Reporter Proteins in Whole-Cell Optical Bioreporter Detection Systems, Biosensor Integrations, and Biosensing Applications

Abstract
:1. Introduction
2. Bioreporter Immobilization Methods
3. Whole-Cell Optical Bioreporters and Biosensor Integration
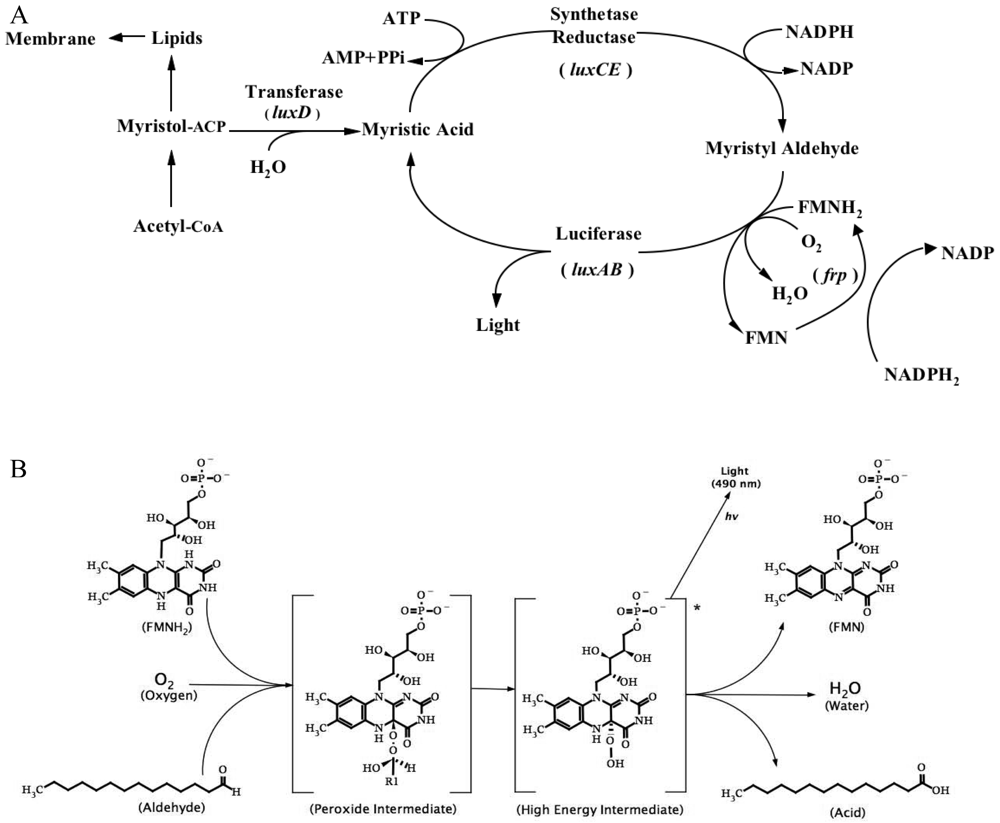
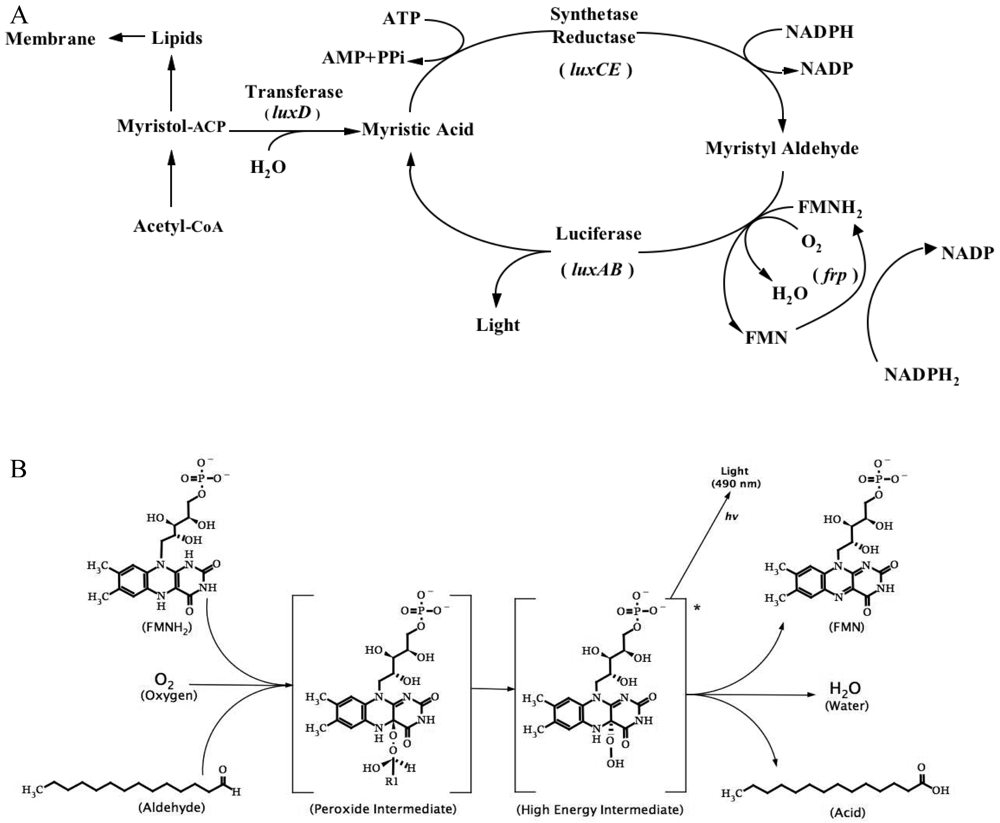
3.1. Bacterial Luciferase (Lux)
3.1.1. Lux biosensors and applications
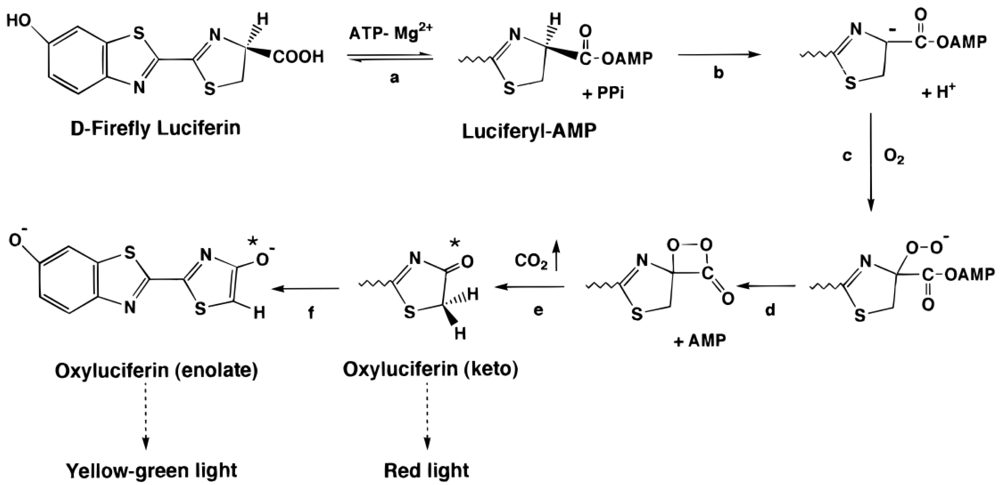
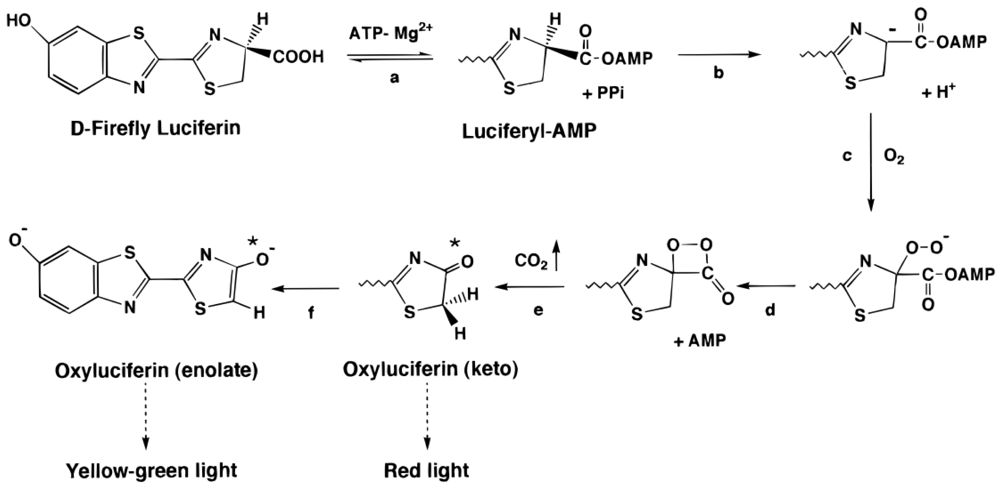
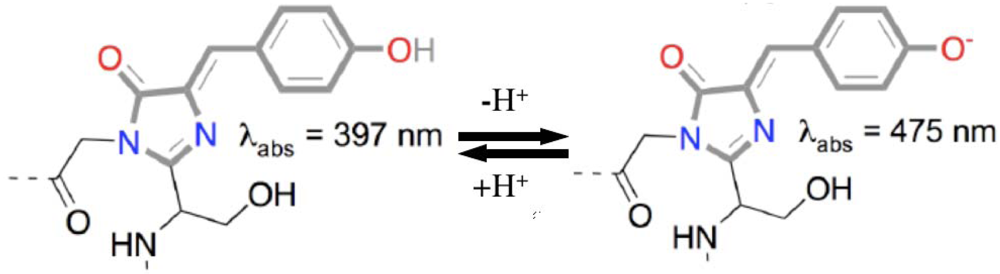
3.2. Firefly Luciferase (Luc)
3.2.1. Luc biosensors and applications
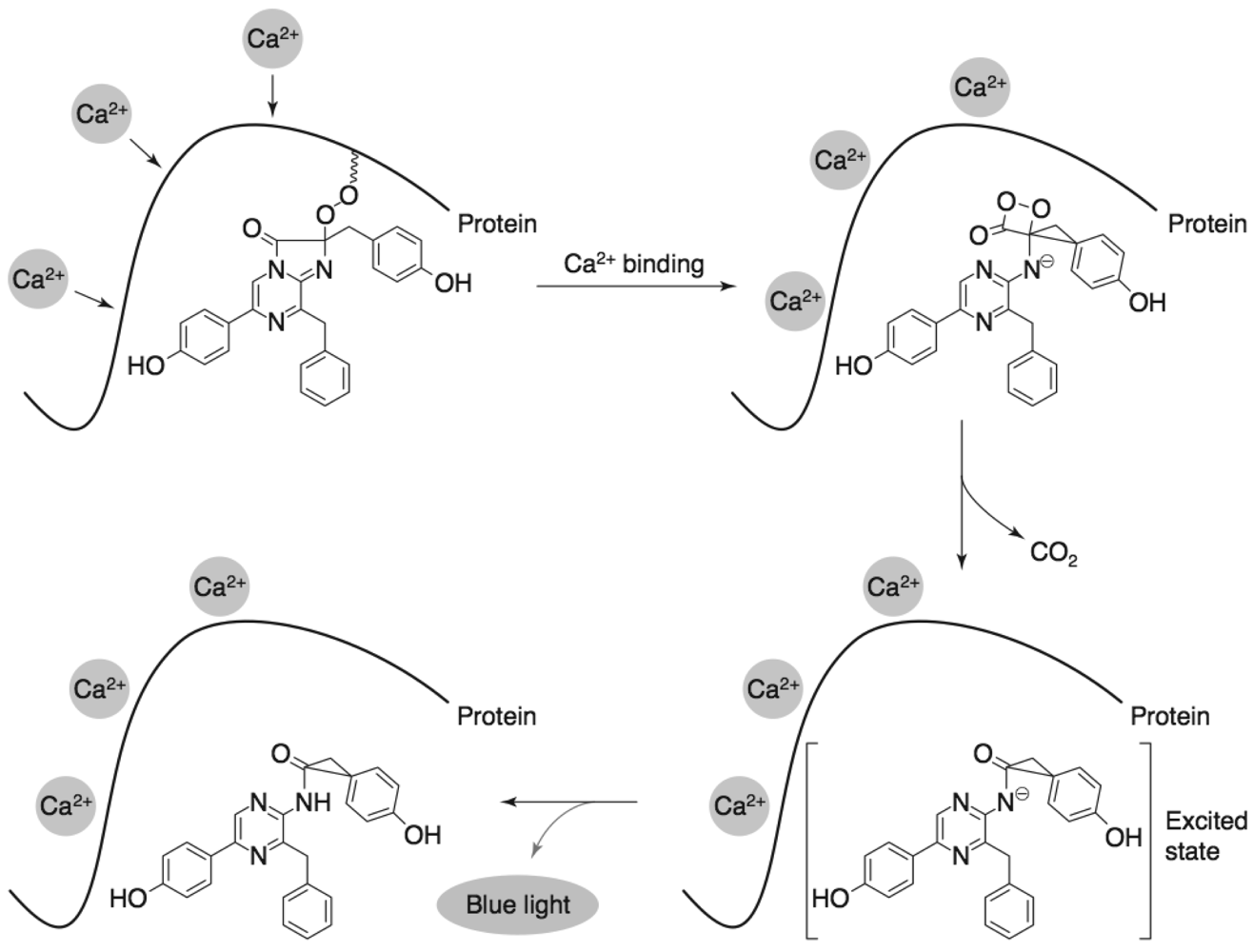
3.3. Aequorin
3.3.1. Aequorin biosensors and applications
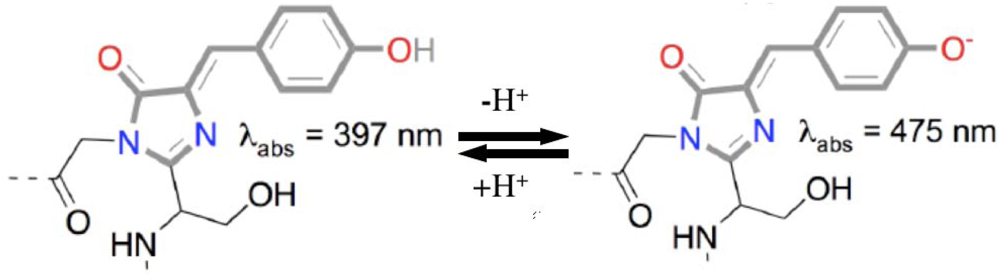
3.4. Green Fluorescent Protein (GFP)
3.4.1. GFP biosensors and applications
3.5. Alternative Bioreporter Systems
Acknowledgments
References and Notes
- Bjerketorp, J.; Hakansson, S.; Belkin, S.; Jansson, J.K. Advances in preservation methods: keeping biosensor microorganisms alive and active. Curr. Opin. Biotechnol. 2006, 17, 43–49. [Google Scholar]
- Premkumar, J.R.; Lev, O.; Marks, R.S.; Polyak, B.; Rosen, R.; Belkin, S. Antibody-based immobilization of bioluminescent bacterial sensor cells. Talanta 2001, 55, 1029–1038. [Google Scholar]
- Kandimalla, V.B.; Tripathi, V.S.; Ju, H.X. Immobilization of biomolecules in sol-gels: biological and analytical applications. Crit. Rev. Anal. Chem. 2006, 36, 73–106. [Google Scholar]
- Kuncova, G.; Podrazky, O.; Ripp, S.; Trogl, J.; Sayler, G.; Demnerova, K.; Vankova, R. Monitoring the viability of cells immobilized by the sol-gel process. J. Sol Gel Sci. Technol. 2004, 31, 335–342. [Google Scholar]
- Flickinger, M.C.; Schottel, J.L.; Bond, D.R.; Aksan, A.; Scriven, L.E. Painting and printing living bacteria: engineering nanoporous biocatalytic coatings to preserve microbial viability and intensify reactivity. Biotechnol. Prog. 2007, 23, 2–17. [Google Scholar]
- Yoo, S.K.; Lee, J.H.; Yun, S.S.; Gu, M.B. Fabrication of a bio-MEMS based cell-chip for toxicity monitoring. Biosens. Bioelectron. 2007, 22, 1586–1592. [Google Scholar]
- Weibel, D.B.; DiLuzio, W.R.; Whitesides, G.M. Microfabrication meets microbiology. Nat. Rev. Microbiol. 2007, 5, 209–218. [Google Scholar]
- Salalha, W.; Kuhn, J.; Dror, Y.; Zussman, E. Encapsulation of bacteria and viruses in electrospun nanofibres. Nanotechnology. 2006, 17, 4675–4681. [Google Scholar]
- Chu, Y.F.; Hsu, C.H.; Soma, P.K.; Lo, Y.M. Immobilization of bioluminescent Escherichia coli cells using natural and artificial fibers treated with polyethyleneimine. Bioresour. Technol. 2009, 100, 3167–3174. [Google Scholar]
- Yeni, F.; Odaci, D.; Timur, S. Use of eggshell membrane as an immobilization platform in microbial sensing. Anal. Lett. 2008, 41, 2743–2758. [Google Scholar]
- Lee, J.H.; Gu, M.B. An integrated mini biosensor system for continuous water toxicity monitoring. Biosens. Bioelectron. 2005, 20, 1744–1749. [Google Scholar]
- Wu, J. Interactions of electrical fields with fluids: laboratory-on-a-chip applications. IET Nanobiotechnol. 2008, 2, 14–27. [Google Scholar]
- Hunt, T.P.; Issadore, D.; Westervelt, R.M. Integrated circuit/microfluidic chip to programmably trap and move cells and droplets with dielectrophoresis. Lab Chip 2008, 8, 81–87. [Google Scholar]
- Date, A.; Pasini, P.; Daunert, S. Construction of spores for portable bacterial whole-cell biosensing systems. Anal. Chem. 2007, 79, 9391–9397. [Google Scholar]
- Mehrvar, M.; Abdi, M. Recent developments, characteristics, and potential applications of electrochemical biosensors. Anal. Sci. 2004, 20, 1113–1126. [Google Scholar]
