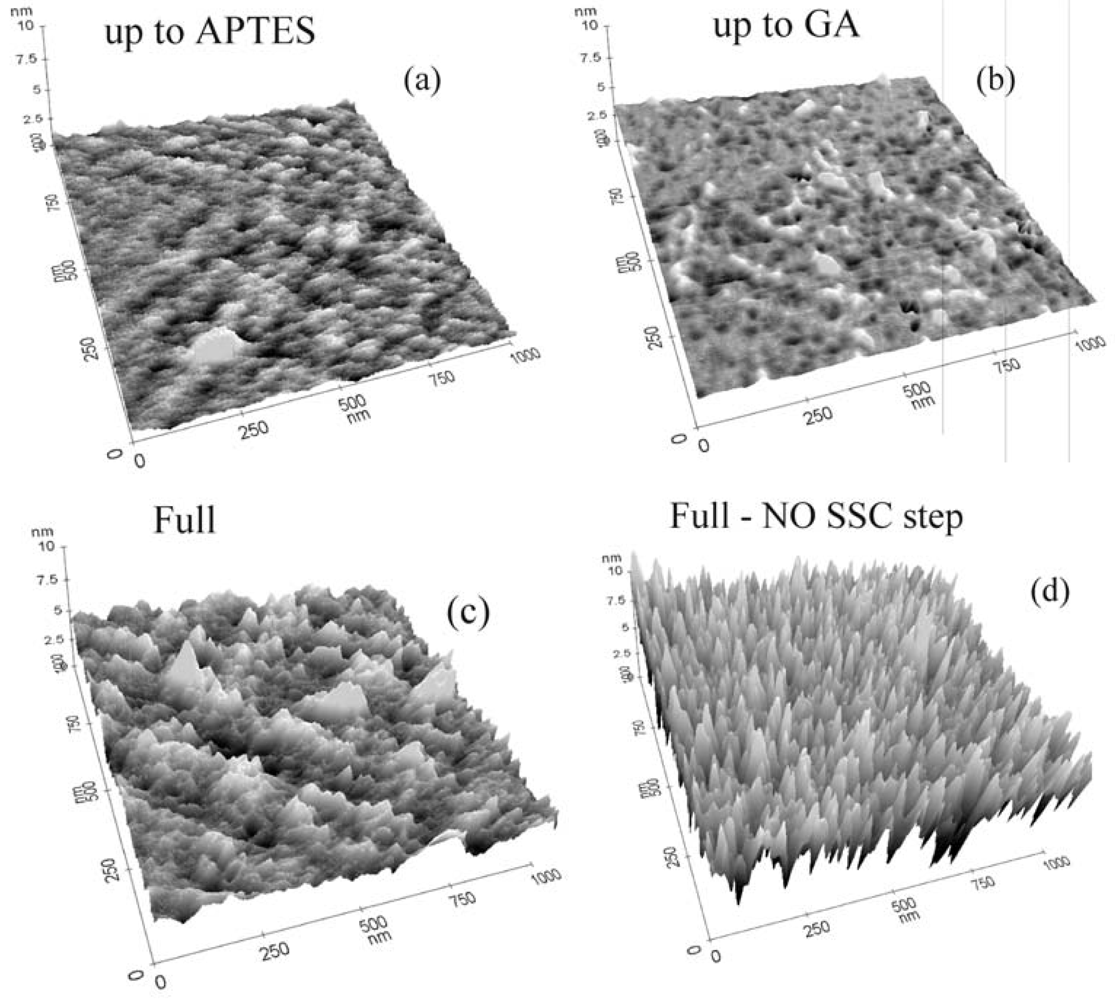
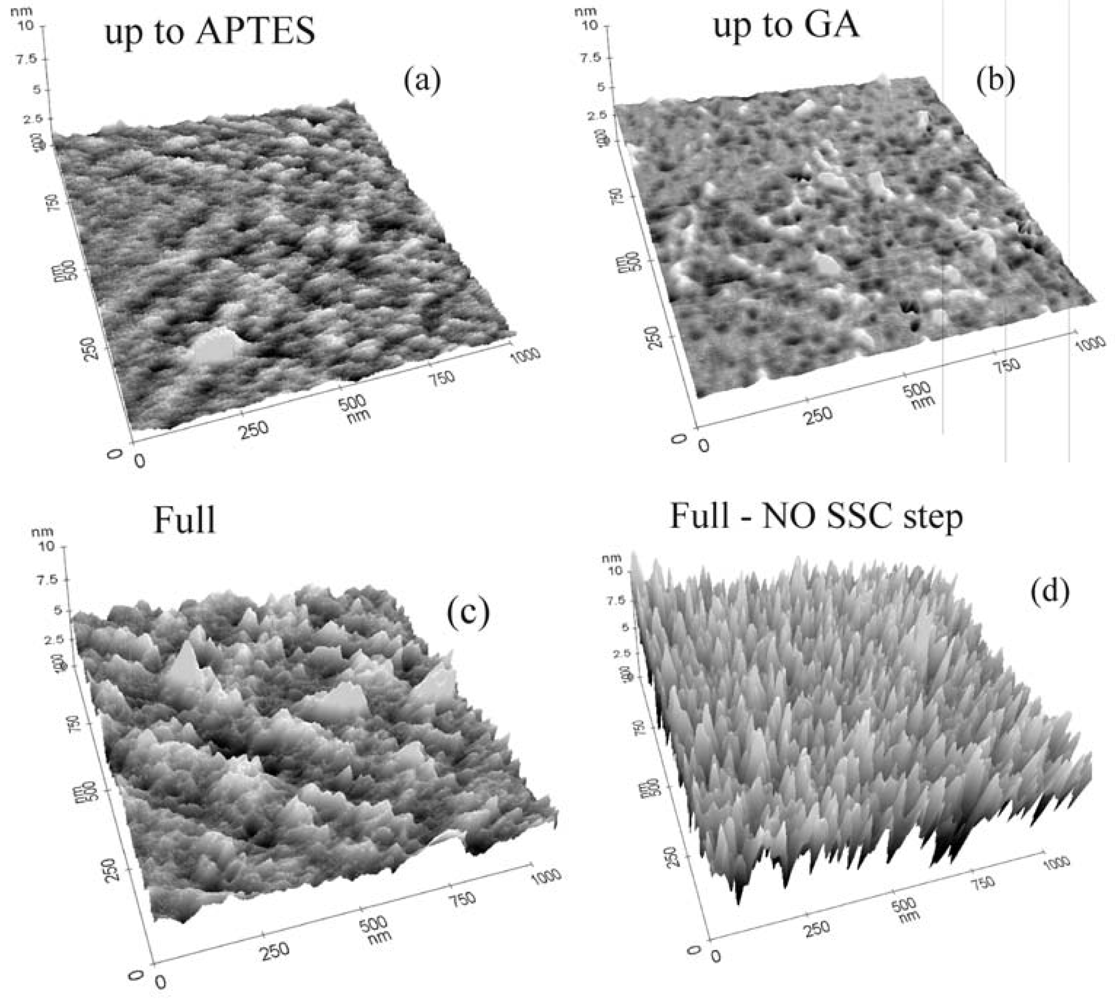
The protocol effectiveness was tested using AFM and XPS measurements. In particular, the AFM analyses for all the intermediate steps of the immobilization process for GOx are reported in
Figure 2. It should be stressed that the immobilization steps are the same for all the biomolecules reported in this work.
Figure 2a shows the result on the sample that underwent oxide activation and silanization. The AFM measurements of this sample indicate a surface rms value of 0.22 nm. We used the same vertical scale in all three-dimensional images to allow a direct comparison of the samples. It should be noted that the APTES layer, if the surface is properly functionalized, has a physical thickness of 0.8±0.1 nm [
43]. The AFM results suggested a good sample coverage. The highest peaks observed in
Figure 2c are due to APTES polymerization. Samples that did not undergo oxide activation showed more polymerized sites (see ref.
40), indicating that a lower number of active sites was available for GA immobilization.
When the GA was deposited on the sample surface, only small modifications of the surface morphology occurred (
Figure 2b). In fact, the surface rms, as measured by AFM, was 0.19 nm, barely distinguishable from the data extracted from the previous sample measurements. The data clearly indicated that the surface did not undergo relevant modifications, detectable by AFM. Measurements carried out on a sample that underwent the full immobilization process with GOx molecules are shown in
Figure 2c. A clear modification of the surface occurred due to enzyme immobilization, with the appearance of peaks having the same height and good distribution on the sample surface. The rms value for this sample was 0.59 nm. Finally, the comparison of
Figures 2c and d allowed us to make some considerations on the immobilization protocol, particularly on the importance of the first two immobilization steps: oxide activation and APTES functionalization. The data showed that oxide activation is fundamental for achieving the best surface coverage. If the sample did not undergo this step, the surface coverage was not uniform. The rms roughness of the sample not subjected to oxide activation (1.46 nm) was much higher than the rms roughness of the fully processed sample (0.59 nm). This indicated that surface coverage was not uniform and the peak heights were not all the same, indicating that there were regions where, probably, the GOx did not bind to the surface. Our results demonstrated that the oxide activation had a double effect: it reduced the surface contaminations and allowed the formation of more surface sites available for silanization (OH terminations).
