1. Introduction
Diabetes is a metabolic disease in which the body does not produce or properly use insulin. Insulin is a hormone that is needed to convert sugar, starches and other food into energy needed for daily life. In 2006, according to the World Health Organization, at least 171 million people worldwide suffer from diabetes. Close control of blood glucose is essential to avoid the long-term adverse consequences of elevated blood glucose, including neuropathies, blindness and other sequelae [
1]. Non-invasive measurements of blood glucose have been a long-standing research goal. Such a capability would immediately allow the development of a variety of devices for diabetic health care, including continuous painless glucose monitoring, control of an insulin pump, and warning systems for hyper and hypoglycemic conditions. At present, the only reliable method to measure blood glucose is by a finger stick and subsequent glucose measurement, typically by glucose oxidase [
2]. This procedure is painful and even the most compliant individuals, with good understanding and motivation for glucose control, are not willing to finger stick themselves more than a few times per day [
3]. An alternative approach to glucose sensing is based on the development of a biosensor based on D-glucose/D-galactose binding protein (GGBP) from
E. coli, which bind glucose.
The GGBP of
E. coli serves as an initial component for both chemotaxis towards D-galactose and D-glucose and high-affinity active transport of the two sugars. GGBP is a monomer of molecular weight of about 32 kDa that binds glucose and galactose with micromolar affinity [
4]. Refined X-ray structures that have been determined by Vyas et al. [
5] and recently by Borrok et al. [
6] for GGBP in the absence and in the presence of D-glucose providing a view of the sugar-binding site at the molecular level. This protein has a monomeric structure that folds in two main domains linked by three strands commonly referred to as the ‘hinge region’, and the sugar-binding site is located in the cleft between the two lobes of the bilobate protein. Conformational changes involving the hinge are thought to be necessary for sugars to enter and/or exit the substrate-binding site [
7-
8]. Differences in the structure of the ligand-bound and ligand-free protein are essential for their proper recognition by the membrane components [
9]. This property of GGBP makes it good candidate for biological-recognition element for the development of glucose biosensor. In fact, Salins et al [