- Johnson, B.T. Microtox acute toxicity test. In Small-Scale Freshwater Toxicity Investigations; Blaise, C., Ferard, J.F., Eds.; Springer: Dordrecht, The Netherlands, 2005; pp. 69–105. [Google Scholar]
- Choi, O.; Deng, K.K.; Kim, N.J.; Ross, L.; Surampalli, R.Y.; Hu, Z.Q. The inhibitory effects of silver nanoparticles, silver ions, and silver chloride colloids on microbial growth. Water Res. 2008, 42, 3066–3074. [Google Scholar]
- Ripp, S. Bacteriophage-based pathogen detection. In Advances in Biochemical Engineering/Biotechnology: Whole Cell Sensing Systems; Belkin, S., Gu, M.B., Eds.; Springer: New York, NY, 2009. [Google Scholar] [CrossRef]
- Shetty, R.S.; Ramanathan, S.; Badr, I.H.A.; Wolford, J.L.; Daunert, S. Green fluorescent protein in the design of a living biosensing system for L-arabinose. Anal. Chem. 1999, 71, 763–768. [Google Scholar]
- Eldridge, M.L.; Sanseverino, J.; Layton, A.C.; Easter, J.P.; Schultz, T.W.; Sayler, G.S. Saccharomyces cerevisiae BLYAS, a new bioluminescent bioreporter for detection of androgenic compounds. Appl. Environ. Microbiol. 2007, 73, 6012–6018. [Google Scholar]
- Knight, A.W.; Goddard, N.J.; Fielden, P.R.; Barker, M.G.; Billinton, N.; Walmsley, R.M. Development of a flow-through detector for monitoring genotoxic compounds by quantifying the expression of green fluorescent protein in genetically modified yeast cells. Meas. Sci. Technol. 1999, 10, 211–217. [Google Scholar]
- Daniel, R.; Almog, R.; Ron, A.; Belkin, S.; Diamand, Y.S. Modeling and measurement of a whole-cell bioluminescent biosensor based on a single photon avalanche diode. Biosens. Bioelectron. 2008, 24, 882–887. [Google Scholar]
- Rothert, A.; Deo, S.K.; Millner, L.; Puckett, L.G.; Madou, M.J.; Daunert, S. Whole-cell-reporter-gene-based biosensing systems on a compact disk microfluidics platform. Anal. Biochem. 2005, 342, 11–19. [Google Scholar]
- Engebrecht, J.; Nealson, K.; Silverman, M. Bacterial bioluminescence: isolation and genetic analysis of functions from Vibrio fischeri. Cell 1983, 32, 773–781. [Google Scholar]
- Patterson, S.S.; Dionisi, H.M.; Gupta, R.K.; Sayler, G.S. Codon optimization of bacterial luciferase (lux) for expression in mammalian cells. J. Ind. Microbiol. Biotechnol. 2005, 32, 115–123. [Google Scholar]
- Zhang, Y.; Phillips, G.J.; Yeung, E.S. Quantitative imaging of gene expression in individual bacterial cells by chemiluminescence. Anal. Chem. 2008, 80, 597–605. [Google Scholar]
- Chang, S.T.; Lee, H.J.; Gu, M.B. Enhancement in the sensitivity of an immobilized cell-based soil biosensor for monitoring PAH toxicity. Sens. Actuat. B 2004, 97, 272–276. [Google Scholar]
- Ikariyama, Y.; Nishiguchi, S.; Koyama, T.; Kobatake, E.; Aizawa, M.; Tsuda, M.; Nakazawa, T. Fiber-optic-based biomonitoring of benzene derivatives by recombinant E. coli bearing luciferase gene-fused TOL-plasmid immobilized on the fiber optic end. Anal. Chem. 1997, 69, 2600–2605. [Google Scholar]
- Leth, S.; Maltoni, S.; Simkus, R.; Mattiasson, B.; Corbisier, P.; Klimant, I.; Wolfbeis, O.S.; Csoregi, E. Engineered bacteria based biosensors for monitoring bioavailable heavy metals. Electroanalysis 2002, 14, 35–42. [Google Scholar]
- Maehana, K.; Tani, H.; Kamidate, T. On-chip genotoxic bioassay based on bioluminescence reporter system using three-dimensional microfluidic network. Anal. Chim. Acta 2006, 560, 24–29. [Google Scholar]
- Ripp, S.; Daumer, K.A.; McKnight, T.; Levine, L.H.; Garland, J.L.; Simpson, M.L.; Sayler, G.S. Bioluminescent bioreporter integrated circuit sensing of microbial volatile organic compounds. J. Ind. Microbiol. Biotechnol. 2003, 30, 636–642. [Google Scholar]