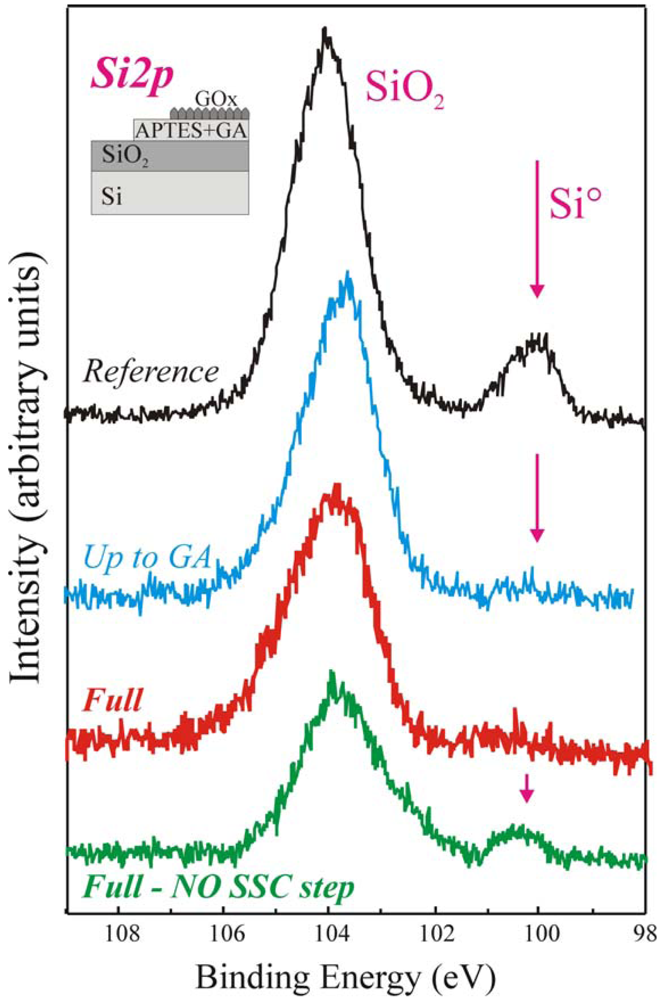
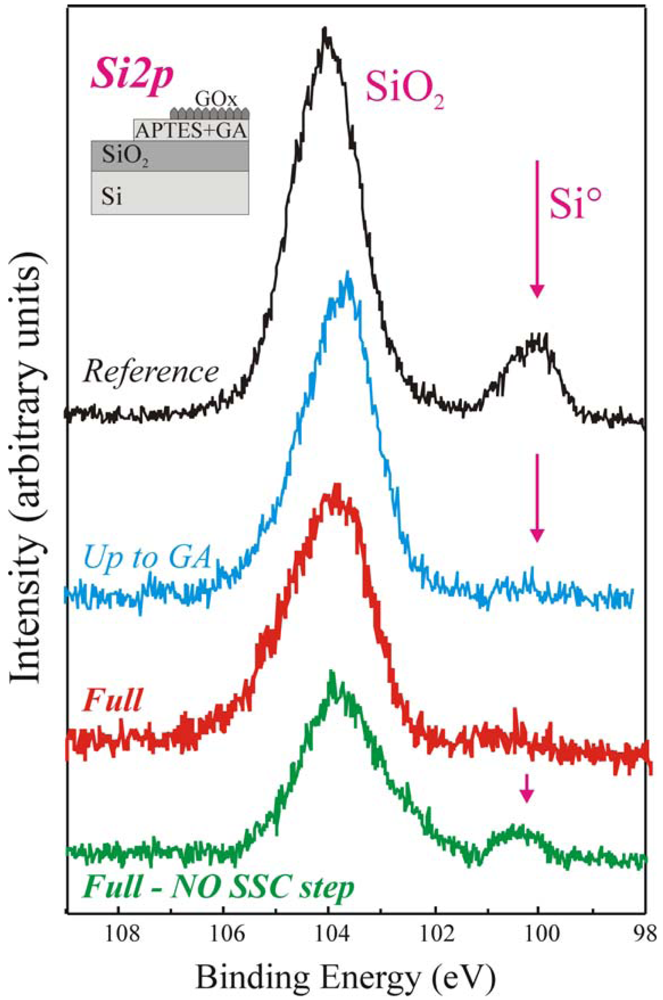
Similar conclusions can be drawn measuring the samples by XPS. In particular, the Si 2
p peaks of the same samples, shown in
Figure 3, provide interesting information. The reference sample (in black) exhibited two components having binding energies of 99.7 eV and 104 eV, assigned to Si° and SiO
2 respectively [
44,
45]. The SiO
2 component is centred at 104 eV instead of 103.4 eV, as expected, due to differential charging between the SiO
2 layer and the Si substrate [
45]. The Si° component is visible since the oxide thickness is small (6.5 nm) and the substrate signal succeeds to pass the oxide and reach the detector. Our results [
40-
42] demonstrate that if the organic film deposited on the sample surface is uniform the substrate Si° signal is fully shield. In fact, the up-to-GA sample, processed using our protocol, does not exhibit such signal (blue line). The same result is, clearly, obtained for the Full+SSC sample (red spectrum), since a further layer is deposited on the surface (see schematic). The situation is completely different for the Full-NO SSC step sample (green spectrum in
Figure 3). Even if a thick layer is deposited on the oxide it still exhibits the Si° component.
This result clearly indicated that the immobilization procedure did not produce a uniform film for the full-NO SSC step sample, despite of the fact that the enzyme was correctly immobilized. The results confirmed that the immobilization procedure without oxide activation was not the best achievable on our SiO
2 surfaces [
40,
41,
46]. The great improvement in film uniformity measured on the samples that underwent Full+SSC protocol can be explained considering that the oxide activation, we introduced as the first step, allowed us to increase the number of available sites for enzyme bonding and to obtain a final uniform deposition. The enzymatic activity measurements confirmed these results (see after) [
46].
It should be mentioned that a correct silanization procedure is fundamental for the uniform coverage of the surface. We demonstrated that if silanization is not properly carried out, APTES polymerization occurs as immediately observed by AFM measurements [
40].
