Integrated Electrochemical Analysis System with Microfluidic and Sensing Functions

Abstract
:1. Introduction
2. Experimental
2.1. Reagents and materials
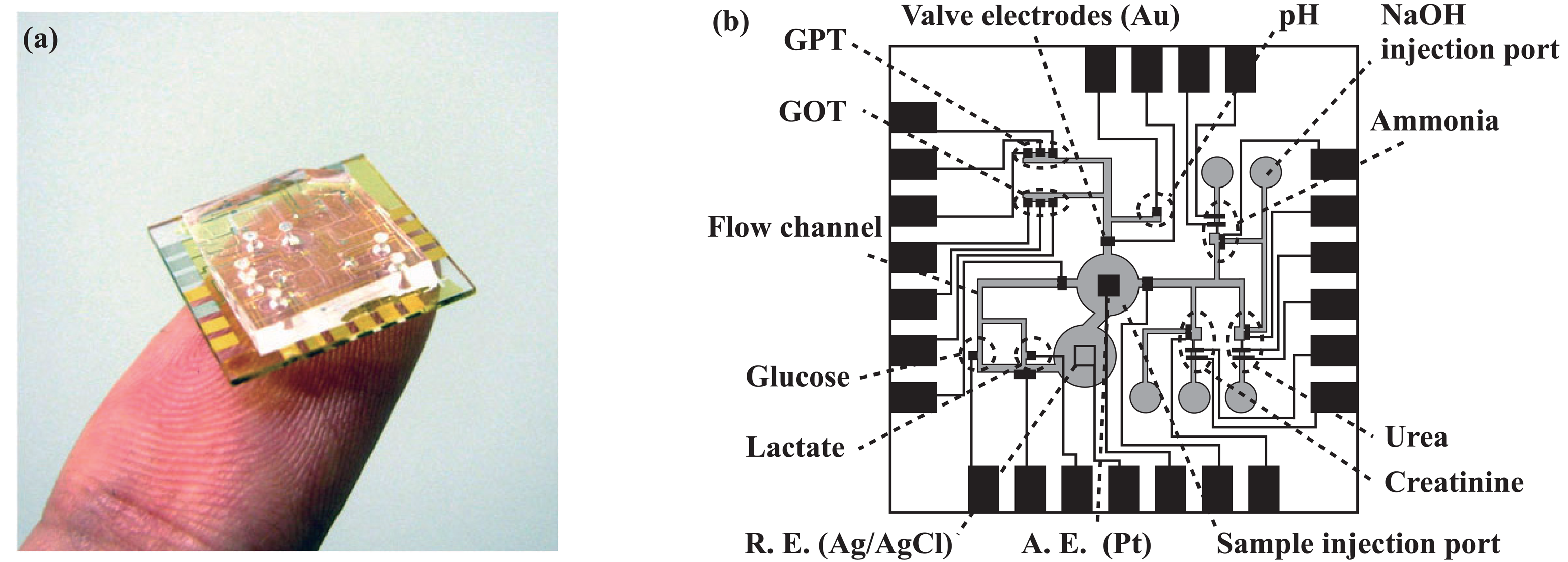
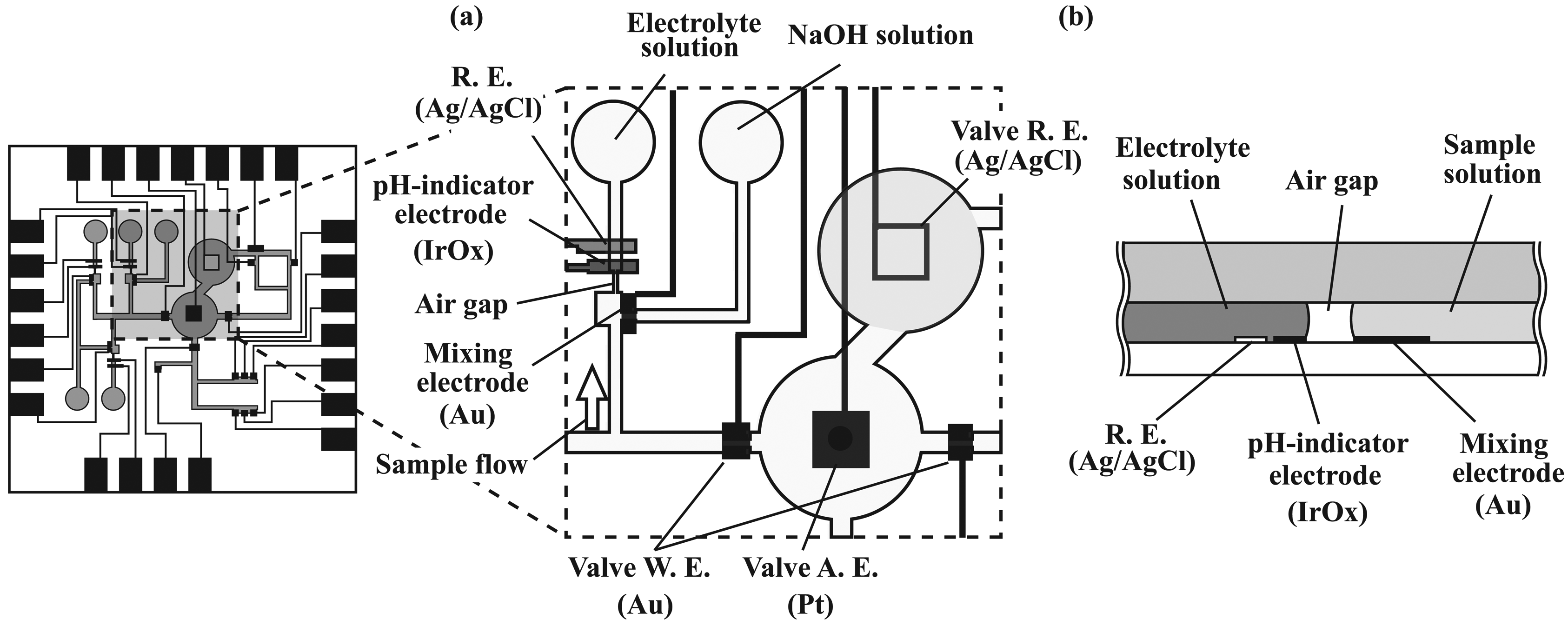
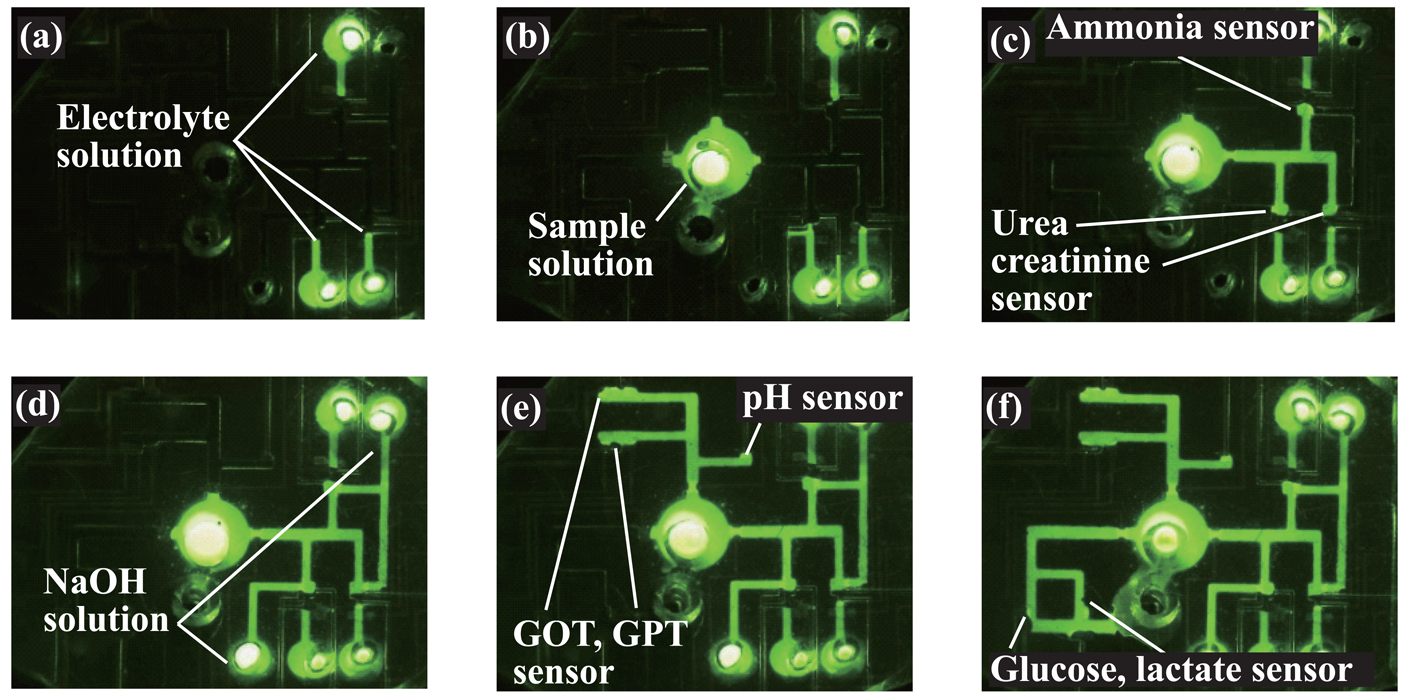
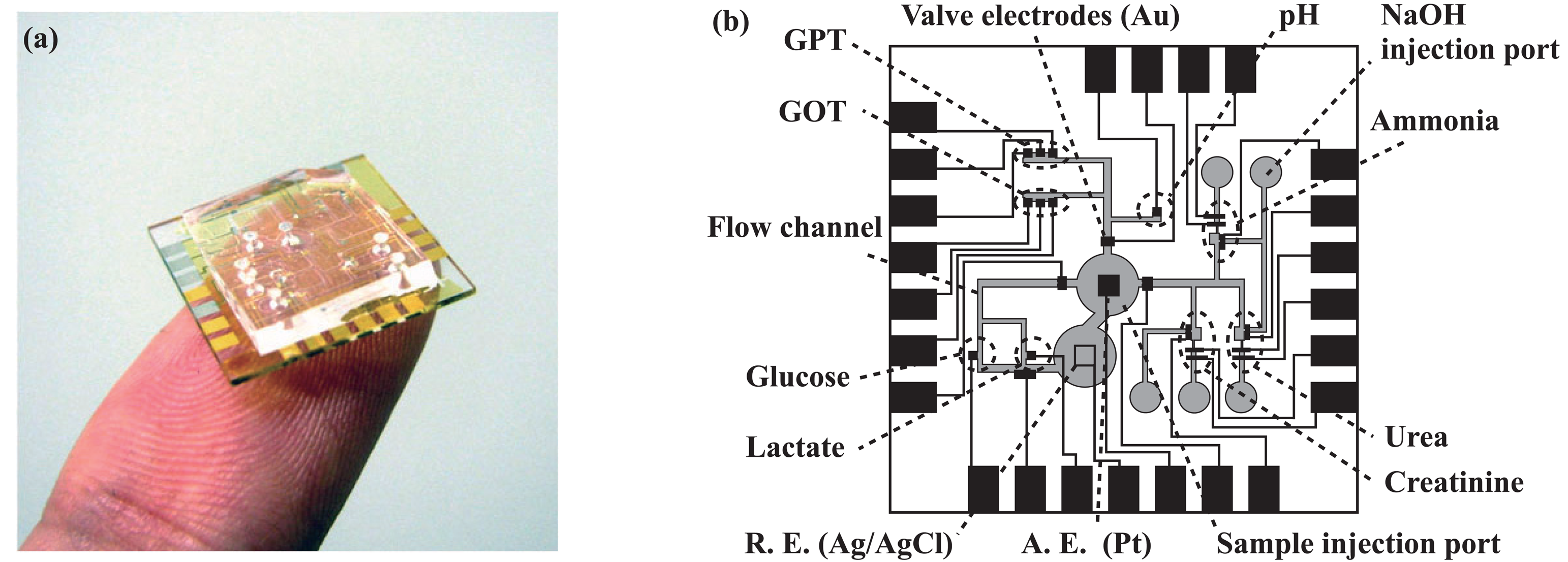
2.2. Overall structure of the microsystem
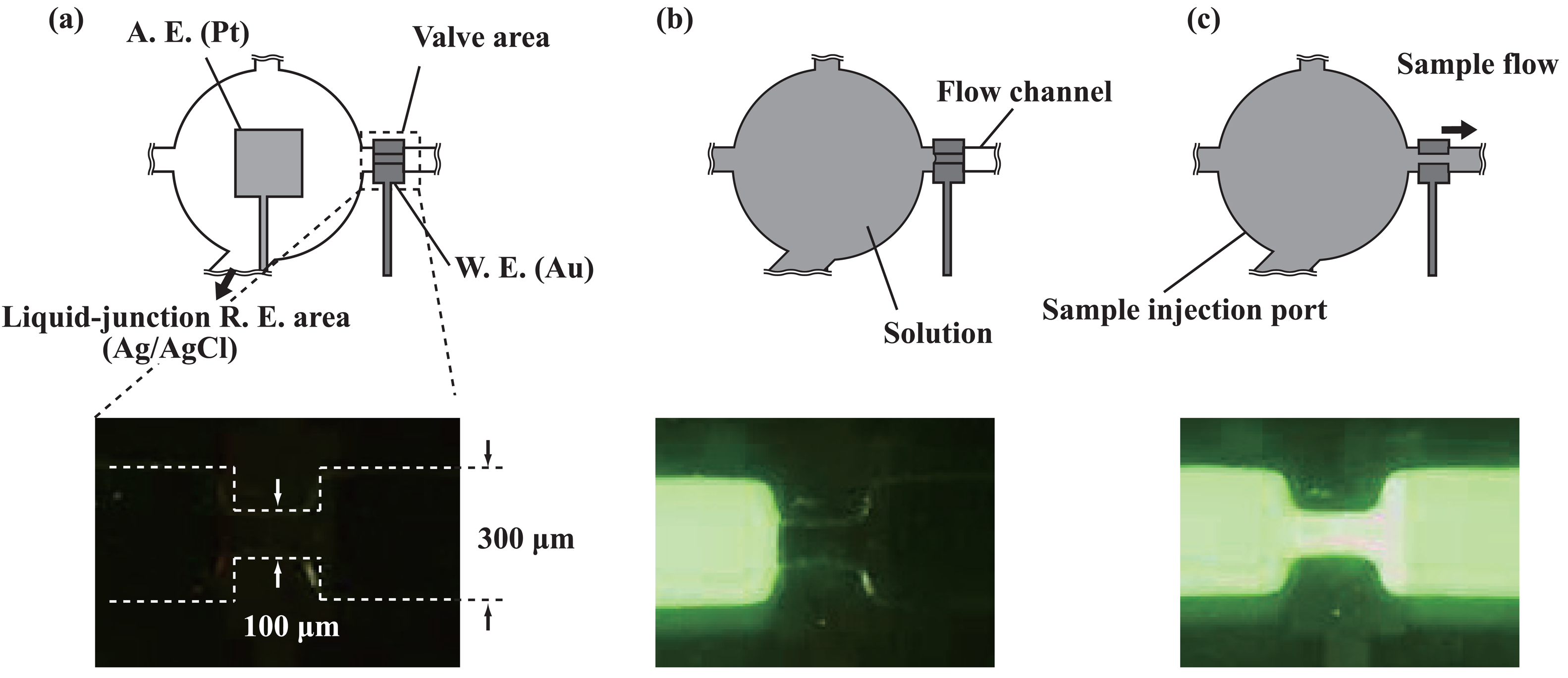
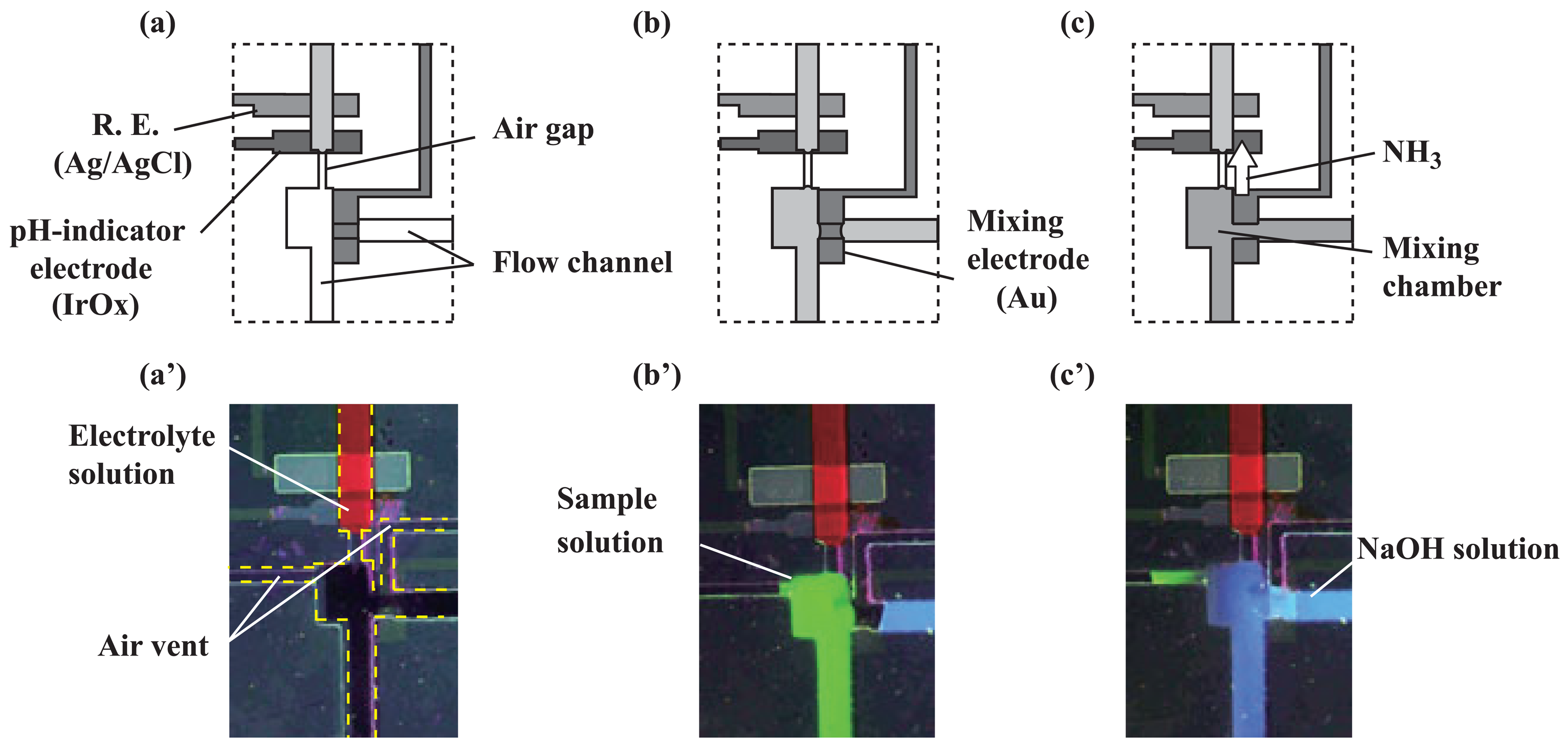
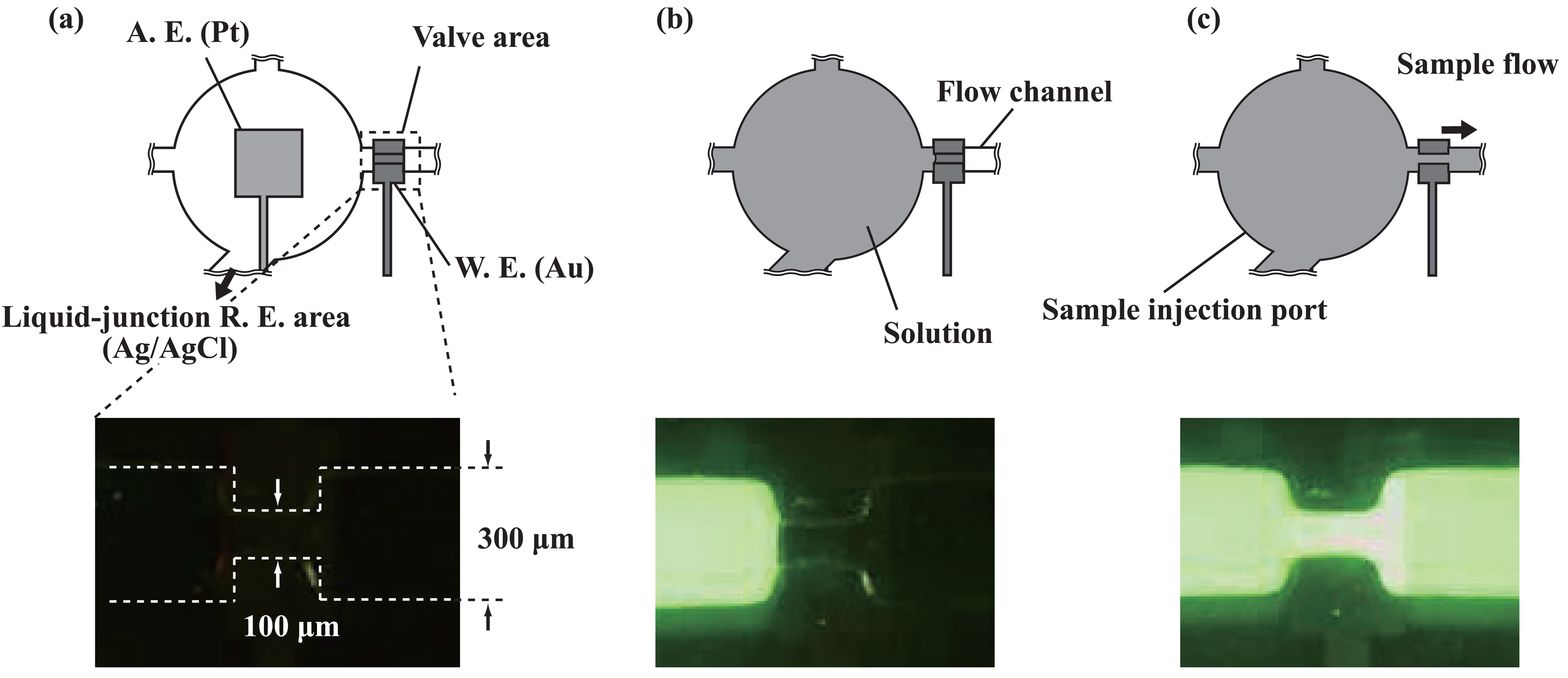
2.3. Transport of solutions
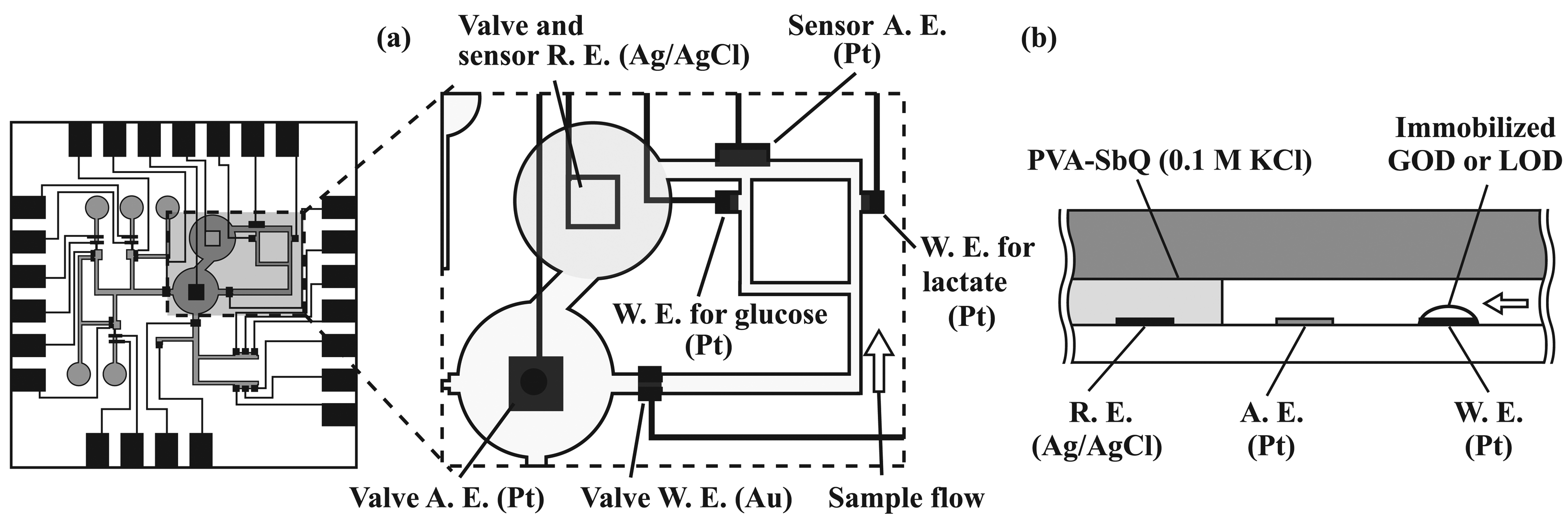
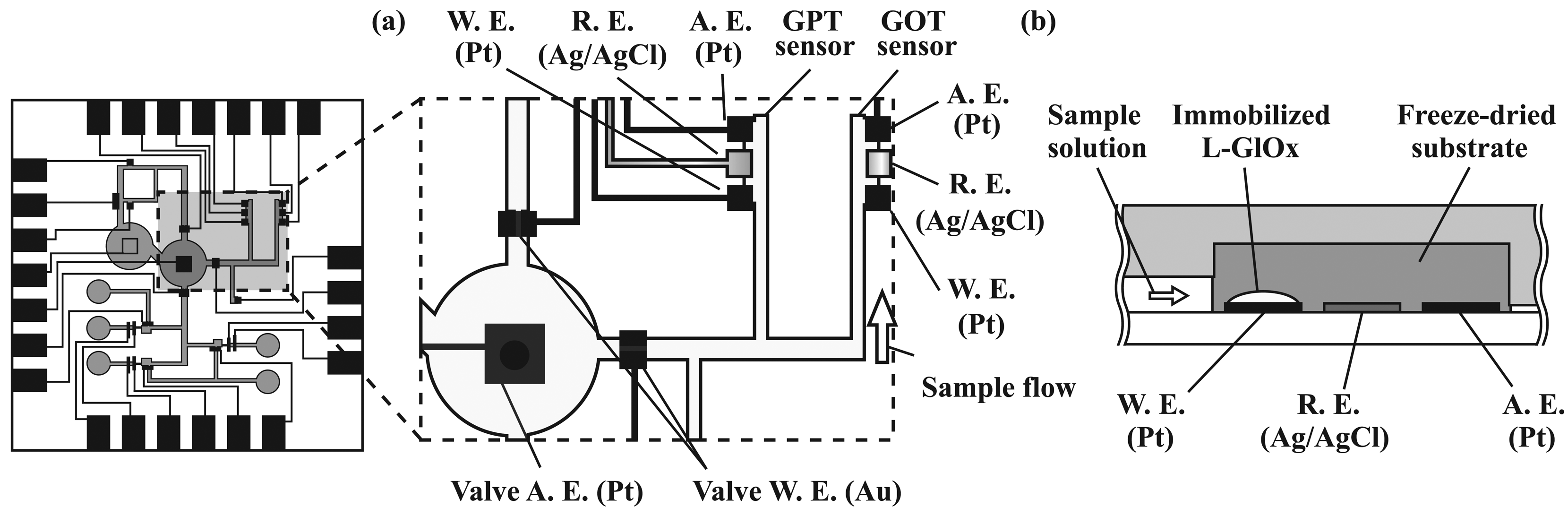
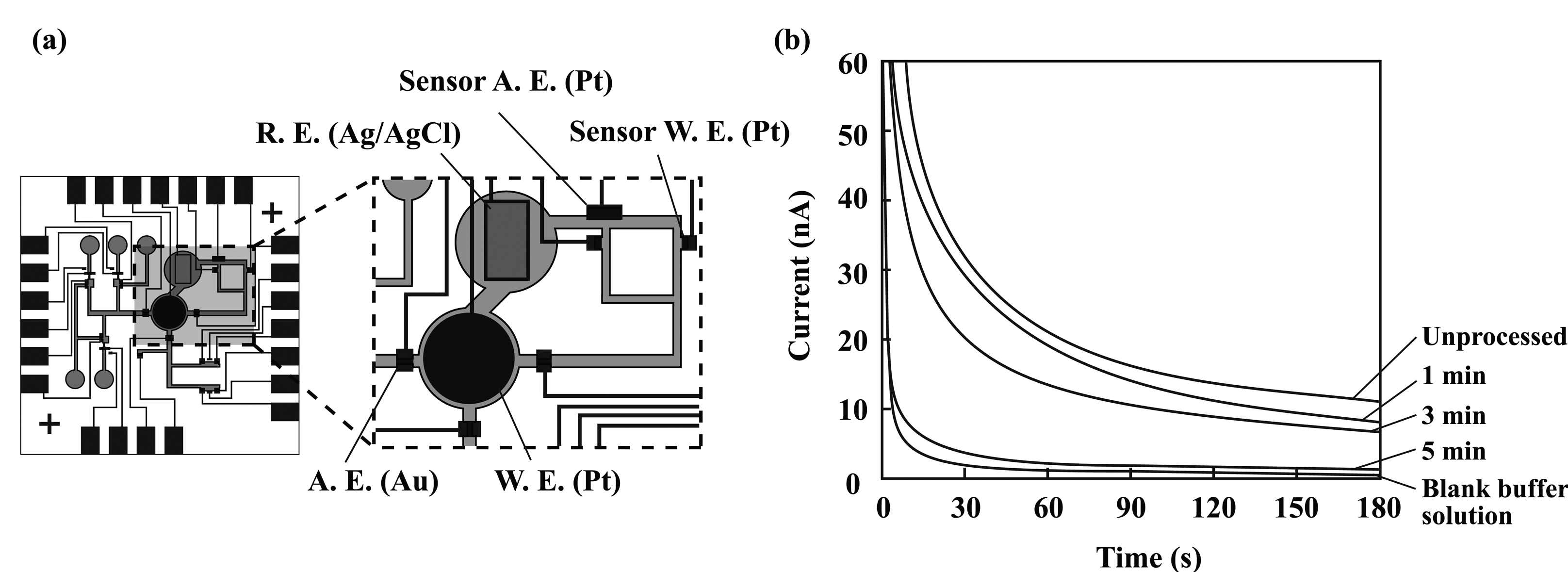
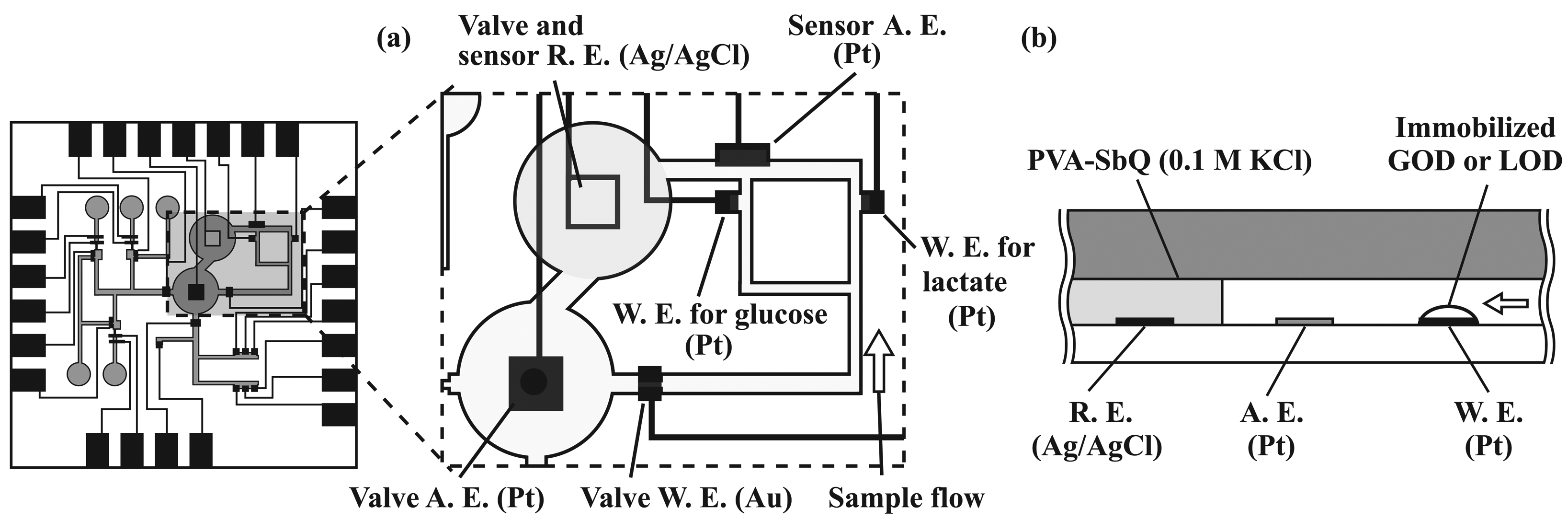
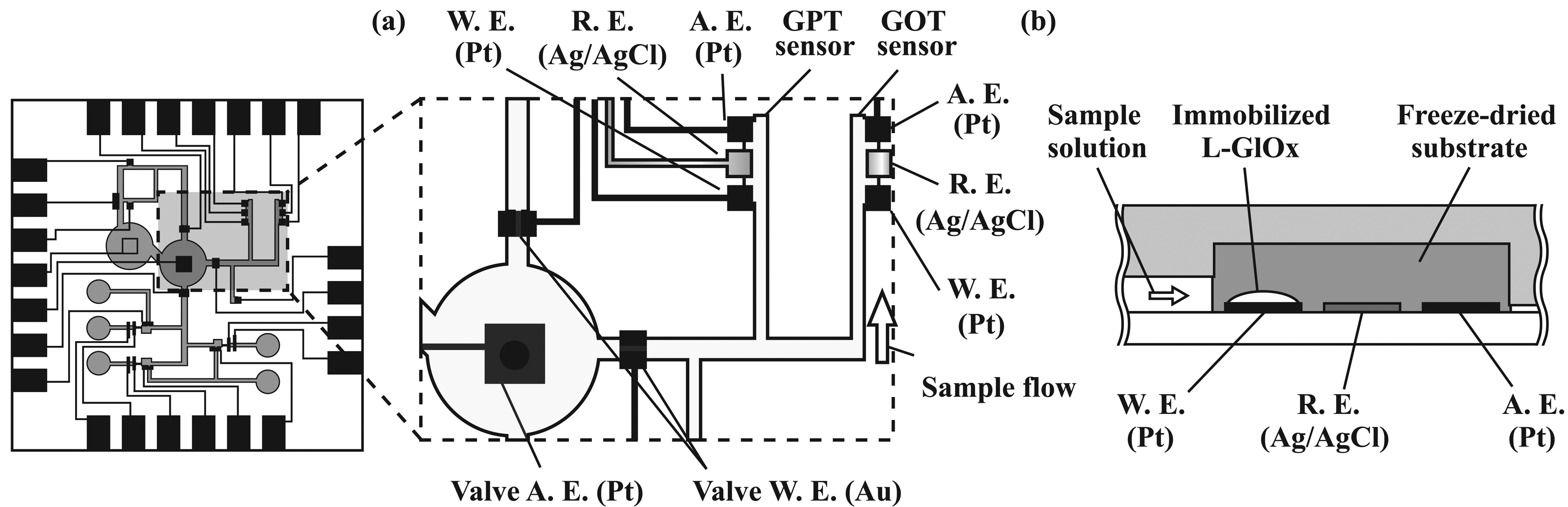
2.4. Formation of amperometric sensors
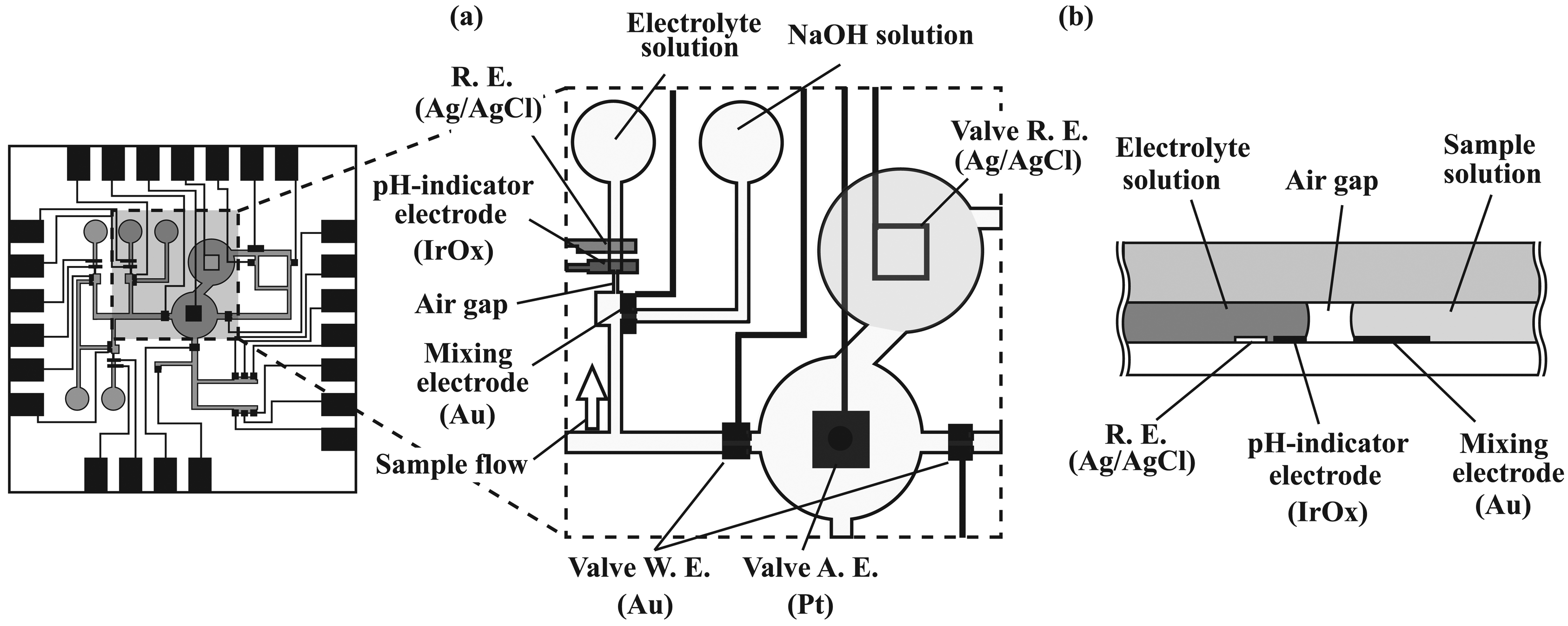
2.5. Formation of potentiometric sensors
2.6. Immobilization of enzymes
2.7. Procedures and instruments
3. Results and discussion
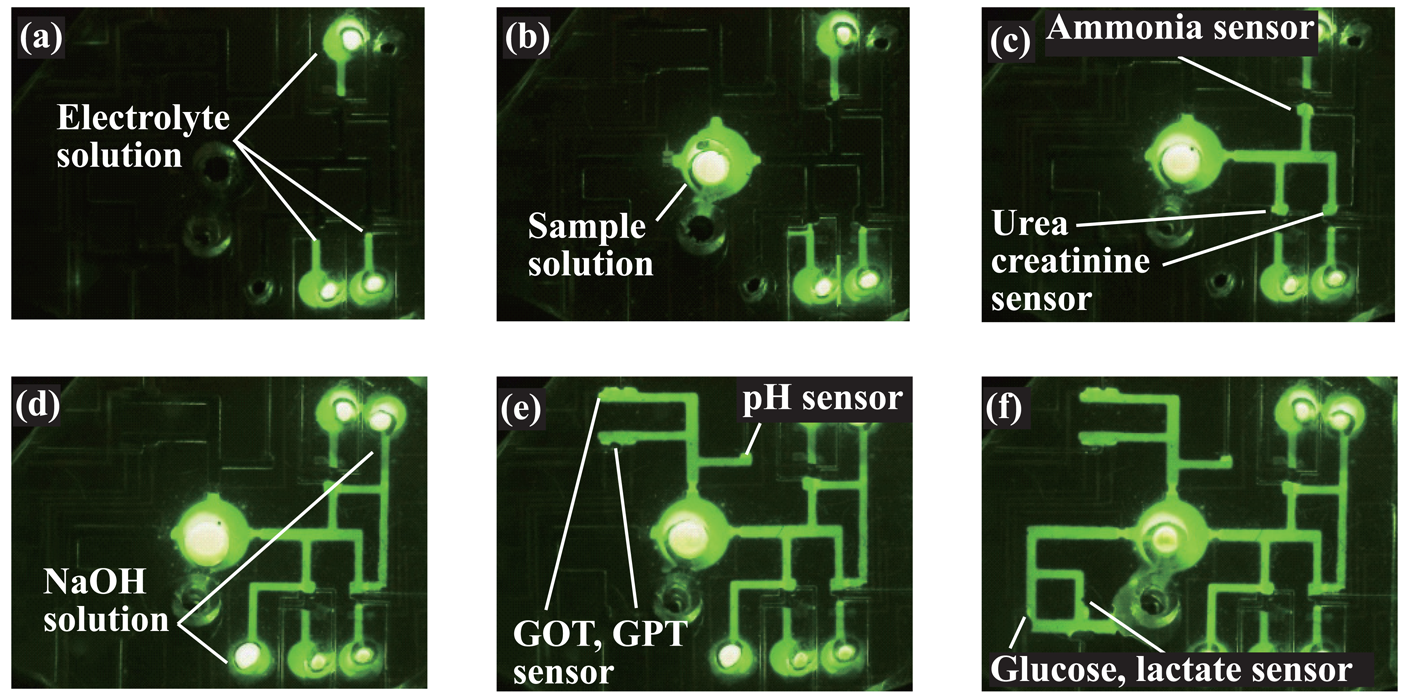