- Naseem, R.; Holland, I.B.; Jacq, A.; Wann, K.T.; Campbell, A.K. pH and monovalent cations regulate cytosolic free Ca2+ in E. coli. BBA-Rev. Biomembranes 2008, 1778, 1415–1422. [Google Scholar]
- Heitzer, A.; Malachowsky, K.; Thonnard, J.E.; Bienkowski, P.R.; White, D.C.; Sayler, G.S. Optical biosensor for environmental on-line monitoring of naphthalene and salicylate bioavailability with an immobilized bioluminescent catabolic reporter bacterium. Appl. Environ. Microbiol. 1994, 60, 1487–1494. [Google Scholar]
- Rider, T.H.; Petrovick, M.S.; Nargi, F.E.; Harper, J.D.; Schwoebel, E.D.; Mathews, R.H.; Blanchard, D.J.; Bortolin, L.T.; Young, A.M.; Chen, J.; Hollis, M.A. A B cell-based sensor for rapid identification of pathogens. Science 2003, 301, 213–215. [Google Scholar]
- Polyak, B.; Bassis, E.; Novodvorets, A.; Belkin, S.; Marks, R.S. Bioluminescent whole cell optical fiber sensor to genotoxicants: system optimization. Sens. Actuat. B-Chem. 2001, 74, 18–26. [Google Scholar]
- Meighen, E.A. Molecular biology of bacterial bioluminescence. Microbiol. Rev. 1991, 55, 123–142. [Google Scholar]
- Boyle, R. New experiments concerning the relation between light and air (in shining wood and fish). Philos. T. R. Soc. 1666, 2, 581–600. [Google Scholar]
- McElroy, W.D.; Strehler, B.L. Bioluminescence. Bacteriol. Rev. 1954, 18, 177–194. [Google Scholar]
- Doudoroff, M. Studies on the luminous bacteria II. Some observations on the anaerobic metabolism of facultatively anaerobic species. J. Bacteriol. 1942, 44, 461–467. [Google Scholar]
- McElroy, W.D.; Hastings, J.W.; Sonnenfeld, V.; Coulombre, J. The requirement of riboflavin phosphate for bacterial luminescence. Science 1953, 118, 385–386. [Google Scholar]
- Strehler, B.L.; Harvey, E.N.; Chang, J.J.; Cormier, M.J. The luminescent oxidation of reduced riboflavin or reduced riboflavin phosphate in the bacterial luciferin-luciferase reaction. Proc. Natl. Acad. Sci. U. S. A. 1954, 40, 10–12. [Google Scholar]
- Belas, R.; Mileham, A.; Cohn, D.; Hilmen, M.; Simon, M.; Silverman, M. Bacterial bioluminescence—isolation and expression of the luciferase genes from Vibrio harveyi. Science 1982, 218, 791–793. [Google Scholar]
- Fisher, A.J.; Raushel, F.M.; Baldwin, T.O.; Rayment, I. Three-dimensional structure of bacterial luciferase from Vibrio harveyi at 2.4. ANG. resolution. Biochemistry 1995, 34, 6581–6586. [Google Scholar]
- Thouand, G.; Daniel, P.; Horry, H.; Picart, P.; Durand, M.J.; Killham, K.; Knox, O.G.G.; DuBow, M.S.; Rousseau, M. Comparison of the spectral emission of lux recombinant and bioluminescent marine bacteria. Luminescence 2003, 18, 145–155. [Google Scholar]
- Campbell, Z.T.; Weichsel, A.; Montfort, W.R.; Baldwin, T.O. Crystal structure of the bacterial luciferase/flavin complex provides insight into the function of the beta subunit. Biochemistry 2009, 48, 6085–6094. [Google Scholar]
- Baldwin, T.O.; Christopher, J.A.; Raushel, F.M.; Sinclair, J.F.; Ziegler, M.M.; Fisher, A.J.; Rayment, I. Structure of bacterial luciferase. Curr. Opin. Struct. Biol. 1995, 5, 798–809. [Google Scholar]
- Vervoort, J.; Muller, F.; Okane, D.J.; Lee, J.; Bacher, A. Bacterial luciferase—A C-13, N-15, and P-31 nuclear magnetic resonance investigation. Biochemistry 1986, 25, 8067–8075. [Google Scholar]
- Nemtseva, E.V.; Kudryasheva, N.S. The mechanism of electronic excitation in the bacterial bioluminescent reaction. Russ. Chem. Rev. 2007, 76, 91–100. [Google Scholar]
- Baldwin, T.O.; Chen, L.H.; Chlumsky, L.J.; Devine, J.H.; Johnston, T.C.; Lin, J.W.; Sugihara, J.; Waddle, J.J.; Ziegler, M.M. Structural analysis of bacterial luciferase. In Flavins and Flavoproteins; Walter de Gruyter & Co.: Berlin, Germany, 1987; pp. 621–631. [Google Scholar]