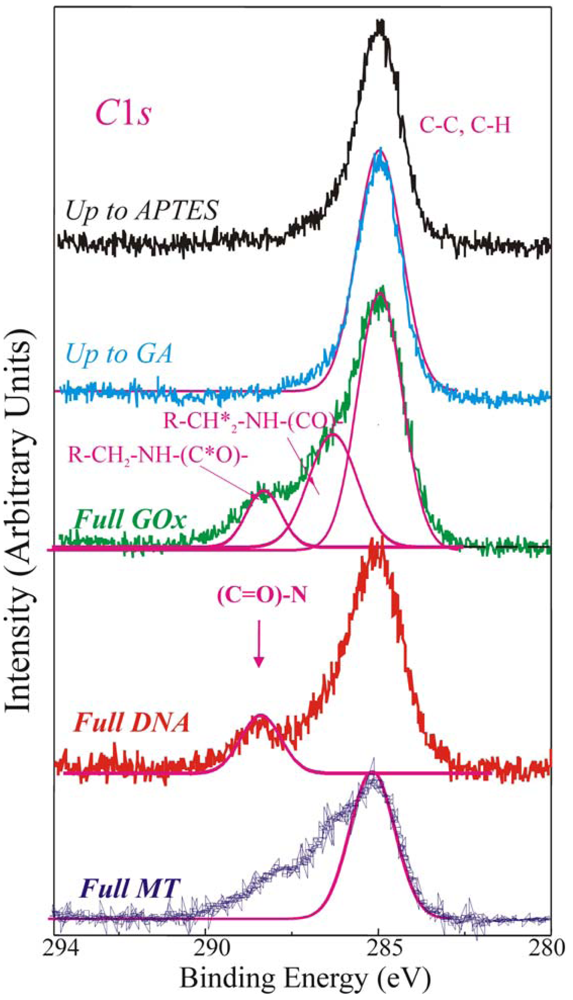
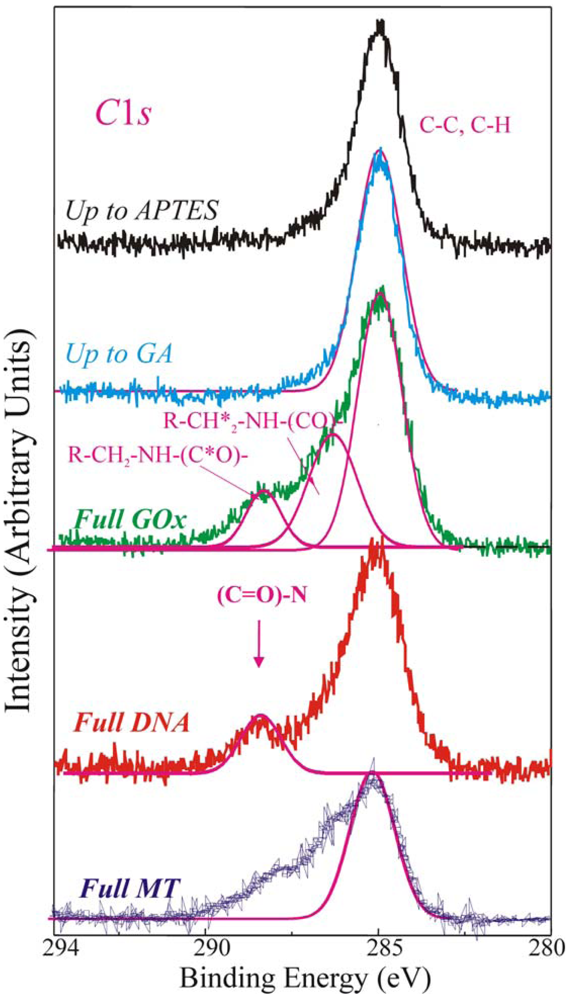
The direct evidence of the biomolecules immobilization on the sample surface was provided by the analysis of the XPS C1
s spectra reported in
Figure 4, of a sample stopped after the APTES step (up to APTES, black line), after the GA (up to GA, light blue line), fully processed with GOx (green line, Full GOx), fully processed with DNA (red line, Full DNA) and fully processed with MT (blue line, Full MT) samples.
The C 1s component, centred at 284.8–285eV and clearly observed in the first two samples (up to APTES and up to GA), is assigned to C-C and C-H bonds. The magenta line superimposed to the experimental spectrum was a simulation of the C-C and C-H XPS peaks. The up-to-GA sample showed only this peak, and an additional weak shoulder at about 287 eV attributed to the R-CHO groups of GA. It should be reminded (see introduction) that the GA is a linear molecule (CHO-(CH2)3-CHO) with one aldehydic group (CHO) at each end. The other samples showed other components at higher binding energies.
The Full GOx samples exhibited at least other two components at 286.3–286.5eV, assigned to R-CH
2*-NH-(CO)-, and at 288.3eV, assigned to R-CH
2-NH-(C*O)-chemical groups respectively. The magenta lines in
Figure 4 are the simulated peaks superimposed to the experimental data to allow one an easier identification of the different peaks. They are characteristic of the proteins and expected if the enzyme is deposited on the sample [
31,
40,
42,
44-
46].
When DNA strands are immobilized on the sample (Full DNA), besides of the C1
s peak at 285 eV, the XPS spectrum exhibited a component at about 288 eV. It has been assigned to –(C=O)-N chemical groups. They are characteristic of the DNA nitrogen bases and their presence is expected only when the DNA is deposited on the sample [
41,
42,
44,
45], as demonstrated by their absence in the other spectra compared in the same Figure. These results confirmed the DNA presence on the sample, in fact the component of C1
s at 288 eV we assigned to the nitrogen bases of DNA is specific of DNA presence in our samples.
