Silicon Wafer-Based Platinum Microelectrode Array Biosensor for Near Real-Time Measurement of Glutamate in Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Reagents
2.2. Instrumentation
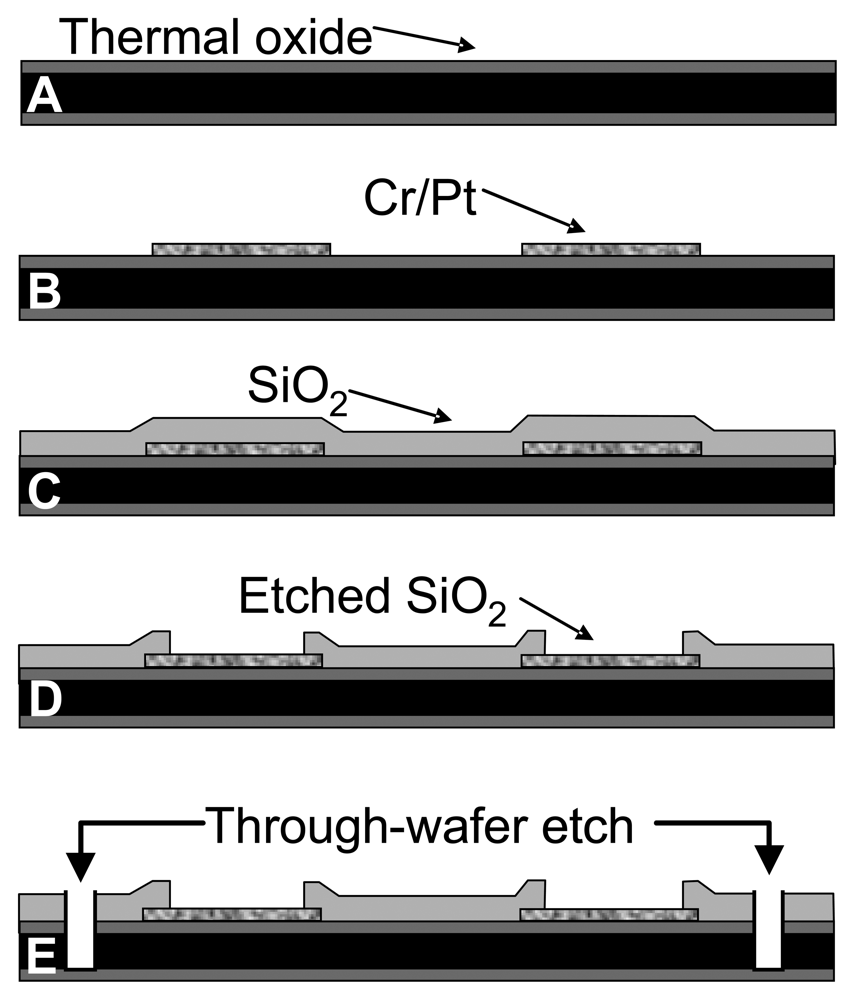
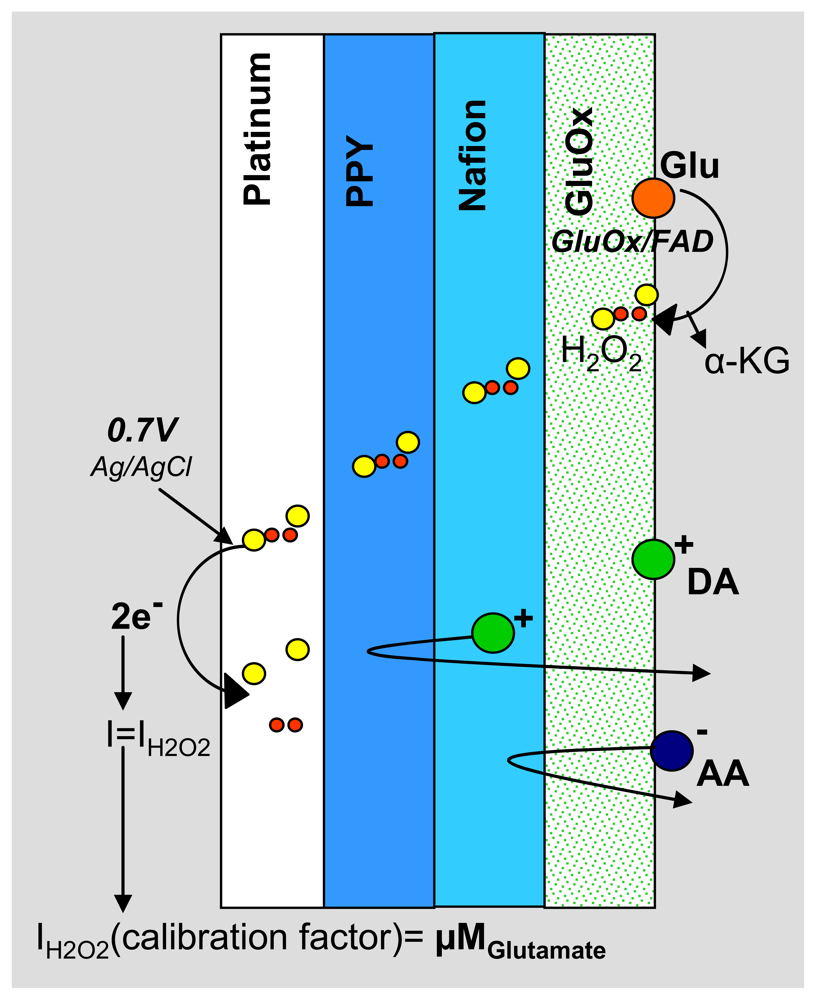
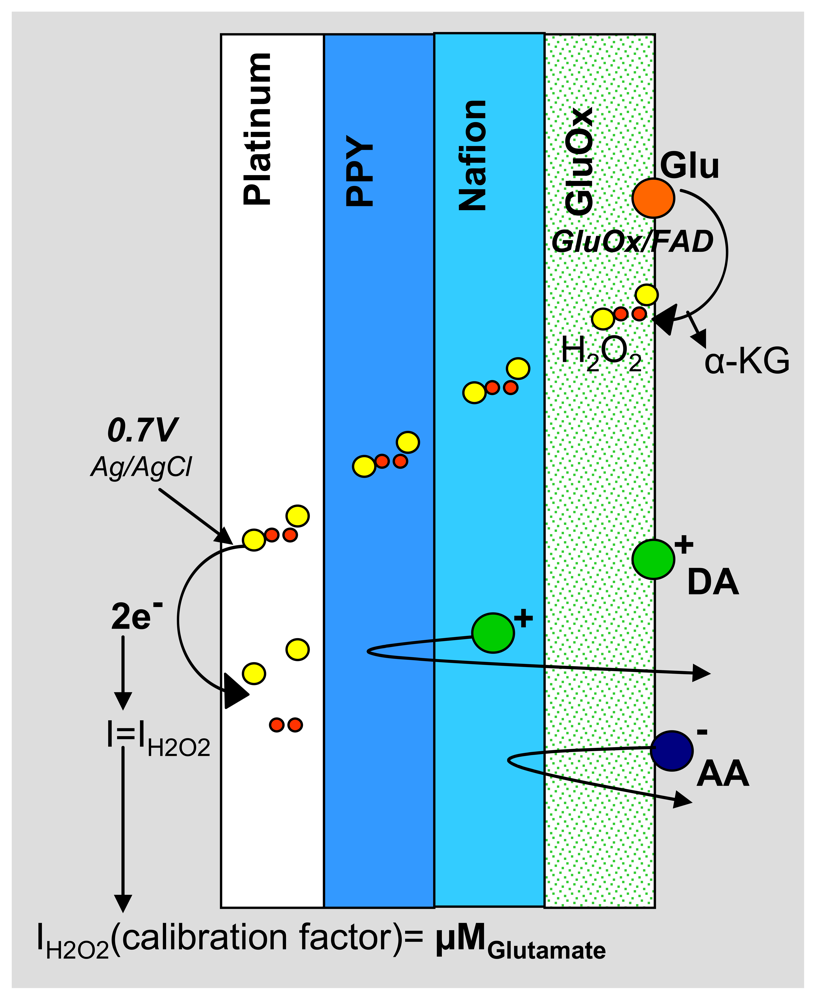
2.3. Electrode Fabrication and Polymer Modification
2.4. Electrode Characterization and Data Analysis
2.5. In Vivo Electrode Characterization and Data Analysis
3. Results and Discussion
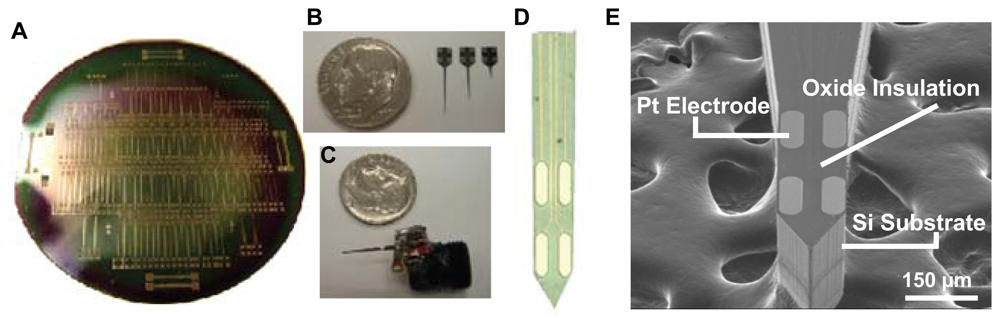
3.1. Silicon Wafer-Based Platinum Microelectrode Array Glutamate Biosensors
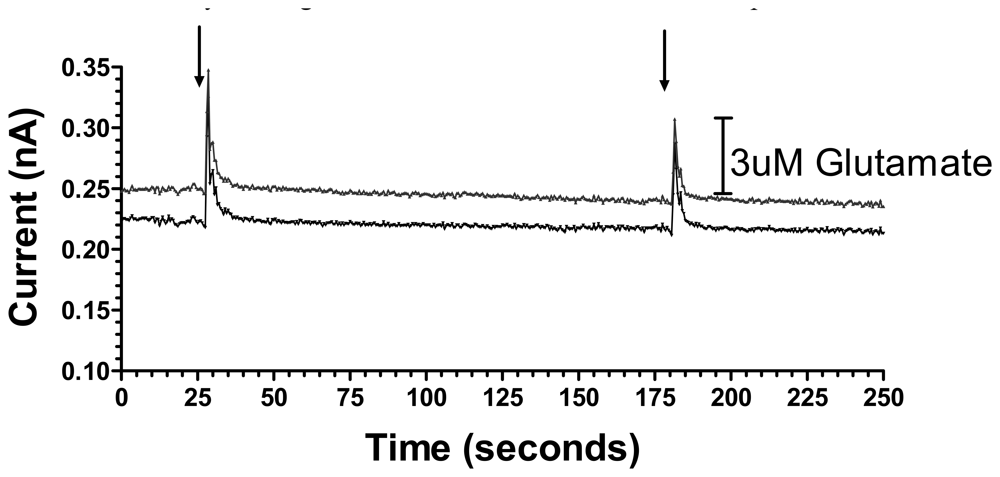
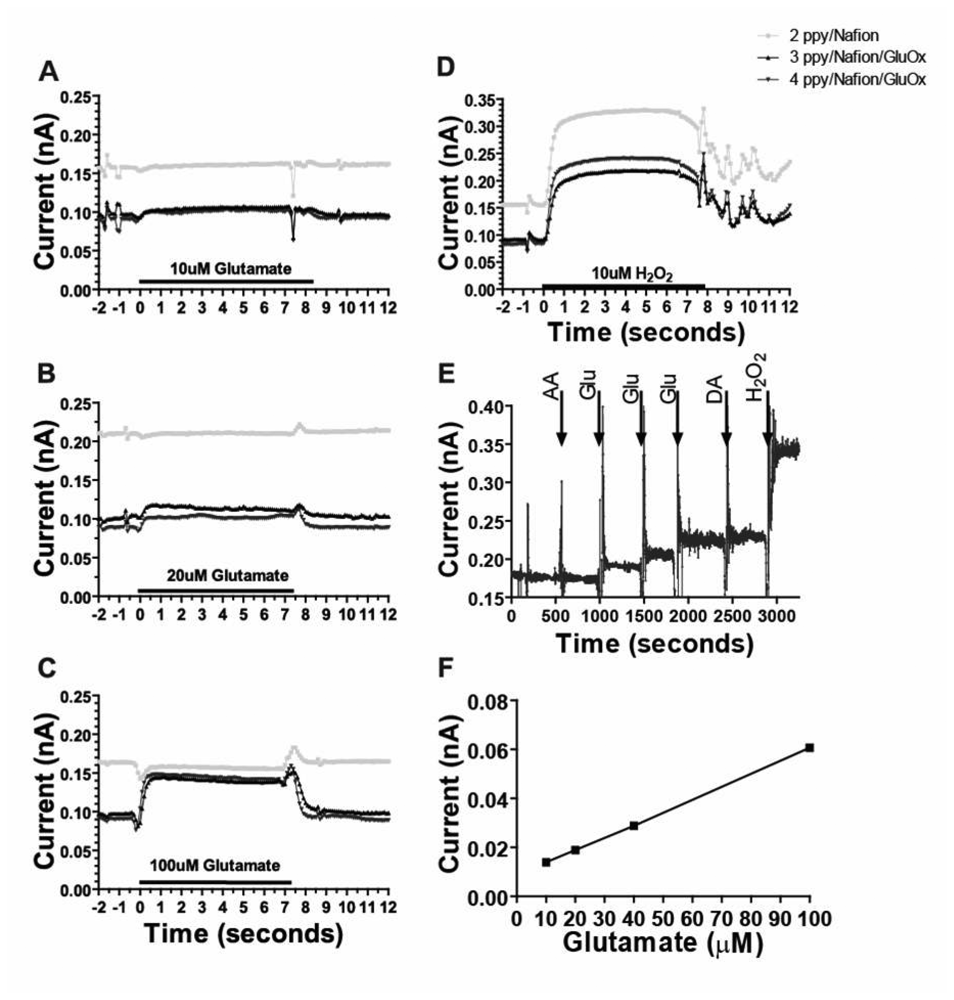
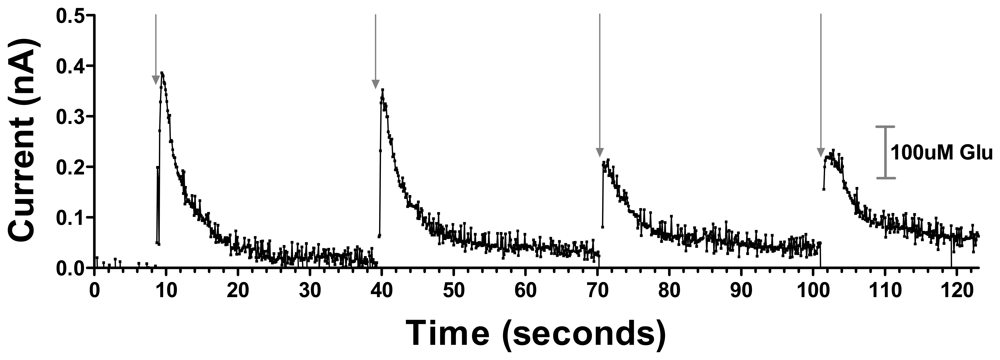
3.2. Sensitive, Selective and Rapid Detection of Glutamate In Vitro
3.3. In Vivo Cortical Stimulation-Evoked Glutamate Release in the Ventral Striatum
3.4. Stress-Induced Striatal Glutamate Release in Awake Freely Moving Rat
4. Conclusions
Acknowledgments
References
- Gass, J.T.; Olive, M.F. Glutamatergic substrates of drug addiction and alcoholism. Biochem. Pharmacol. 2008, 75, 218–265. [Google Scholar]
- Blandini, F.; Porter, R.H.; Greenamyre, J.T. Glutamate and Parkinson's disease. Mol. Neurobiol. 1996, 12, 73–94. [Google Scholar]