- Hastings, J.W.; Nealson, K.H. Bacterial bioluminescence. Annu. Rev. Microbiol. 1977, 31, 549–595. [Google Scholar]
- Kelly, C.J.; Lajoie, C.A.; Layton, A.C.; Sayler, G.S. Bioluminescent reporter bacterium for toxicity monitoring in biological wastewater treatment systems. Water Environ. Res. 1999, 71, 31–35. [Google Scholar]
- Ripp, S.; DiClaudio, M.L.; Sayler, G.S. Biosensors as environmental monitors. In Environmental Microbiology, 2nd ed.; Mitchell, R., Gu, J.D., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 219–239. [Google Scholar]
- Allen, M.S.; Wilgus, J.R.; Chewning, C.S.; Sayler, G.S.; Simpson, M.L. A destabilized bacterial luciferase for dynamic gene expression studies. Syst. Synth. Biol. 2007, 1, 3–9. [Google Scholar]
- Nivens, D.E.; McKnight, T.E.; Moser, S.A.; Osbourn, S.J.; Simpson, M.L.; Sayler, G.S. Bioluminescent bioreporter integrated circuits: potentially small, rugged and inexpensive whole-cell biosensors for remote environmental monitoring. J. Appl. Microbiol. 2004, 96, 33–46. [Google Scholar]
- Ivask, A.; Green, T.; Polyak, B.; Mor, A.; Kahru, A.; Virta, M.; Marks, R. Fibre-optic bacterial biosensors and their application for the analysis of bioavailable Hg and As in soils and sediments from Aznalcollar mining area in Spain. Biosens. Bioelectron. 2007, 22, 1396–1402. [Google Scholar]
- Hakkila, K.; Green, T.; Leskinen, P.; Ivask, A.; Marks, R.; Virta, M. Detection of bioavailable heavy metals in EILATox-Oregon samples using whole-cell luminescent bacterial sensors in suspension or immobilized onto fibre-optic tips. J. Appl. Toxicol. 2004, 24, 333–342. [Google Scholar]
- Horry, H.; Charrier, T.; Durand, M.J.; Vrignaud, B.; Picart, P.; Daniel, P.; Thouand, G. Technological conception of an optical biosensor with a disposable card for use with bioluminescent bacteria. Sens. Actuat. B-Chem. 2007, 122, 527–534. [Google Scholar]
- Dorn, J.G.; Mahal, M.K.; Brusseau, M.L.; Maier, R.M. Employing a novel fiber optic detection system to monitor the dynamics of in situ lux bioreporter activity in porous media: system performance update. Anal. Chim. Acta 2004, 525, 63–74. [Google Scholar]
- Vijayaraghavn, R.; Islam, S.K.; Zhang, M.; Ripp, S.; Caylor, S.; Weathers, B.; Moser, S.; Terry, S.; Blalock, B.; Sayler, G.S. A bioreporter bioluminescent integrated circuit for very low-level chemical sensing in both gas and liquid environments. Sens. Actuat. B 2007, 123, 922–928. [Google Scholar]
- Elman, N.M.; Ben-Yoav, H.; Sternheim, M.; Rosen, R.; Krylov, S.; Shacham-Diamand, Y. Towards toxicity detection using a lab-on-chip based on the integration of MOEMS and whole-cell sensors. Biosens. Bioelectron. 2008, 23, 1631–1636. [Google Scholar]
- Eltoukhy, H.; Salama, K.; El Gamal, A. A 0.18-um CMOS bioluminescence detection lab-on-chip. IEEE J. Solid-State Circuit 2006, 41, 651–662. [Google Scholar]
- Yotter, R.A.; Wilson, D.M. A review of photodetectors for sensing light-emitting reporters in biological systems. IEEE Sens. J. 2003, 3, 288–303. [Google Scholar]
- Daniel, R.; Almog, R.; Sverdlov, Y.; Yagurkroll, S.; Belkin, S.; Shacham-Diamand, Y. Development of a quantitative optical biochip based on a double integrating sphere system that determines absolute photon number in bioluminescent solution: application to quantum yield scale realization. Appl. Optics 2009, 48, 3216–3224. [Google Scholar]
- Viviani, V.R. The origin, diversity, and structure function relationships of insect luciferases. Cell. Mol. Life Sci. 2002, 59, 1833–1850. [Google Scholar]
- Fraga, H. Firefly luminescence: A historical perspective and recent developments. Photochem. Photobiol. Sci. 2008, 7, 146–158. [Google Scholar]