3.1. Microfluidic transport
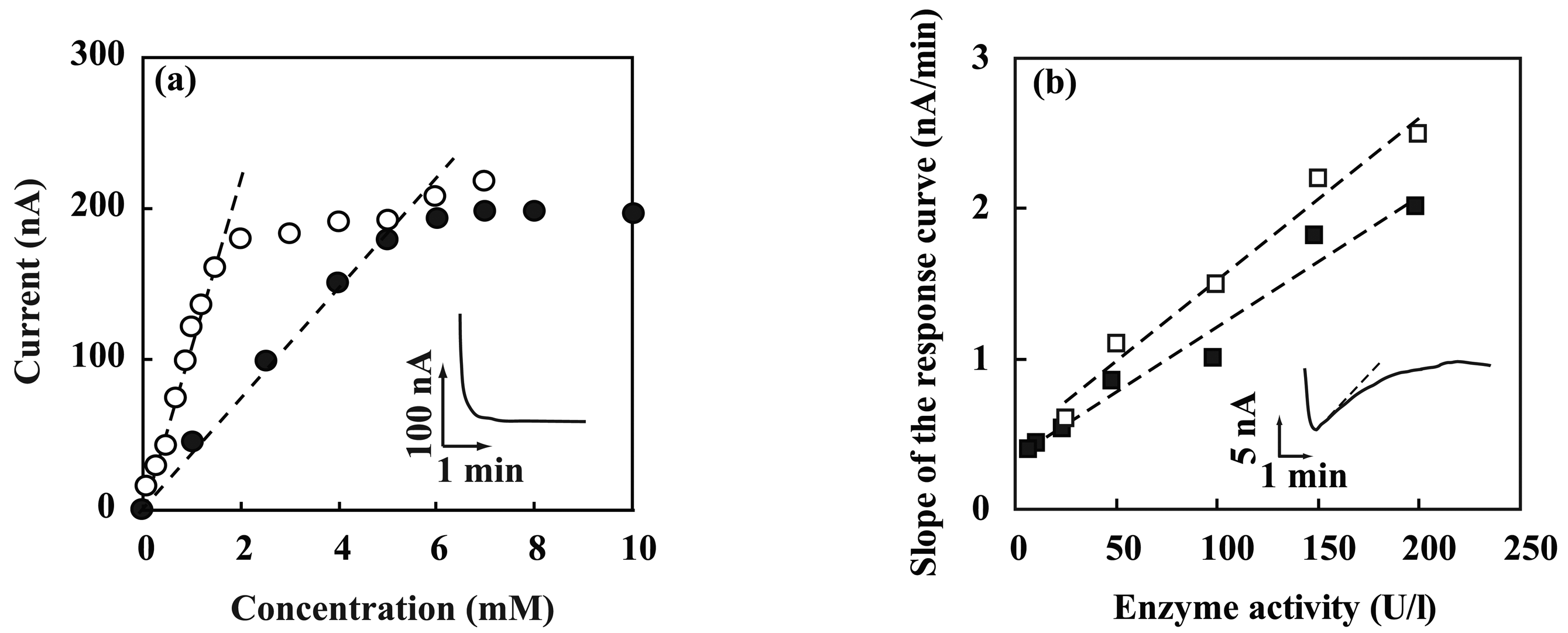
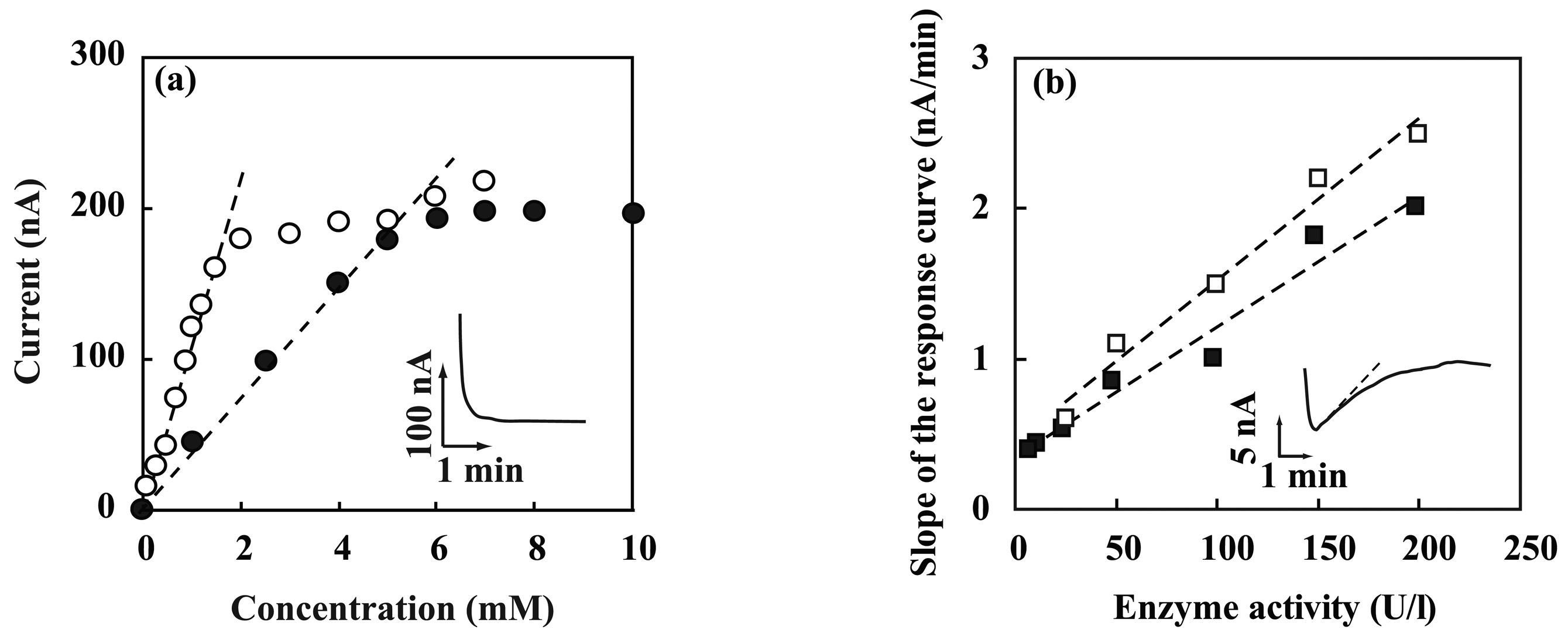
3.2. Performance characterization of amperometric sensors
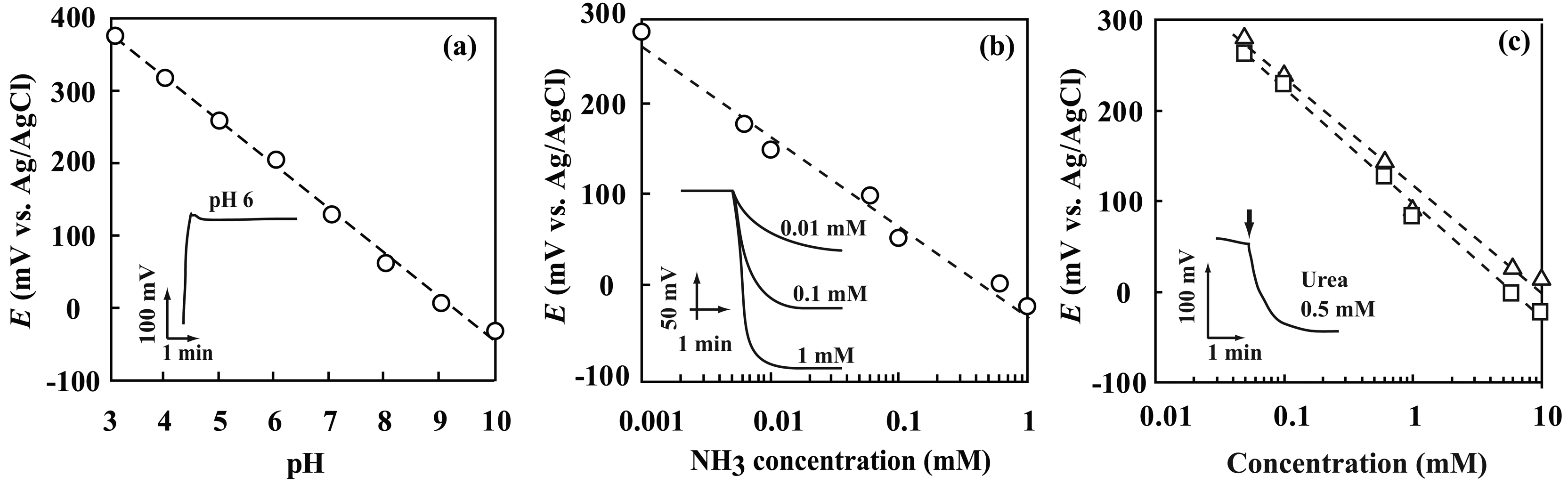
3.3. Performance characterization of potentiometric sensors
3.4. Comparison between the on-chip measurement and the conventional method
3.5. Preprocessing in the sample injection port
Conclusions
Acknowledgments
References and Notes
- Song, H.; Chen, D.L.; Ismagilov, R. F. Reactions in droplets in microfluidic channels. Angew. Chem. Int. Ed. 2006, 45, 7336–7356. [Google Scholar]
- Delamarche, E.; Juncker, D.; Schmid, H. Microfluidics for processing surfaces and miniaturizing biological assays. Adv. Mater. 2005, 17, 2911–2933. [Google Scholar]
- Suzuki, H. Advances in the microfabrication of electrochemical sensors and systems. Electroanalysis 2000, 12, 703–715. [Google Scholar]