Finally, the Full MT sample shows at least other two additional components at 286.3 eV assigned to R–CH
2*–NH–(CO)–, and 288.3 eV, assigned to R–CH
2–NH–(C*O)– chemical groups, characteristic of the proteins, as already observed for the Full GOx sample. In order to understand if the immobilization procedure has affected the bio-molecules' characteristics, the enzymatic activity of the fully processed GOx samples was monitored using a simple spectrophotometric assay [
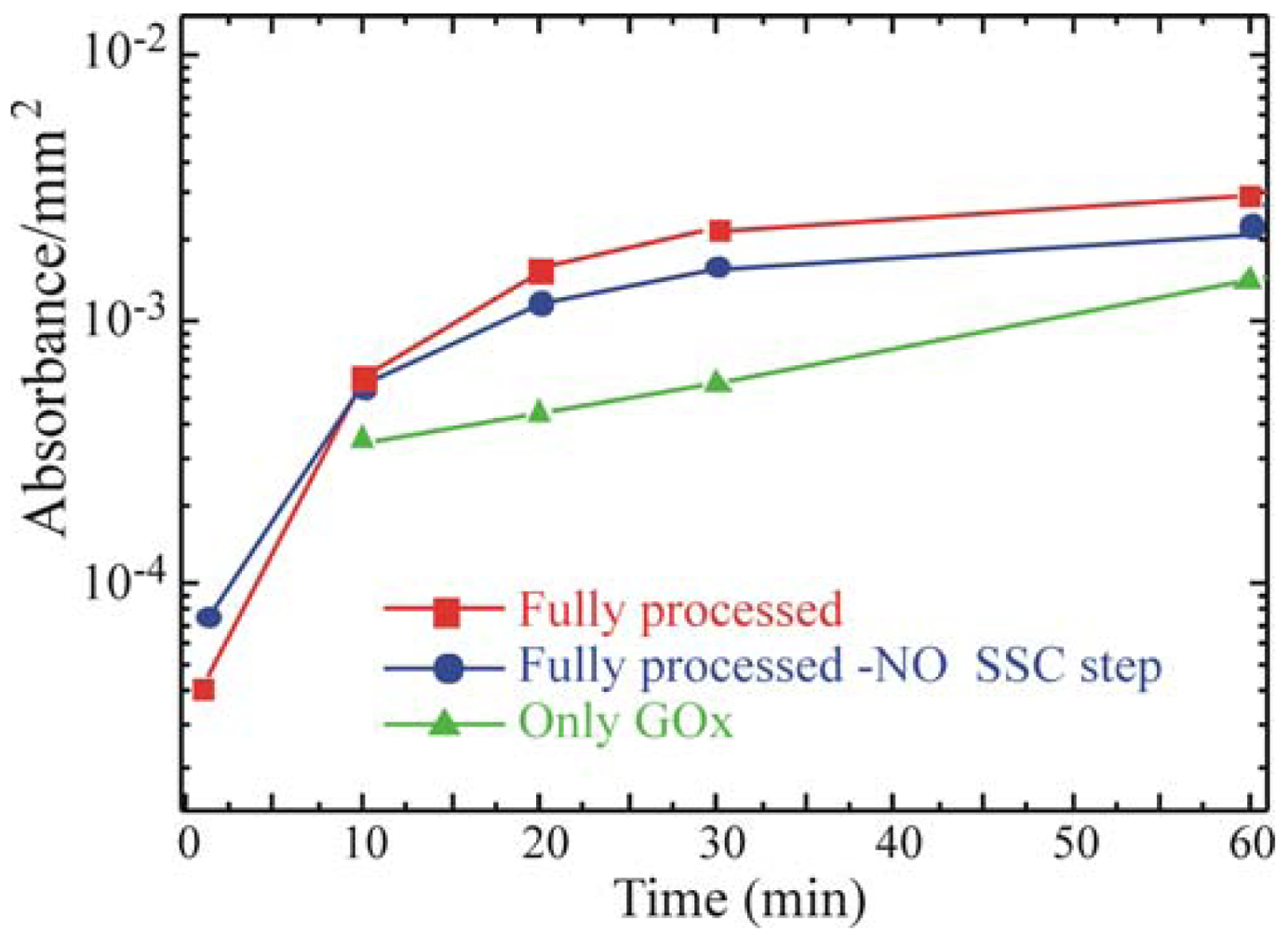
46]. GOx activity was monitored on the Full+SSC sample, on the Full-NO SSC step sample and on the sample that underwent only GOx deposition without previous surface functionalization (only GOx). The results are summarized in
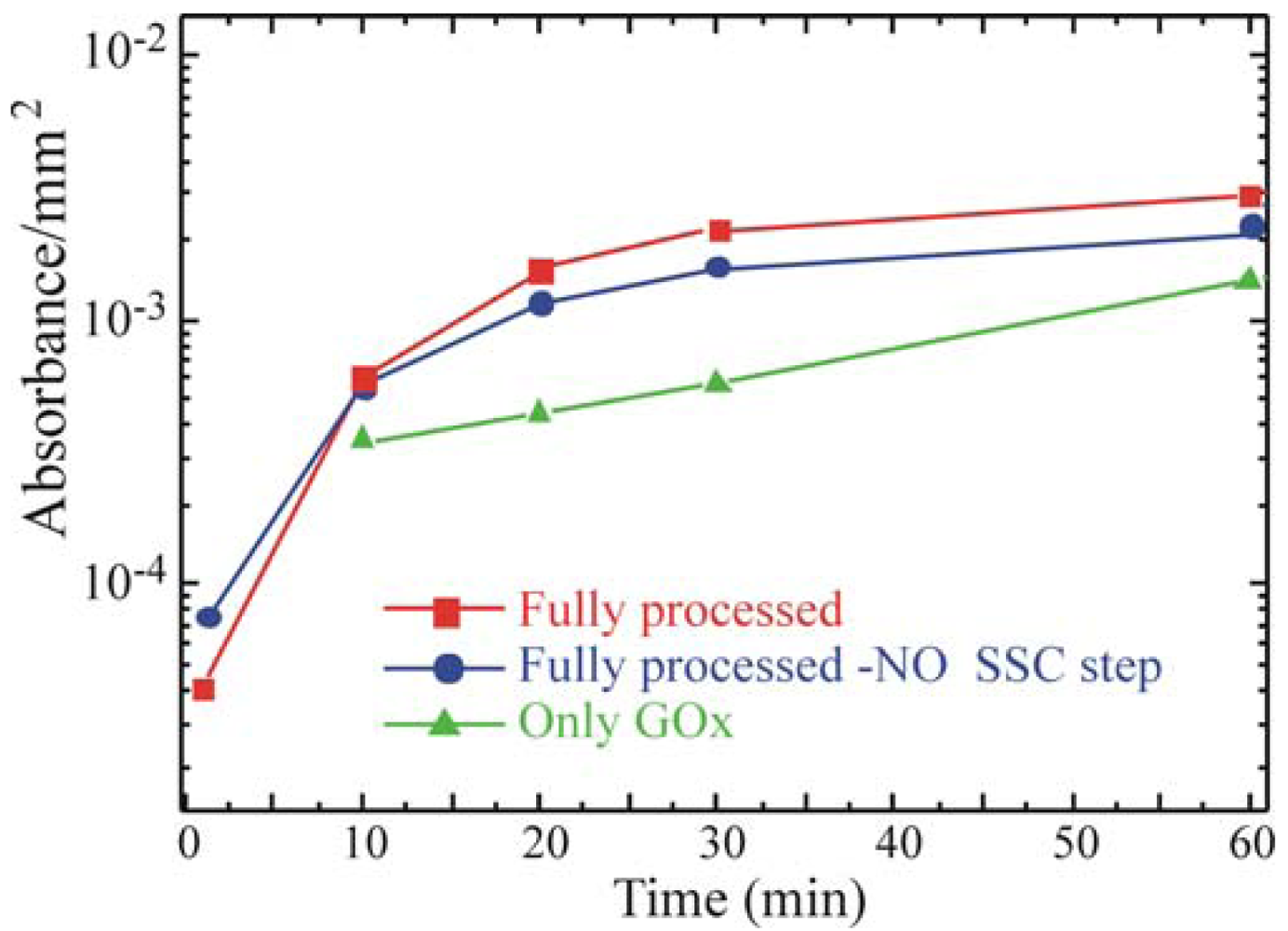
Figure 5. All data were normalized to the real area of the sample.
They show an increase in the enzymatic activity when the oxide activation is carried out before silanization (red squares) with respect to the Full-NO SSC step sample (blue circles). The only-GOx sample (green triangles) exhibited an enzymatic activity lower than the one measured for both the samples that underwent chemical immobilization. The comparison of the absorbance values with those obtained from the free enzymes in solution, allowed us to estimate a concentration of active GOx on the SiO2 samples of about 0.002 U mL-1.
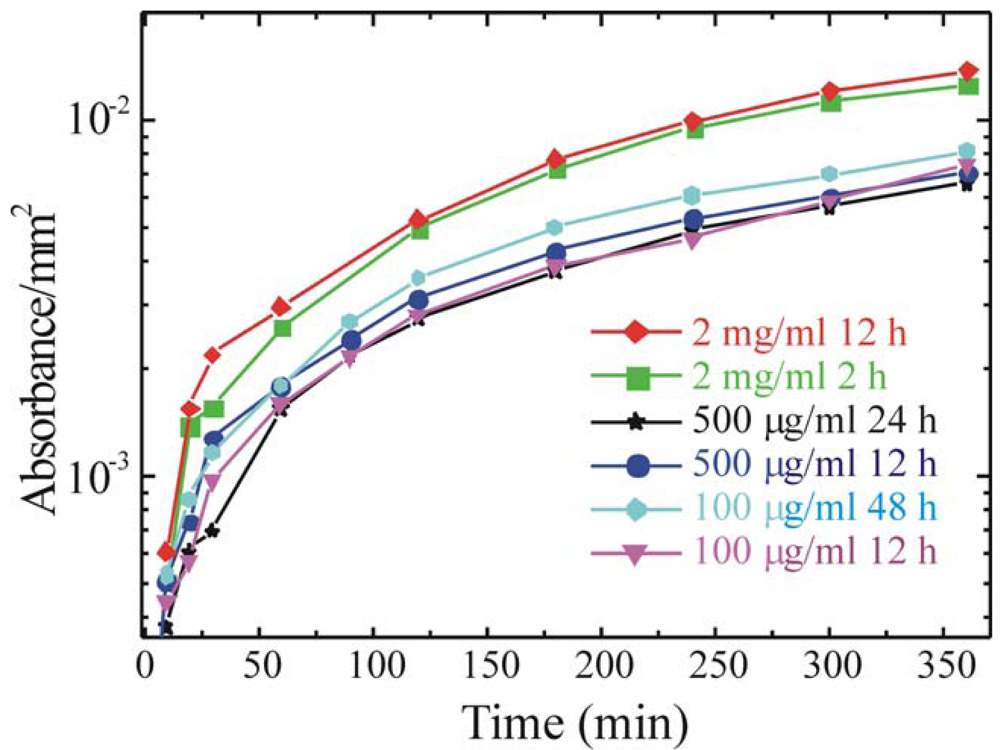
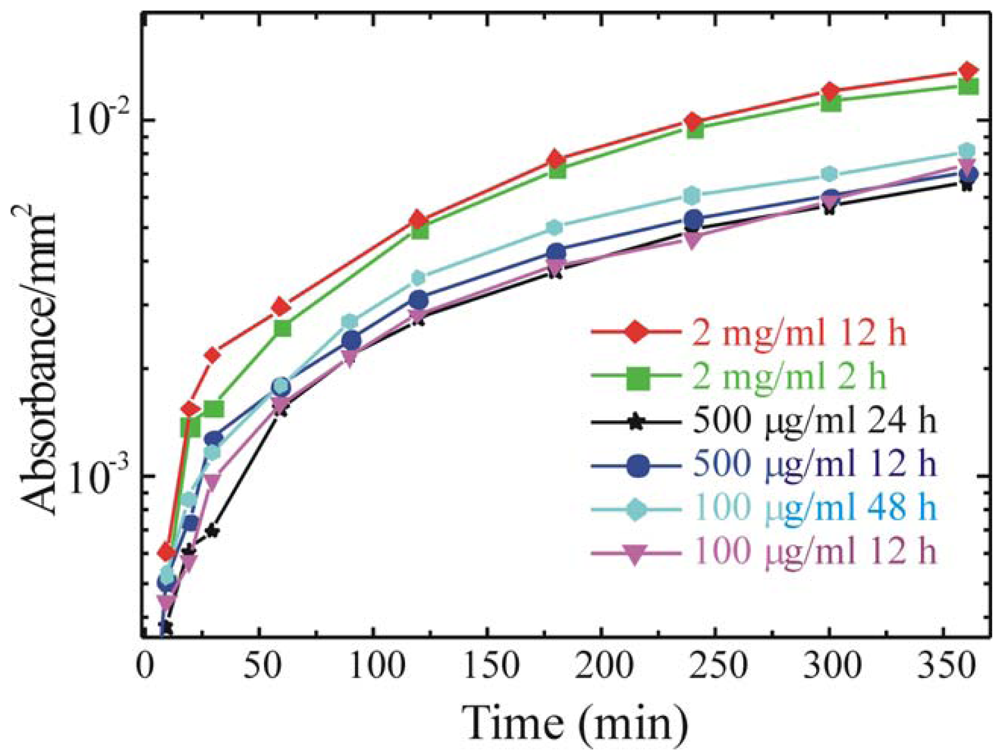
In order to optimize the immobilization protocol also the immobilization step parameters must be studied. In particular, we tested solutions differing for the enzyme concentration in a range from 100 μg/mL up to 2 mg/mL and immersion times ranging from 2 h to 48 h. These parameters have a great importance when a large scale production is performed. In fact, to immobilize the enzyme on large surfaces, larger amounts of solution must be prepared. Moreover, immobilization for times longer than needed will causes a reduction in the production rate. The results, obtained measuring the enzymatic activity are summarized in
Figure 6.