- Greenamyre, J.T.; Porter, R.H. Anatomy and physiology of glutamate in the CNS. Neurology 1994, 44, S7–13. [Google Scholar]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar]
- Bordi, F.; Ugolini, A. Group I metabotropic glutamate receptors: implications for brain diseases. Prog. Neurobiol. 1999, 59, 55–79. [Google Scholar]
- Drew, K.L.; Pehek, E.A.; Rasley, B.T.; Ma, Y.L.; Green, T.K. Sampling glutamate and GABA with microdialysis: suggestions on how to get the dialysis membrane closer to the synapse. J. Neurosci. Methods 2004, 140, 127–131. [Google Scholar]
- Phillips, P.E.; Stuber, G.D.; Heien, M.L.; Wightman, R.M.; Carelli, R.M. Subsecond dopamine release promotes cocaine seeking. Nature 2003, 422, 614–618. [Google Scholar]
- Robinson, D.L.; Heien, M.L.; Wightman, R.M. Frequency of dopamine concentration transients increases in dorsal and ventral striatum of male rats during introduction of conspecifics. J. Neurosci. 2002, 22, 10477–10486. [Google Scholar]
- Robinson, D.L.; Phillips, P.E.; Budygin, E.A.; Trafton, B.J.; Garris, P.A.; Wightman, R.M. Sub-second changes in accumbal dopamine during sexual behavior in male rats. Neuroreport 2001, 12, 2549–2552. [Google Scholar]
- Heien, M.L.; Khan, A. S.; Ariansen, J.L.; Cheer, J.F.; Phillips, P.E.; Wassum, K.M.; Wightman, R.M. Real-time measurement of dopamine fluctuations after cocaine in the brain of behaving rats. Proc. Natl. Acad. Sci. USA 2005, 102, 10023–10028. [Google Scholar]