- Suzuki, H.; Upadhyay, S.; Loughran, M. Microfabrication of electrochemical sensors. In Encyclopedia of Sensors; Grimes, C. A., Dickey, E. C., Pishko, M. V., Eds.; American Scientific Publishers: Stevenson Ranch, 2006; Volume 6, pp. 161–187. [Google Scholar]
- Gravesen, P.; Branebjerg, J.; Jensen, O.S. Microfluidics - a review. J. Micromech. Microeng. 1993, 3, 168–182. [Google Scholar]
- Shoji, S.; Esashi, M. Microflow devices and systems. J. Micromech. Microeng. 1994, 4, 157–171. [Google Scholar]
- Laser, D.J.; Santiago, J. G. A review of micropumps. J. Micromech. Microeng. 2004, 14, R35–R64. [Google Scholar]
- Woias, P. Micropumps - past, progress and future prospects. Sensor. Actuat. B 2005, 105, 28–38. [Google Scholar]
- Bockris, J. O. M.; Reddy, A. K. N.; Gamboa-Aldeco, M. Modern Electrochemistry; Kluwer Academic/Plenum Publishers: New York, 2000. [Google Scholar]
- Quilliet, C.; Berge, B. Electrowetting: a recent outbreak. Curr. Opin. Colloid Interface Sci. 2001, 6, 34–39. [Google Scholar]