10] already showed the high potential applications of this protein by introducing a single cysteine residue at positions 148, 152, and 182 of the protein GGBP in order to monitor the effect of glucose binding on the protein structure.
In the present study a biosensor for convenient optical measurement of glucose concentration was obtained by genetically engineering the GGBP. The protein was mutated to introduce a single cysteine residue in one of the two domains of the protein by labeling it with a fluorescent probe sensitive to the protein environmental changes. Alternatively, two fluorescent probes were introduced in the protein structure for fluorescence resonance energy transfer (RET) measurements.
2. Results and Discussion
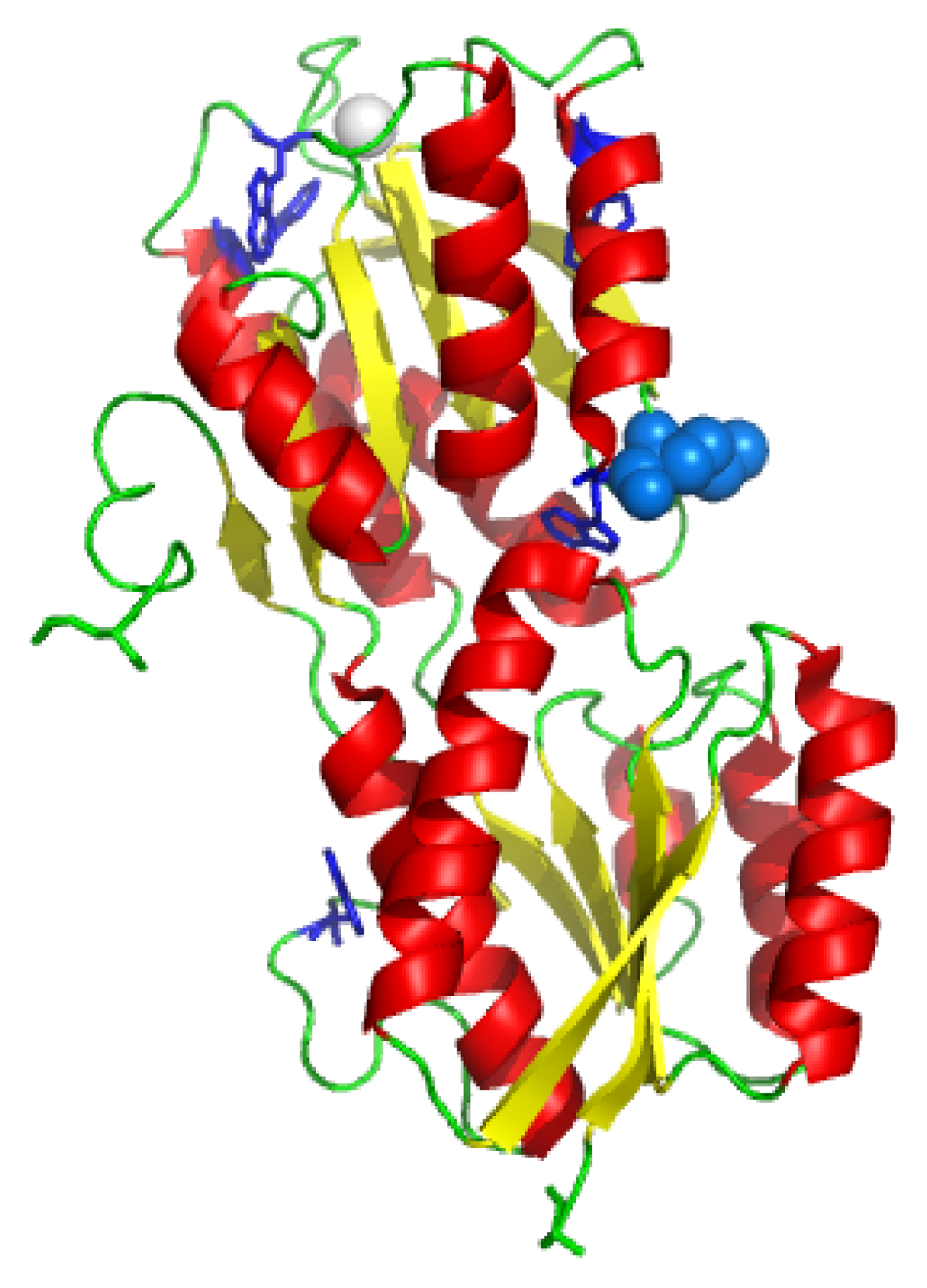
The GGBP (
Figure 1) is a monomer of 32 kDa consisting of two main domains, the N-terminus domain and the C-terminus domain. The sugar-binding site is located in the cleft between these two domains. A
KD value of 0.21 mM for D-glucose dissociation from GGBP has been reported by Zukin et al. [
11] at pH 8.0 and 4 °C. The mutated protein GBP-M182C contains a single cysteine residue at 182 amino acid position in the proximity of the sugar-binding site. Fluorescence, CD, FI-TR and MD data obtained for GGBP-M182C [
12], suggested that the protein structure undergoes no important alteration upon substitution of the methionine 182 with a cysteine residue. The presence of a single cysteine GGBP gave us the possibility to obtain more detailed informations about the glucose-binding site. This is important to deeply understand the biological and physical phenomena of biological recognition of the ligand in order to use GGBP as a probe for the development of a glucose biosensor for diabetic patients.
In the present work, GGBP-M182C was used as a model to design a biosensor that employs fluorescence spectroscopy to detect the glucose-induced protein changes. Since the intrinsic fluorescence from proteins is usually not useful for sensing because of the need for complex or bulky light sources as well as for the presence of numerous contaminant proteins in most biological samples, we decided to label the protein with an extrinsic fluorescence probe. For this reason, we used acrylodan and rhodamine as probes in an attempt to obtain a glucose response in the visible region of light.
In order to perform glucose titration by using an extrinsic fluorophore, we labeled the cysteine 182 of the engineered GGBP mutant with acrylodan, a suitable fluorescence probe that is able to sense environmental changes upon glucose binding.