- Pomerleau, F.; Day, B.K.; Huettl, P.; Burmeister, J.J.; Gerhardt, G. A. Real time in vivo measures of L-glutamate in the rat central nervous system using ceramic-based multisite microelectrode arrays. Ann. N. Y. Acad. Sci. 2003, 1003, 454–457. [Google Scholar]
- Lowry, J.P.; Ryan, M.R.; O'Neill, R.D. Behaviourally induced changes in extracellular levels of brain glutamate monitored at 1s resolution with an implanted biosensor. Anal. Commun. 1998, 35, 87–89. [Google Scholar]
- Hamdi, N.W.J.; Walker, E.; Maidment, N.T.; Monbouquette, H.G. An electroenzymatic L-glutamate microbiosensor selective against dopamine. J. Electroanal. Chem. 2006, 591, 33–40. [Google Scholar]
- Kusakabe, H.; M., Y.; Fujishima, T.; Kuninaka, A.; Yoshino, H. Purification and properties of a new enzyme, L-glutamate oxidase, from Streptomyces sp. X-119-6 grown on wheat bran. Agr. Biol. Chem. 1983, 47, 1323–1328. [Google Scholar]
- Ryan, M.R.; Lowry, J.P.; O'Neill, R.D. Biosensor for neurotransmitter L-glutamic acid designed for efficient use of L-glutamate oxidase and effective rejection of interference. Analyst 1997, 122, 1419–1424. [Google Scholar]