- Mugele, F.; Baret, J.-C. Electrowetting: from basics to applications. J. Phys.: Condens. Matter 2005, 17, R705–774. [Google Scholar]
- Cheng, J.-Y.; Hsiung, L.-C. Electrowetting (EW)-based valve combined with hydrophilic Teflon microfluidic guidance in controlling continuous fluid flow. Biomed. Microdevices 2004, 6, 341–347. [Google Scholar]
- Satoh, W.; Loughran, M.; Suzuki, H. Microfluidic transport based on direct electrowetting. J. Appl. Phys. 2004, 96, 835–841. [Google Scholar]
- Shiu, J.-Y.; Chen, P. Active patterning using an addressable microfluidic network. Adv. Mater. 2005, 17, 1866–1869. [Google Scholar]
- Satoh, W.; Hosono, H.; Suzuki, H. On-chip microfluidic transport and mixing using electrowetting and incorporation of sensing functions. Anal. Chem. 2005, 77, 6857–6863. [Google Scholar]
- Morimoto, K.; Suzuki, H. Micro analysis system for pH and protease activities with an integrated sample injection mechanism. Biosens. Bioelectron. 2006, 22, 86–93. [Google Scholar]
- Nashida, N.; Satoh, W.; Fukuda, J.; Suzuki, H. Electrochemical immunoassay on a microfluidic device with sequential injection and flushing functions. Biosens. Bioelectron. 2007, 22, 3167–3173. [Google Scholar]
- Srinivasan, V.; Pamula, V.K.; Fair, R. B. An integrated digital microfluidic lab-on-a-chip for clinical diagnosis on human physiological fluids. Lab Chip 2004, 4, 310–315. [Google Scholar]
- Chung, K.H.; Hong, J. W.; Lee, D.-S.; Yoon, H. C. Microfluidic chip accomplishing self-fluid replacement using only capillary force and its bioanalytical application. Anal. Chim. Acta. 2007, 585, 1–10. [Google Scholar]
- Washburn, E.W. The dynamics of capillary flow. Phys. Rev. 1921, 17, 273–283. [Google Scholar]
- Kim, E.; Xia, Y.; Whitesides, G.M. Polymer microstructures formed by moulding in capillaries. Nature 1995, 376, 581–584. [Google Scholar]