The data clearly show that similar absorbance values are obtained regardless of the immobilization time for solutions of 500 μg/mL and 100 μg/mL, while more than a factor two increase is obtained for 2mg/mL solutions. For this concentration is interesting to observe that there is no difference for immersion times of 2 and 12 h, thus suggesting that a strong reduction in the immobilization time can be obtained.
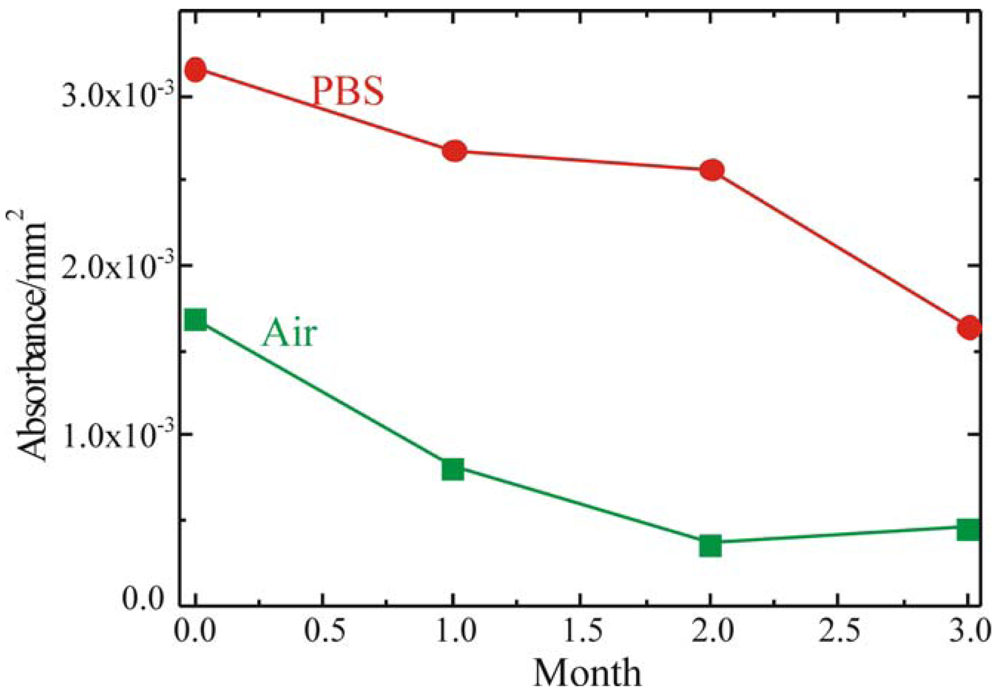
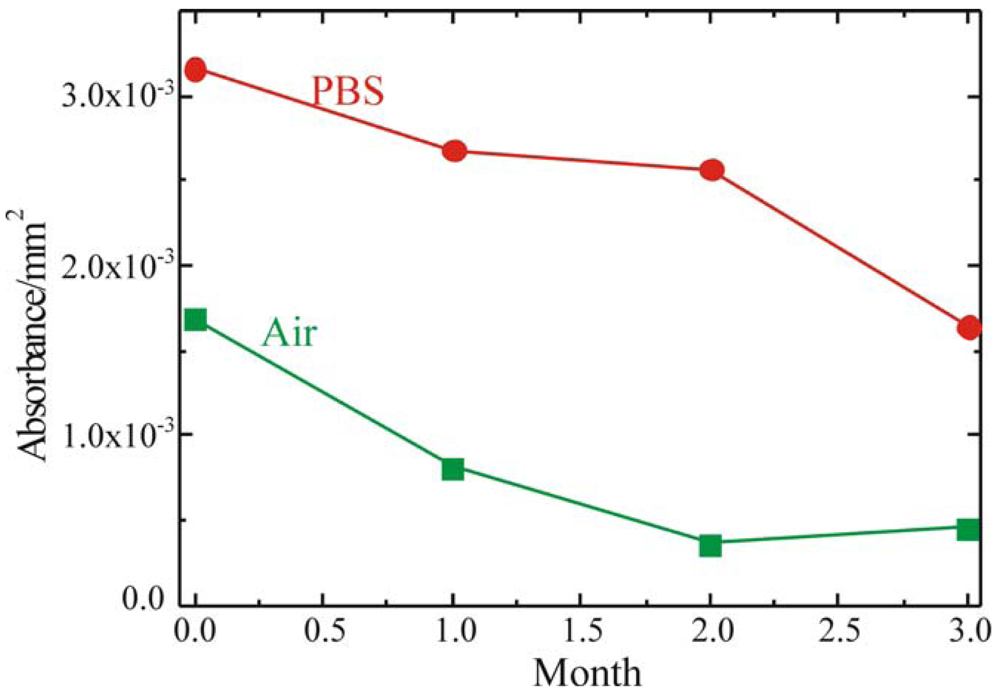
To fabricate a sensing layer using GOx as sensitive element, it is fundamental to understand the device shelf life and to determine the best storage conditions. To this purpose the enzymatic activity of the fully processed sample, stored either in buffer solution (PBS, 0.1M pH 6.5) at 4°C or in air at RT, was monitored. The results of these measurements are summarized in
Figure 7 where the red circles represent the sample stored in PBS, while the green squares represent the sample stored in air. Both samples were monitored immediately after immobilization and after 1, 2 and 3 months. The absorbance values reported in Figure were detected 60 min after the reaction started. The samples stored in PBS retained their activity over longer periods of time. On the other hand, the samples stored in air showed an immediate decrease in the enzymatic activity, already after the first month of aging.
Another important test is the dependence of the layer efficiency on the SiO2 surface characteristics. To this purpose, we thermally grow a thin Si layer (see Section 3 for the fabrication details) on various Si substrates having different doping and impurity concentrations and on quartz. The results, not shown, indicate that the surface immobilization efficiency is not affected by the doping and impurity concentration of the Si substrate.
A modified surface can be used to improve the device performances (in particular its sensitivity). As an example porous silicon dioxide can be fabricated. Measurements we performed on porous SiO
2 fully processed samples demonstrate the enzyme presence within the pores and an enhanced enzymatic activity, due to the higher surface/volume ratio. An increase in sensitivity of one order of magnitude was obtained using a 3 μm thick layer of porous SiO
2. The results are reported in [
46]. A further improvement in the device sensitivity can be obtained by micropatterning of the surface. In particular, we demonstrated that a strong increase in the enzymatic activity was observed by ink-jet printing the GOx on a planar SiO
2 surface [
47]. The result so far reported indicated that the final device performances will strongly depend on the fabrication method and on the final device structure, hence it is not possible to infer the final device sensitivity only from the results here reported.
Finally, to check that both DNA strands and MTs keep their characteristics once immobilized, different measurements were performed. DNA single strands were tested electrically monitoring their ability to hybridize the complementary strands (see later).