- Lewis, S.M.; Cratsley, C.K. Flash signal evolution, mate choice, and predation in fireflies. Annu. Rev. Entomol. 2008, 53, 293–321. [Google Scholar]
- McElroy, W.D. The energy source for bioluminescence in an isolated system. Proc. Natl. Acad. Sci. USA 1947, 33, 342–345. [Google Scholar]
- Hastings, J.W.; McElroy, W.D.; Coulombre, J. The effect of oxygen upon the immobilization reaction in firefly luminescence. J. Cell. Compar. Physl. 1953, 42, 137–150. [Google Scholar]
- White, E.H.; Field, G.F.; McElroy, W.D.; McCapra, F. Structure and synthesis of firefly luciferin. J. Am. Chem. Soc. 1961, 83, 2402–2403. [Google Scholar]
- White, E.H.; Rapaport, E.; Hopkins, T.A.; Seliger, H.H. Chemi- and bioluminescence of firefly luciferin. J. Am. Chem. Soc. 1969, 91, 2178–2180. [Google Scholar]
- McCapra, F. Chemical mechanisms in bioluminescence. Acc. Chem. Res. 1976, 9, 201–208. [Google Scholar]
- White, E.H.; Steinmetz, M.G.; Miano, J.D.; Wildes, P.D.; Morland, R. Chemi-luminescence and bioluminescence of firefly luciferin. J. Am. Chem. Soc. 1980, 102, 3199–3208. [Google Scholar]
- Conti, E.; Franks, N.P.; Brick, P. Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure 1996, 4, 287–298. [Google Scholar]
- Gould, S.J.; Subramani, S. Firefly luciferase as a tool in molecular and cell biology. Anal. Biochem. 1988, 175, 5–13. [Google Scholar]
- Ugarova, N.N. Luciferase of Luciola mingrelica fireflies—kinetics and regulation mechanism. J. Biolumin. Chemilumin. 1989, 4, 406–418. [Google Scholar]
- McCapra, F.; Chang, Y.C.; Francois, V.P. Chemiluminescence of a firefly luciferin analogue. Chem. Commun. 1968, 22–23. [Google Scholar]
- Branchini, B.R.; Magyar, R.A.; Murtiashaw, M.H.; Anderson, S.M.; Zimmer, M. Site-directed mutagenesis of histidine 245 in firefly luciferase: A proposed model of the active site. Biochemistry 1998, 37, 15311–15319. [Google Scholar]
- DeLuca, M.; Wannlund, J.; McElroy, W.D. Factors affecting the kinetics of light emission from crude and purified firefly luciferase. Anal. Biochem. 1979, 95, 194–198. [Google Scholar]
- Denburg, J.L.; Lee, R.T.; McElroy, W.D. Substrate-binding properties of firefly luciferase. I. Luciferin binding site. Arch. Biochem. Biophys. 1969, 134, 381–394. [Google Scholar]
- Fontes, R.; Ortiz, B.; de Diego, A.; Sillero, A.; Sillero, M.A.G. Dehydroluciferyl-AMP is the main intermediate in the luciferin dependent synthesis of Ap(4)A catalyzed by firefly luciferase. FEBS Lett. 1998, 438, 190–194. [Google Scholar]
- Airth, R.L.; Rhodes, W.C.; McElroy, W.D. The function of coenzyme-A in luminescence. Biochim. Biophys. Acta 1958, 27, 519–532. [Google Scholar]
- Wood, K.V.; Lam, Y.A.; Seliger, H.H.; McElroy, W.D. Complementary DNA coding click beetle luciferase can elicit bioluminescence of different colors. Science 1989, 244, 700–702. [Google Scholar]
- DeLuca, M. Hydrophobic nature of active site of firefly luciferase. Biochemistry 1969, 8, 160–166. [Google Scholar]
- White, E.H.; Branchini, B.R. Modification of firefly luciferase with a luciferin analog - red light producing enzyme. J. Am. Chem. Soc. 1975, 97, 1243–1245. [Google Scholar]
- McCapra, F.; Gilfoyle, D.J.; Young, D.W.; Church, N.J.; Spencer, P. The chemical origin of color differences in beetle bioluminescence. In Bioluminescence and Chemiluminescence: Fundamental and Applied Aspects; Campbell, A.K., Kricka, L.J., Stanley, P.E., Eds.; Wiley: Chichester, UK, 1994. [Google Scholar]
- Branchini, B.R.; Murtiashaw, M.H.; Magyar, R.A.; Portier, N.C.; Ruggiero, M.C.; Stroh, J.G. Yellow-green and red firefly bioluminescence from 5,5-dimethyloxyluciferin. J. Am. Chem. Soc. 2002, 124, 2112–2113. [Google Scholar]