- Hu, Y.; Mitchell, K.M.; Albahadily, F.N.; Michaelis, E.K.; Wilson, G.S. Direct measurement of glutamate release in the brain using a dual enzyme-based electrochemical sensor. Brain Res. 1994, 659, 117–125. [Google Scholar]
- Alvarez-Crespo, S.L.; Lobo-Castanon, M.J.; Miranda-Ordieres, A.J.; Tunon-Blanco, P. Amperometric glutamate biosensor based on poly(o-phenylenediamine) film electrogenerated onto modified carbon paste electrodes. Biosens. Bioelectron. 1997, 12, 739–747. [Google Scholar]
- Burmeister, J.J.; Gerhardt, G.A. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal. Chem. 2001, 73, 1037–1042. [Google Scholar]
- Garguilo, M.G.; Huynh, N.; Proctor, A.; Michael, A.C. Amperometric sensors for peroxide, choline, and acetylcholine based on electron transfer between horseradish peroxidase and a redox polymer. Anal. Chem. 1993, 65, 523–528. [Google Scholar]
- Kelley, A.E. Functional specificity of ventral striatal compartments in appetitive behaviors. Ann. N. Y. Acad. Sci. 1999, 877, 71–90. [Google Scholar]
- Lu, Y.; Peters, J.L.; Michael, A.C. Direct comparison of the response of voltammetry and microdialysis to electrically evoked release of striatal dopamine. J. Neurochem. 1998, 70, 584–593. [Google Scholar]
- Floresco, S.B.; McLaughlin, R.J.; Haluk, D.M. Opposing roles for the nucleus accumbens core and shell in cue-induced reinstatement of food-seeking behavior. J. Neurosci. 2008. [Google Scholar]