- Delamarche, E.; Bernard, A.; Schmid, H.; Bietsch, A.; Michel, B.; Biebuyck, H. Microfluidic networks for chemical patterning of substrates: design and application to bioassays. J. Am. Chem. Soc. 1998, 120, 500–508. [Google Scholar]
- Janshoff, A.; Künneke, S. Micropatterned solid-supported membranes formed by micromolding in capillaries. Eur. Biophys. J. 2000, 29, 549–554. [Google Scholar]
- Satoh, W.; Yokomaku, H.; Hosono, H.; Ohnishi, N.; Suzuki, H. Electrowetting-based valve for the control of the capillary flow. J. Appl. Phys. 2008, 103, 034903. [Google Scholar]
- Morimoto, K.; Upadhyay, S.; Higashiyama, T.; Ohgami, N.; Kusakabe, H.; Fukuda, J.; Suzuki, H. Electrochemical microsystem with porous matrix packed-beds for enzyme analysis. Sensor. Actuat. B 2007, 124, 477–485. [Google Scholar]
- Suzuki, H.; Shiroishi, H.; Sasaki, S.; Karube, I. Microfabricated liquid junction Ag/AgCl reference electrode and its application to a one-chip potentiometric sensor. Anal. Chem. 1999, 71, 5069–5075. [Google Scholar]