- Chiu, N.H.L.; Christopoulos, T.K. Two-site expression immunoassay using a firefly luciferase-coding DNA label. Clin. Chem. 1999, 45, 1954–1959. [Google Scholar]
- Gomi, K.; Kajiyama, N. Oxyluciferin, a luminescence product of firefly luciferase, is enzymatically regenerated into luciferin. J. Biol. Chem. 2001, 276, 36508–36513. [Google Scholar]
- Doyle, T.C.; Nawotka, K.A.; Purchio, A.F.; Akin, A.R.; Francis, K.P.; Contag, P.R. Expression of firefly luciferase in Candida albicans and its use in the selection of stable transformants. Microb. Pathog. 2006, 40, 69–81. [Google Scholar]
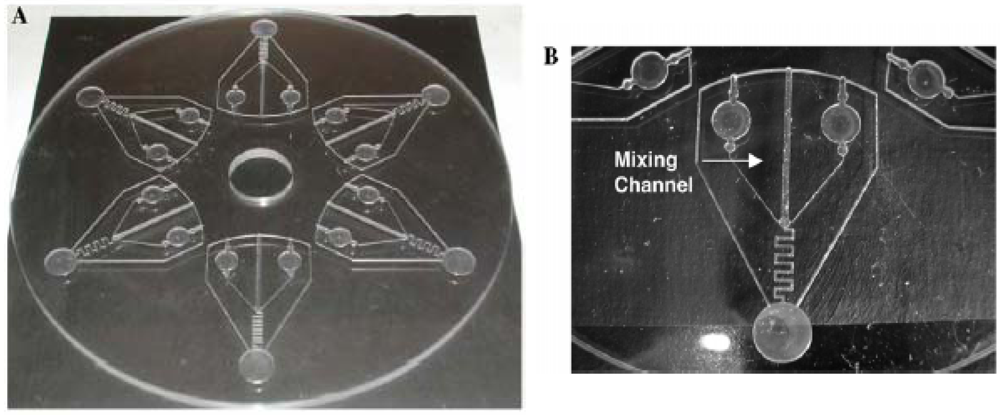
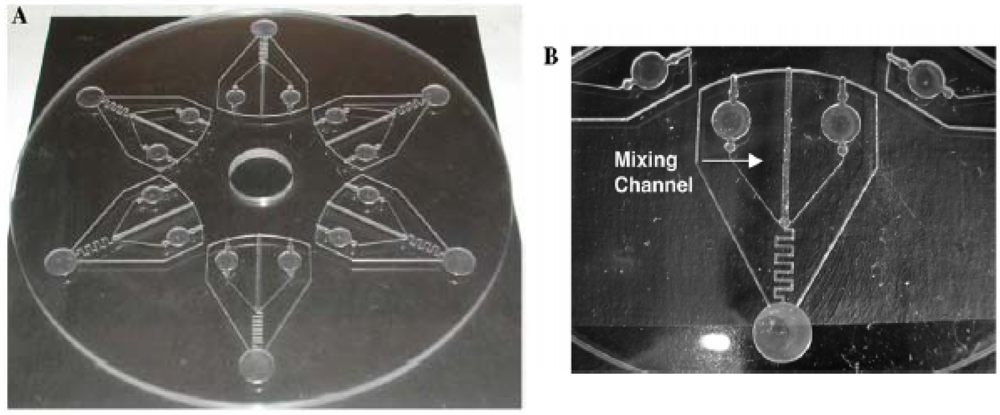
- Mei, Q.; Xia, Z.; Xu, F.; Soper, S.A.; Fan, Z.H. Fabrication of microfluidic reactors and mixing studies for luciferase detection. Anal. Chem. 2008, 80, 6045–6050. [Google Scholar]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from luminous Hydromedusan, Aequorea. J. Cell. Compar. Physl. 1962, 59, 223–239. [Google Scholar]
- Shimomura, O. The discovery of aequorin and green fluorescent protein. J. Microsc.-Oxf. 2005, 217, 3–15. [Google Scholar]
- Ridgway, E.B.; Ashley, C.C. Calcium transients in single muscle fibers. Biochem. Biophys. Res. Commun. 1967, 29, 229–234. [Google Scholar]
- Jones, K.; Hibbert, F.; Keenan, M. Glowing jellyfish, luminescence and a molecule called coelenterazine. Trends Biotechnol. 1999, 17, 477–481. [Google Scholar]
- Shimomura, O.; Johnson, F.H. Regeneration of photoprotein aequorin. Nature 1975, 256, 236–238. [Google Scholar]
- Inoue, S.; Sugiura, S.; Kakoi, H.; Hasizume, K.; Goto, T.; Iio, H. Squid bioluminescence. 2. Isolation from Watasenia scintillans and synthesis of 2-(para-hydroxybenzyl)-6-(para-hydroxyphenyl)-3,7-dihydroimidazo[1,2-A]pyrazin-3-one. Chem. Lett. 1975, 141–144. [Google Scholar]
- Prasher, D.; McCann, R.O.; Cormier, M.J. Cloning and expression of the cDNA coding for aequorin, a bioluminescent calcium binding protein. Biochem. Biophys. Res. Commun. 1985, 126, 1259–1268. [Google Scholar]
- Inouye, S.; Sakaki, Y.; Goto, T.; Tsuji, F.I. Expression of apoaequorin complementary DNA in Escherichia coli. Biochemistry 1986, 25, 8425–8429. [Google Scholar]
- Head, J.F.; Inouye, S.; Teranishi, K.; Shimomura, O. The crystal structure of the photoprotein aequorin at 2.3 angstrom resolution. Nature 2000, 405, 372–376. [Google Scholar]
- Shimomura, O.; Johnson, F.H. Peroxidized coelenterazine, the active group in the photoprotein aequorin. Proc. Natl. Acad. Sci. USA 1978, 75, 2611–2615. [Google Scholar]
- Vysotski, E.S.; Lee, J. Ca2+-regulated photoproteins: Structural insight into the bioluminescence mechanism. Accounts Chem. Res. 2004, 37, 405–415. [Google Scholar]
- McCapra, F.; Chang, Y.C. Chemiluminescence of a Cypridina luciferin analogue. Chem. Commun. 1967, 1011–1012. [Google Scholar]
- Ohmiya, Y.; Hirano, T. Shining the light: The mechanism of the bioluminescence reaction of calcium-binding photoproteins. Chem. Biol. 1996, 3, 337–347. [Google Scholar]
- Chiesa, A.; Rapizzi, E.; Tosello, V.; Pinton, P.; de Virgilio, M.; Fogarty, K.E.; Rizzuto, R. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signalling. Biochem. J. 2001, 355, 1–12. [Google Scholar]
- Prendergast, F.G.; Allen, D.G.; Blinks, J.R. Calcium-binding proteins and calcium function. In Properties of the Calcium-Sensitive Bioluminescent Protein Aequorin; Wasserman, R.H., Corradiano, R.A., Carafoil, E., Kretsinger, R.H., MacLennan, D.H., Siegel, F.L., Eds.; North-Holland: New York, NY, USA, 1977; pp. 469–480. [Google Scholar]