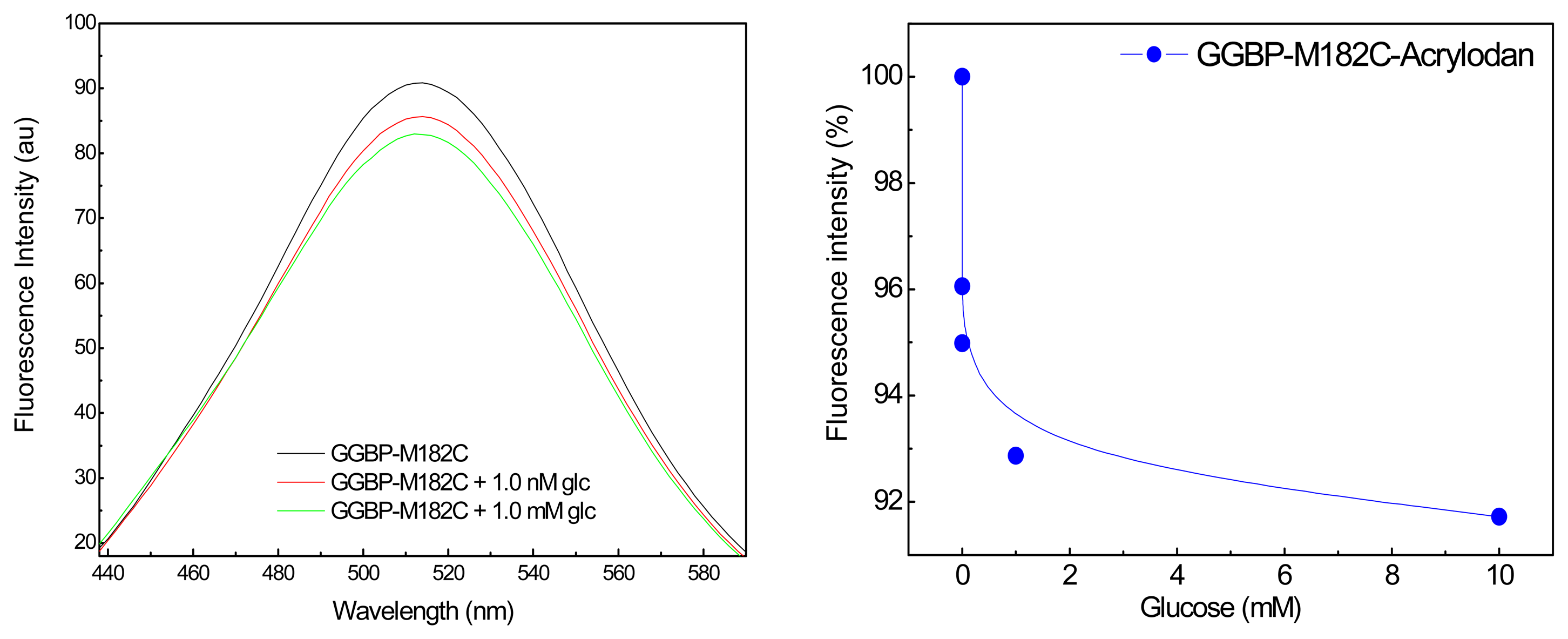
In
Figure 2 (Left) are shown the emission fluorescence spectra of the covalently labeled GGBP-M182C-Acrylodan. The first important observation is that the intensity of the acrylodan emission was sensitive to the additions of glucose. In particular, we can see the first effect of about 5% in the presence of sub-microomolar glucose concentration, and of about 10% in the presence of micromolar glucose concentration. In
Figure 2 (Right) is depicted the effect of the glucose addition on the fluorescence emission maximum of GGBP-M182C-Acrylodan. Acrylodan is known to be a molecule sensitive to its local environment. Thus the results obtained (a decrease in the emission intensity) suggest that the binding of glucose to the GGBP-M182C-Acrylodan displaces the acrylodan into a more polar environment as a result of a conformational change of the protein.
In order to perform fluorescence resonance energy transfer (RET) measurements, acrylodan and rhodamine were selected for labeling the cysteine 182 and the N-terminus of the GGBP-M182C, respectively, as donor–acceptor pair fluorophores, because fluorescence emission of the donor overlaps with the absorption/excitation spectrum of the acceptor. The use of RET between two fluorophores present in the protein allows to monitor fluorescence spectral variations since RET is a through-space interaction that occurs whenever the donor and the acceptor are within the Forster distance (Ro) and does not require change in the probe microenvironment.