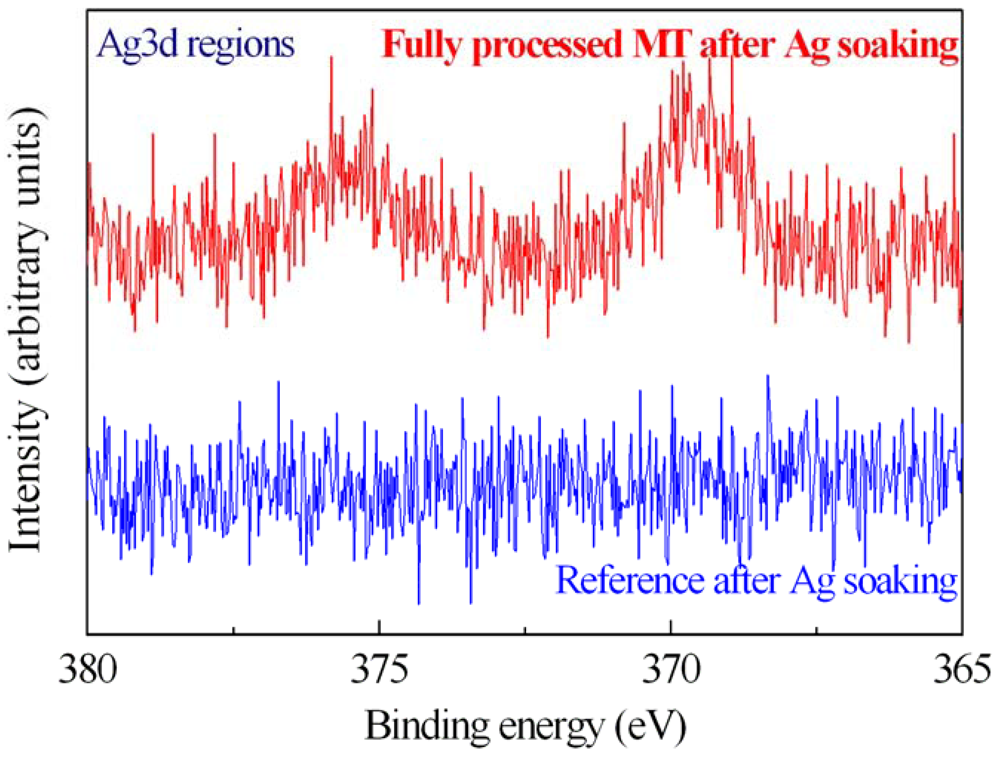
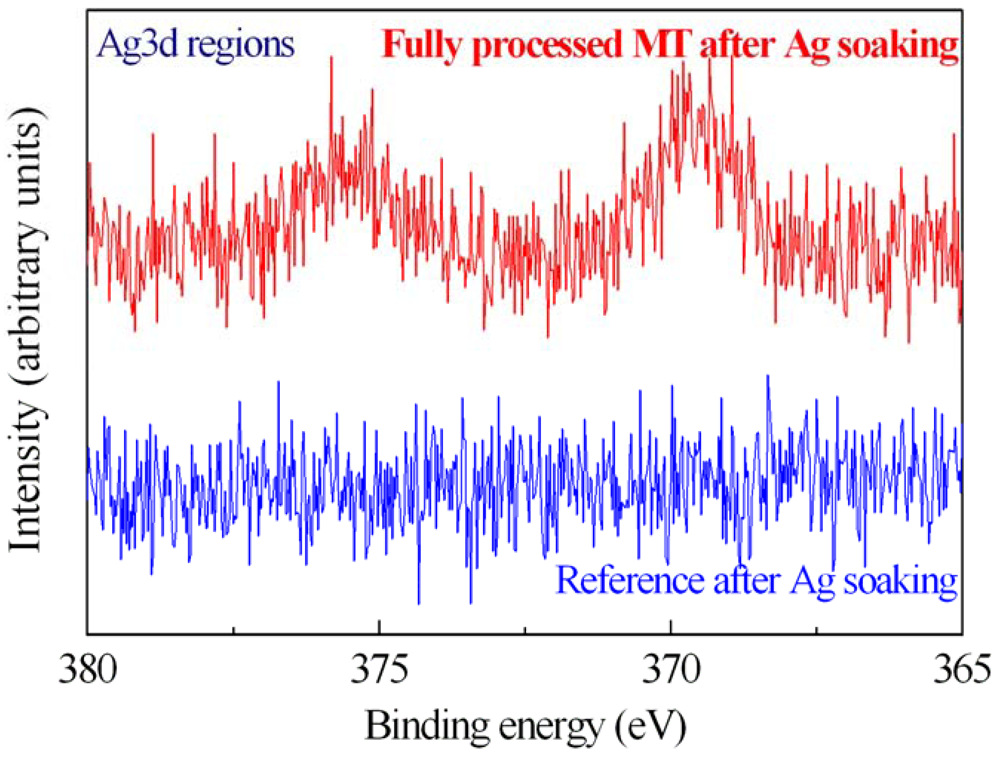
The MT activity on immobilized samples was tested by measuring with XPS the spectra of fully processed samples after Ag salts soaking (
Figure 8). The XPS characterization clearly shows the presence of Ag on the sample surface after immersion. In fact, the Ag 3d spectrum of one of them (500 μg/mL) is shown in
Figure 8 (red line). The reference sample (blue line) doesn't exhibit traces of Ag after immersion in the same solution. This result indicated that the proteins maintained their ability in getter heavy metals also after the immobilization process. A careful description of the method and the results is provided in [
42].
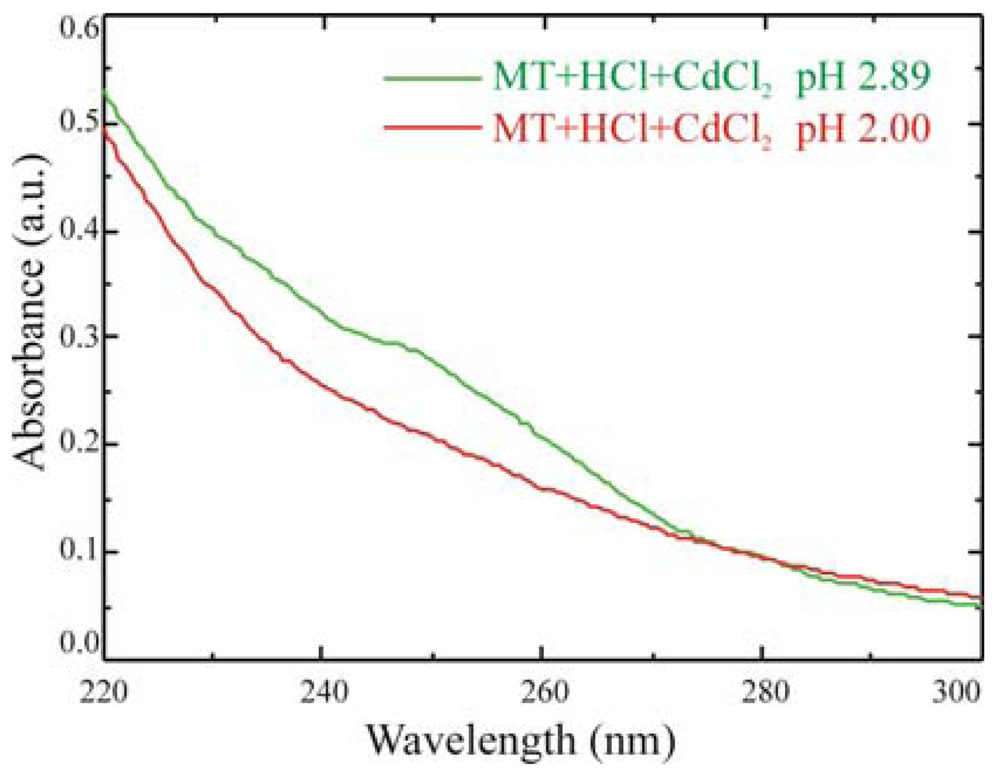
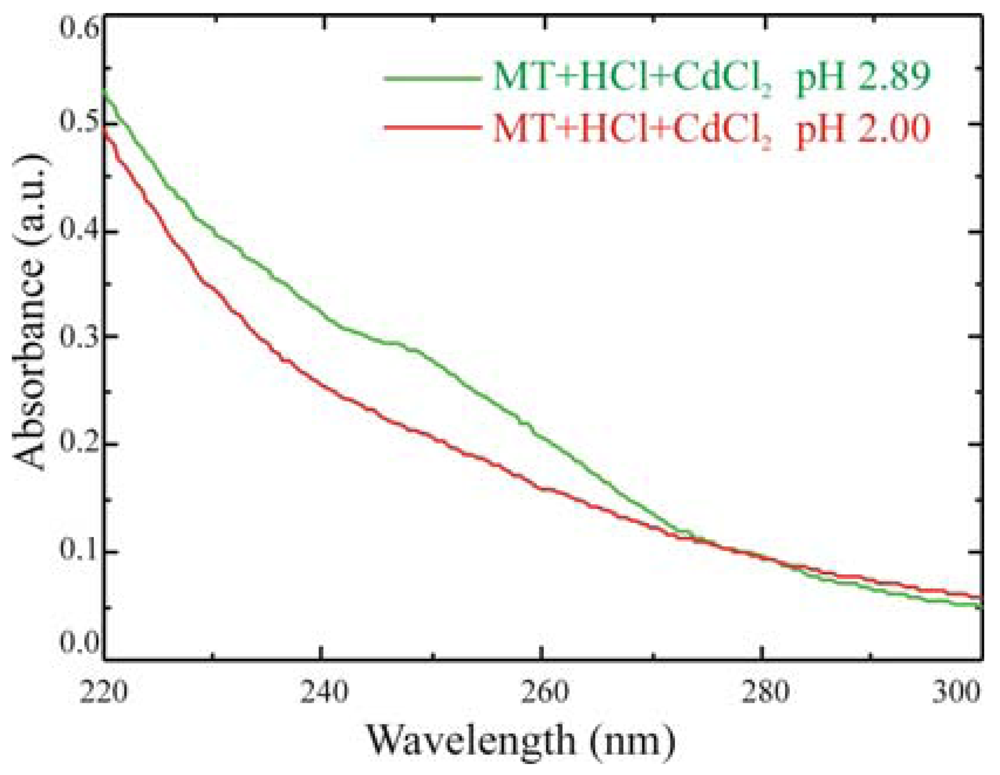
Also spectrophotometric results can provide information on immobilized MT activity, as demonstrated in
Figure 9, where the comparison between two solutions of 100 μg of MT/ml at different pH is reported.