- Lewis, J.C.; Daunert, S. Photoproteins as luminescent labels in binding assays. Fresenius J. Anal. Chem. 2000, 366, 760–768. [Google Scholar]
- Kendall, J.M.; Badminton, M.N. Aequorea victoria bioluminescence moves into an exciting new era. Trends Biotechnol. 1998, 16, 216–224. [Google Scholar]
- Giepmans, B.N.G.; Adams, S.R.; Ellisman, M.H.; Tsien, R.Y. Review - The fluorescent toolbox for assessing protein location and function. Science 2006, 312, 217–224. [Google Scholar]
- Prasher, D.C.; Eckenrode, V.K.; Ward, W.W.; Prendergast, F.G.; Cormier, M.J. Primary structure of the Aequorea victoria green fluorescent protein. Gene 1992, 111, 229–233. [Google Scholar]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar]
- Zimmer, M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem. Rev. 2002, 102, 759–781. [Google Scholar]
- Naylor, L.H. Reporter gene technology: The future looks bright. Biochem. Pharmacol. 1999, 58, 749–757. [Google Scholar]
- Ehrmann, M.A.; Scheyhing, C.H.; Vogel, R.F. In vitro stability and expression of green fluorescent protein under high pressure conditions. Lett. Appl. Microbiol. 2001, 32, 230–234. [Google Scholar]
- Liu, H.S.; Jan, M.S.; Chou, C.K.; Chen, P.H.; Ke, N.J. Is green fluorescent protein toxic to the living cells? Biochem. Biophys. Res. Commun. 1999, 260, 712–717. [Google Scholar]
- Hanazono, Y.; Yu, J.M.; Dunbar, C.E.; Emmons, R.V.B. Green fluorescent protein retroviral vectors: Low titer and high recombination frequency suggest a selective disadvantage. Human Gene Ther. 1997, 8, 1313–1319. [Google Scholar]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar]
- Drepper, T.; Eggert, T.; Circolone, F.; Heck, A.; Krauss, U.; Guterl, J.K.; Wendorff, M.; Losi, A.; Gartner, W.; Jaeger, K.E. Reporter proteins for in vivo fluorescence without oxygen. Nat. Biotechnol. 2007, 25, 443–445. [Google Scholar]
- Patterson, G.H.; Knobel, S.M.; Sharif, W.D.; Kain, S.R.; Piston, D.W. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 1997, 73, 2782–2790. [Google Scholar]
- Niwa, H.; Inouye, S.; Hirano, T.; Matsuno, T.; Kojima, S.; Kubota, M.; Ohashi, M.; Tsuji, F.I. Chemical nature of the light emitter of the Aequorea green fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13617–13622. [Google Scholar]
- Chattoraj, M.; King, B.A.; Bublitz, G.U.; Boxer, S.G. Ultra-fast excited state dynamics in green fluorescent protein: Multiple states and proton transfer. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 8362–8367. [Google Scholar]
- Jung, G.; Wiehler, J.; Zumbusch, A. The photophysics of green fluorescent protein: Influence of the key amino acids at positions 65, 203, and 222. Biophys. J. 2005, 88, 1932–1947. [Google Scholar]
- Cubitt, A.B.; Heim, R.; Adams, S.R.; Boyd, A.E.; Gross, L.A.; Tsien, R.Y. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci. 1995, 20, 448–455. [Google Scholar]
- Yokoe, H.; Meyer, T. Spatial dynamics of GFP-tagged proteins investigated by local fluorescence enhancement. Nat. Biotechnol. 1996, 14, 1252–1256. [Google Scholar]
- Crameri, A.; Whitehorn, E.A.; Tate, E.; Stemmer, W.P.C. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat. Biotechnol. 1996, 14, 315–319. [Google Scholar]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 2002, 296, 913–916. [Google Scholar]
- Heim, R.; Prasher, D.C.; Tsien, R.Y. Wavelength mutations and posttranslational autooxidation of green fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 12501–12504. [Google Scholar]
- Heim, R.; Tsien, R.Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 1996, 6, 178–182. [Google Scholar]
- Ormo, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar]