- Suzuki, H.; Matsugi, Y. Microfabricated flow system for ammonia and creatinine with an air-gap structure. Sensor. Actuat. B 2004, 98, 101–111. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Enzyme | Reaction |
|---|---|---|
| Glucose | Glucose oxidase (GOD), EC 1.1.3.4, from a microorganism | |
| Lactate | Lactate oxidase (LOD), EC 1.1.3.2, from pediococcus species | |
| GOT | Glutamic oxaloacetic transaminase (GOT), EC 2.6.1.1, from a pig heart | |
| GPT | Glutamic pyruvic transaminase (GPT), EC 2.6.1.2, from a pig heart | |
| Urea | Urease, EC 3.5.1.5, from jack bean | |
| Creatinine | Creatinine deiminase, EC 3.5.4.21, from Bacillus sp. |
| On-chip measurement | Conventional method | Adjusted concentration | |
|---|---|---|---|
| Glucose | 5.3 mM | 5.6 mM | 6.0 mM |
| Lactate | 1.7 mM | 2.2 mM | 2.0 mM |
| GOT activity | 27 U/L | 32 U/L | 30 U/L |
| GPT activity | 27 U/L | 28 U/L | 30 U/L |
| pH | 7.3 | 7.4 | 7.4 |
| Ammonia | 58 μM | 61 μM | 50 μM |
| Urea | 6.4 mM | 6.9 mM | 7.0 mM |
| Creatinine | 89 μM | 94 μM | 100 μM |
© 2008 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Satoh, W.; Hosono, H.; Yokomaku, H.; Morimoto, K.; Upadhyay, S.; Suzuki, H. Integrated Electrochemical Analysis System with Microfluidic and Sensing Functions. Sensors 2008, 8, 1111-1127. https://doi.org/10.3390/s8021111
Satoh W, Hosono H, Yokomaku H, Morimoto K, Upadhyay S, Suzuki H. Integrated Electrochemical Analysis System with Microfluidic and Sensing Functions. Sensors. 2008; 8(2):1111-1127. https://doi.org/10.3390/s8021111
Chicago/Turabian StyleSatoh, Wataru, Hiroki Hosono, Hiroomi Yokomaku, Katsuya Morimoto, Sanjay Upadhyay, and Hiroaki Suzuki. 2008. "Integrated Electrochemical Analysis System with Microfluidic and Sensing Functions" Sensors 8, no. 2: 1111-1127. https://doi.org/10.3390/s8021111