At pH above 2.5, MTs getter Cd and a shoulder in the absorbance spectrum at 250 nm is clearly visible. When the pH is reduced below the 2.5 threshold, by adding hydrochloric acid, MTs release the metallic ions captured and the shoulder disappears. Experiments are in progress, on quartz samples, to test the mechanism on immobilized proteins.
2.2. Electrical Testing
As mentioned in the introduction, the possibility of obtaining an electrical signal as a result of the transduction mechanism can greatly expand the miniaturization potentiality of biosensors. In this perspective electrolyte-insulator-semiconductor (EIS) structures are quite intriguing. Their working principle is straightforward: the recognition event, occurring at the insulator/electrolyte interface, causes a change in the charge (potential) at that interface and it can, in principle, be detected. The capacitance in a EIS structure can be described as the built-up of several capacitors in series. The total capacitance (C
tot) is the series of: the semiconductor capacitor (C
Si); the insulator capacitor (C
ox) and the capacitor provided by the layer given by the sensitive element (C
bio) [
19]. The recognition event will produce a change in the capacitance of C
bio, that will produce also a change in the surface potential. Hence, a shift in the flat band voltage (V
FB), associated to the flat band capacitance (C
FB), may be detected.
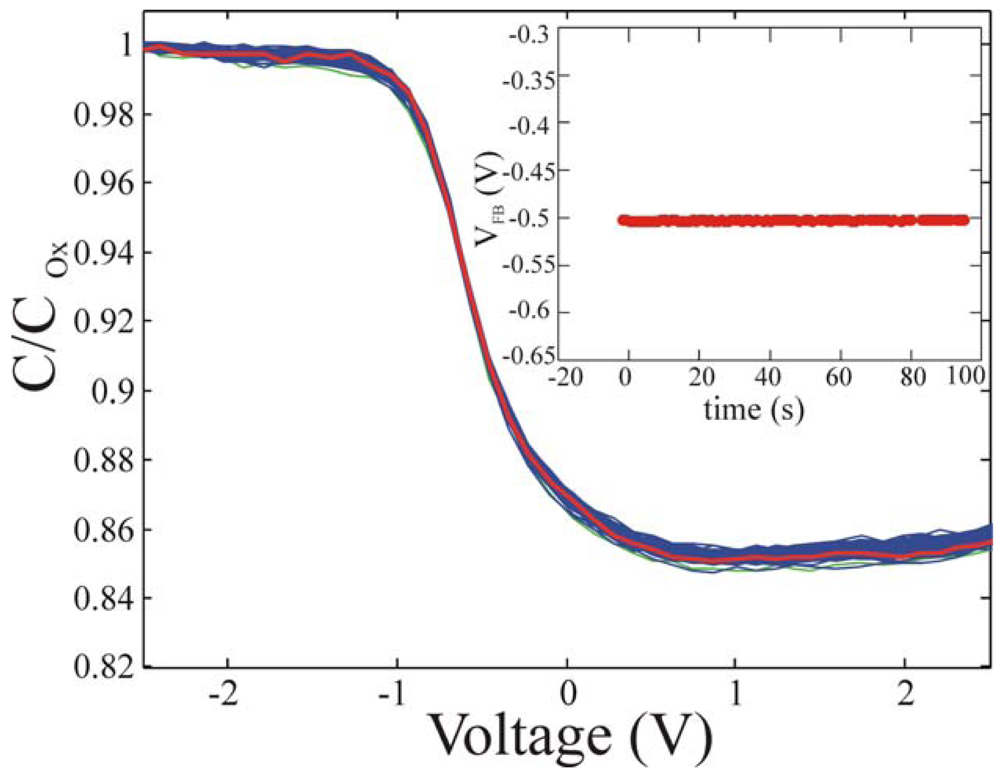
The MOS-like devices were prepared as described in the experimental section, in which all the fabrication details are reported, and the immobilization procedure already described was used. After immobilization, the samples were dried under a gentle Nitrogen flow and measured using capacitance-voltage (CV) measurements at 1 MHz, with a repetition frequency of 10 Hz and the curves were acquired during 100s of measurements. The results are shown in
Figure 10 for the reference sample.
The measurement reproducibility was tested on each set of measurements and on at least three samples per type. The data reproducibility and the absence of oxide aging were tested. If oxide aging was present, a shift in the flat band voltage (V
FB) had to be observed as a function of time, having a direction depending on the trapped charges. The V
FB values as a function of time, as extracted from C-V measurements, and reported in the inset of
Figure 10, did not change during measurements, indicating that no oxide aging was present [
41,
48]. For all measurements, the capacitance was normalized to C
ox, measured in the device accumulation region (negative biases). A constant value of -0.50±0.05 V was detected for the V
FB of the reference samples and used for the comparison with the immobilized samples.
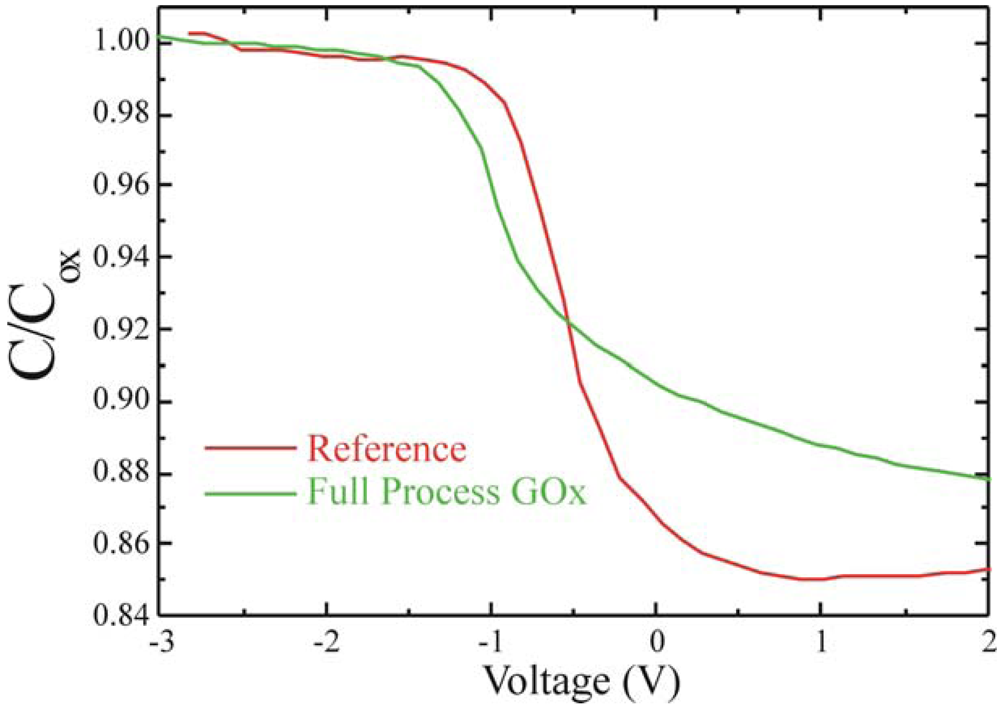
Once verified the measurement reproducibility, GOx immobilized samples were tested. Also in this case no dielectric (C
bio) aging was detected, but a clear shift of V
FB towards negative biases was monitored, as shown in
Figure 11, where the C-V of both the reference (green line) and the GOx fully immobilized (green line) samples are compared. The V
FB approaches to -1.07±0.04 V (average value), hence, a shift of 0.57 V, well above the measurements indetermination (±0.05 V), was detected. This result confirmed that GOx immobilized on the dielectric surface introduced a positive charge as already observed by Wang
et al. [
49]. The most important conclusion that can be drawn from these data is that this device is very sensitive to the presence of organic layers, hence it is a promising candidate for the fabrication of fully electric (MOS-based) glucose sensors.