- Burmeister, J.J.; Pomerleau, F.; Palmer, M.; Day, B.K.; Huettl, P.; Gerhardt, G.A. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J. Neurosci. Methods 2002, 119, 163–171. [Google Scholar]
- Walker, E.; Wang, J.; Hamdi, N.; Monbouquette, H.G.; Maidment, N.T. Selective detection of extracellular glutamate in brain tissue using microelectrode arrays coated with over-oxidized polypyrrole. Analyst 2007, 132, 1107–1111. [Google Scholar]
- McMahon, C.P.; Rocchitta, G.; Serra, P.A.; Kirwan, S.M.; Lowry, J.P.; O'Neill, R.D. Control of the oxygen dependence of an implantable polymer/enzyme composite biosensor for glutamate. Anal. Chem. 2006, 78, 2352–2359. [Google Scholar]
- McMahon, C.P.; Rocchitta, G.; Kirwan, S.M.; Killoran, S.J.; Serra, P.A.; Lowry, J.P.; O'Neill, R.D. Oxygen tolerance of an implantable polymer/enzyme composite glutamate biosensor displaying polycation-enhanced substrate sensitivity. Biosens. Bioelectron. 2007, 22, 1466–1473. [Google Scholar]
- You, Z.B.; Tzschentke, T.M.; Brodin, E.; Wise, R.A. Electrical stimulation of the prefrontal cortex increases cholecystokinin, glutamate, and dopamine release in the nucleus accumbens: an in vivo microdialysis study in freely moving rats. J. Neurosci. 1998, 18, 6492–500. [Google Scholar]