- Matz, M.V.; Fradkov, A.F.; Labas, Y.A.; Savitsky, A.P.; Zaraisky, A.G.; Markelov, M.L.; Lukyanov, S.A. Fluorescent proteins from nonbioluminescent Anthozoa species. Nature Biotechnol. 1999, 17, 969–973. [Google Scholar]
- Gvakharia, B.O.; Bottomley, P.J.; Arp, D.J.; Sayavedra-Soto, L.A. Construction of recombinant Nitrosomonas europaea expressing green fluorescent protein in response to co-oxidation of chloroform. Appl. Microbiol. Biotechnol. 2009, 82, 1179–1185. [Google Scholar]
- Bumann, D.; Valdivia, R.H. Identification of host-induced pathogen genes by differential fluorescence induction reporter systems. Nat. Protocols 2007, 2, 770–777. [Google Scholar]
- Yotter, R.A.; Warren, M.R.; Wilson, D.M. Optimized CMOS photodetector structures for the detection of green luminescent probes in biological applications. Sens. Actuat. B-Chem. 2004, 103, 43–49. [Google Scholar]
- Wells, M. Advances in optical detection strategies for reporter signal measurements. Curr. Opin. Biotechnol. 2006, 17, 28–33. [Google Scholar]
- Daunert, S.; Barrett, G.; Feliciano, J.S.; Shetty, R.S.; Shrestha, S.; Smith-Spencer, W. Genetically engineered whole-cell sensing systems: coupling biological recognition with reporter genes. Chem. Rev. 2000, 100, 2705–2738. [Google Scholar]
- Leveau, J.H.J.; Lindow, S.E. Bioreporters in microbial ecology. Curr. Opin. Microbiol. 2002, 5, 259–265. [Google Scholar]
- Shu, X.K.; Royant, A.; Lin, M.Z.; Aguilera, T.A.; Lev-Ram, V.; Steinbach, P.A.; Tsien, R.Y. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science 2009, 324, 804–807. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Luciferase (Lux) | Green Fluorescent Protein (GFP) | ||
|---|---|---|---|
| V. fischeri | [16] | E. coli | [17] |
| Various toxins | Various toxins | ||
| Bacteriophage | [18] | E. coli | [19] |
| Pathogenic Bacteria | L-arabinose | ||
| Saccharomyces cerevisiae | [20] | Yeast | [21] |
| Androgenic compounds | DNA damage | ||
| E. coli | [22] | E. coli | [23] |
| Nalidixic Acid | Arsenic | ||
| E. coli | [24] | Bacillus sp. | [14] |
| Various stressors | Arsenic/Zinc | ||
| HEK293 mammalian cell line | [25] | Firefly Luciferase (Luc) | |
| Whole animal imaging | E. coli | [26] | |
| Gene expression | |||
| E. coli | [27] | ||
| Polycyclic aromatic hydrocarbons | E. coli | [28] | |
| Benzene | |||
| Alcaligenes eutrophus | [29] | ||
| Heavy metals | E. coli | [30] | |
| Various toxins | |||
| Pseudomonas putida | [31] | ||
| Microbial volatile organics | Aequorin | ||
| E. coli | [32] | ||
| Pseudomonas fluorescens | [33] | Calcium | |
| Naphthalene/salicylate | |||
| B Cells | [34] | ||
| E. coli | [35] | Pathogenic Bacteria | |
| DNA damaging agents | |||
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Close, D.M.; Ripp, S.; Sayler, G.S. Reporter Proteins in Whole-Cell Optical Bioreporter Detection Systems, Biosensor Integrations, and Biosensing Applications. Sensors 2009, 9, 9147-9174. https://doi.org/10.3390/s91109147
Close DM, Ripp S, Sayler GS. Reporter Proteins in Whole-Cell Optical Bioreporter Detection Systems, Biosensor Integrations, and Biosensing Applications. Sensors. 2009; 9(11):9147-9174. https://doi.org/10.3390/s91109147
Chicago/Turabian StyleClose, Dan M., Steven Ripp, and Gary S. Sayler. 2009. "Reporter Proteins in Whole-Cell Optical Bioreporter Detection Systems, Biosensor Integrations, and Biosensing Applications" Sensors 9, no. 11: 9147-9174. https://doi.org/10.3390/s91109147
APA StyleClose, D. M., Ripp, S., & Sayler, G. S. (2009). Reporter Proteins in Whole-Cell Optical Bioreporter Detection Systems, Biosensor Integrations, and Biosensing Applications. Sensors, 9(11), 9147-9174. https://doi.org/10.3390/s91109147