The V
FB behavior when ssDNA is immobilized on the SiO
2 surface is quite different. Two different set of measurements were carried out to discriminate the different effects. In a first set of measurements the dependence of measurement from the solution parameters was tested. In fact, the solution parameters, pH and ionic concentration, can strongly affect the measurement, since they may give a contribution to the value of C
bio, thus reducing the sensor performances [
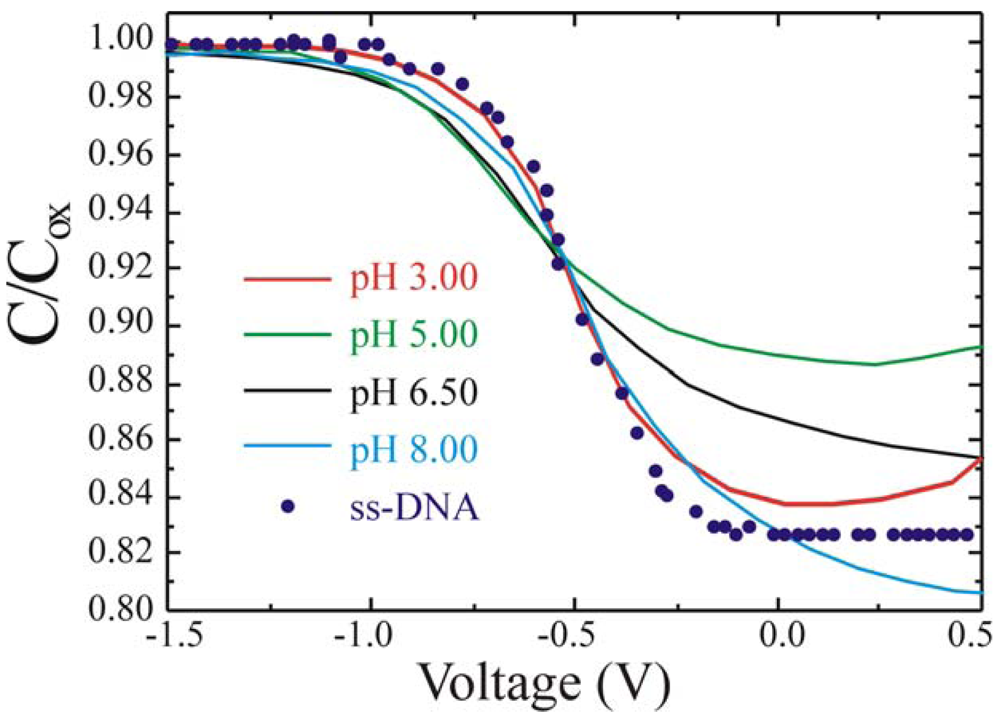
19]. C-V measurements were carried out on the reference samples as a function of the solution pH. An example is shown in
Figure 12. The curves perfectly overlap (within the experimental errors) regardless of the buffer solution pH, from 3.0 (red line) up to pH 8 (light blue line). It should be mentioned we tested solutions ranging from pH 3 to pH 9 (data are not shown) and obtained the same results. Please note that the horizontal scale was expanded in the -0.5 region to better show the curves overlap.
It is known that SiO
2 exhibits a pH dependence of ∼ 30 mV/pH in the observed pH range. Since no shift was observed, within the experimental errors, in the measured curves it is reasonable to assume that the SiO
2 surface was perfectly passivated. In fact, it is known that the pH sensitivity depends on the density of OH groups at the SiO
2/solution surface. The oxidation process employed to prepare the sample used in this work was in a dry ambient, hence the number of OH
- groups is strongly reduced. Moreover, since no permanent V
FB shift was observed as a function of time, also stressing the samples we may conclude that there was no dielectric aging during the measurements [
41,
48]. Finally, a PBS solution with the complementary oligonucleotide was spotted on the reference sample and the C-V measured to verify if the presence of the additional negative charge in solution cold cause a V
FB shift. The data, also shown in
Figure 12 as circles, demonstrate that the C-V was not affected by the various pH or the DNA presence in the PBS solution.
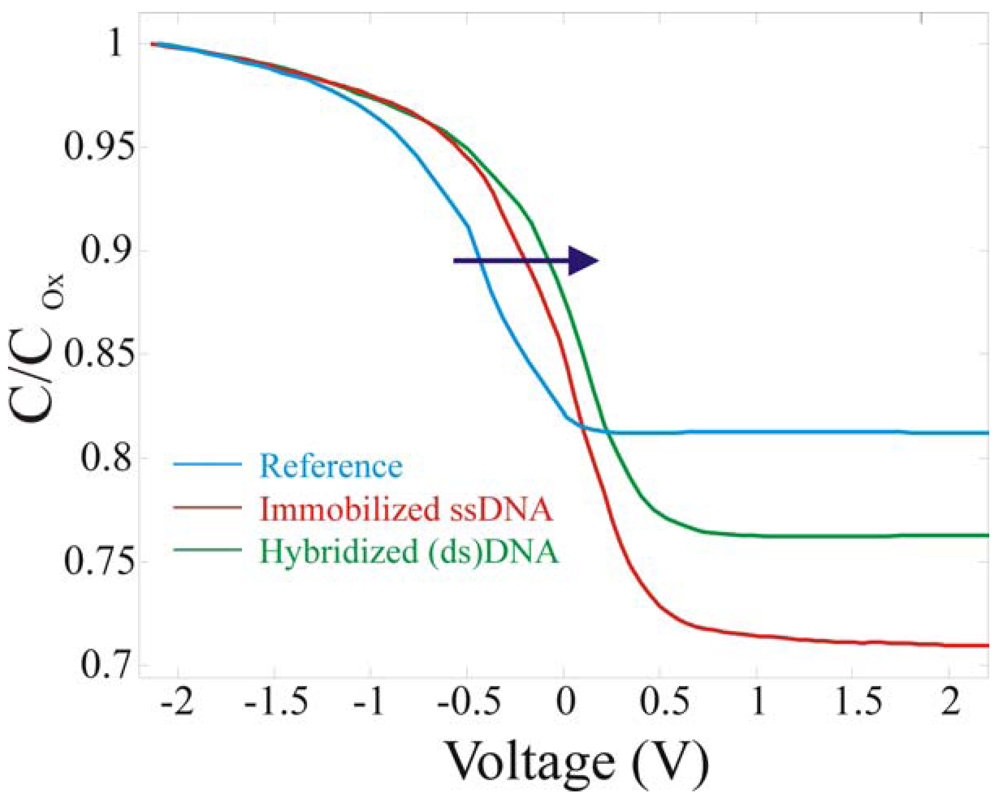
The second set of measurement we performed was aimed to verify if an electrical transduction could be achieved from DNA hybridization. The reference and the two fully processed samples were electrically characterized and the results are compared in
Figure 13. The first test was carried out on reference samples (SiO
2/Si, light blue line) and a V
FB constant value of -0.41±0.03 V was detected during the entire measurement time. Since there was no oxide aging, any shift in V
FB eventually detected must be attributed to the organic layer deposited on the oxide surface. Quite different is the V
FB behaviour when ssDNA is immobilized on the SiO
2 surface. The fully processed ssDNA sample (red line) and the same sample after the hybridization process (green line) exhibited a clear shift in the V
FB values. The data show that V
FB approached to +0.06±0.01 V after the device full processing. DNA molecules have negative charges derived from the phosphate groups in aqueous solutions, hence a negative charge was trapped on the sample surface, as expected. The negative charge presence on the oxide caused positive V
FB shift, with respect to the reference sample, as already observed using different structures [
14]. In particular, a +0.47±0.04 V shift on V
FB was detected.
The DNA immobilization causes a visible difference in the C
Min value. The reduction in the C
Min value (C
DNA) to 0.7 of the C
ox can be attributed to the presence of the extra layer on the SiO
2 surface. Knowing that C
tot is the series of C
Si, C
ox and C
bio and that C
ref/C
ox is 0.8 while C
DNA/C
ox is 0.7, with simple calculations the biological layer thickness can be evaluated, assuming a dielectric constant for the biological layer of 3 [
12]. A value of ∼5.6 nm was obtained, in good agreement with the ssDNA length (6.8 nm). The final set of measurements of
Figure 13 was aimed to demonstrate the device sensitivity to DNA hybridization. The hybridization caused a further shift of 0.07±0.02 V, always in the same direction, since more negative charges are immobilized on the surface. The V
FB approached to 0.13±0.01 V. The results are shown as a solid line. The shifts from the reference did not linearly increase, in fact the hybridized sample did not exhibit a shift double with respect to the ssDNA as one could assume at the first glance. This could be due to two effects: not all the DNA hybridizes and the well known counter-ion effect [
20] takes place.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}