- McFarland, K.; Lapish, C.C.; Kalivas, P.W. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J. Neurosci. 2003, 23, 3531–3537. [Google Scholar]
- LaLumiere, R.T.; Kalivas, P.W. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J. Neurosci. 2008, 28, 3170–3177. [Google Scholar]
- Rutherford, E.C.; Pomerleau, F.; Huettl, P.; Stromberg, I.; Gerhardt, G.A. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J. Neurochem. 2007, 102, 712–722. [Google Scholar]
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wassum, K.M.; Tolosa, V.M.; Wang, J.; Walker, E.; Monbouquette, H.G.; Maidment, N.T. Silicon Wafer-Based Platinum Microelectrode Array Biosensor for Near Real-Time Measurement of Glutamate in Vivo. Sensors 2008, 8, 5023-5036. https://doi.org/10.3390/s8085023
Wassum KM, Tolosa VM, Wang J, Walker E, Monbouquette HG, Maidment NT. Silicon Wafer-Based Platinum Microelectrode Array Biosensor for Near Real-Time Measurement of Glutamate in Vivo. Sensors. 2008; 8(8):5023-5036. https://doi.org/10.3390/s8085023
Chicago/Turabian StyleWassum, Kate M., Vanessa M. Tolosa, Jianjun Wang, Eric Walker, Harold G. Monbouquette, and Nigel T. Maidment. 2008. "Silicon Wafer-Based Platinum Microelectrode Array Biosensor for Near Real-Time Measurement of Glutamate in Vivo" Sensors 8, no. 8: 5023-5036. https://doi.org/10.3390/s8085023