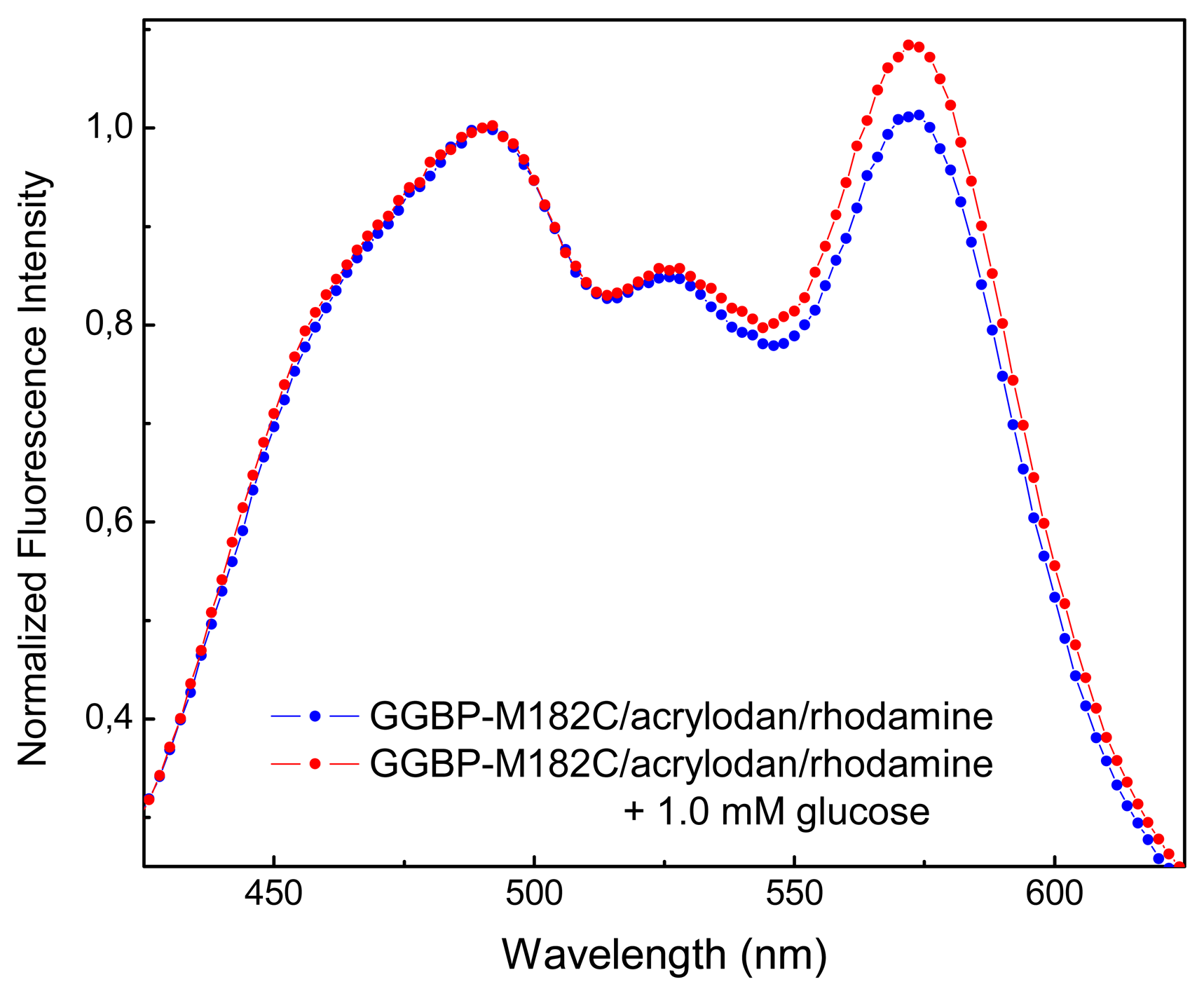
In
Figure 3 are shown the emission spectra of GGBP-M182C labeled with acrylodan and rhodamine, in the absence and in the presence of micromolar concentration of glucose. As we can see, there is an increase in fluorescence emission of rhodamine of about 10% upon glucose binding. Moreover, as we can see from crystal structures observation of the GGBP in the absence and in the presence of glucose [
6], the distance between the cysteine 182 and N-terminus amino acid residue is 46.27 Å in the absence of glucose and 38.27 Å in the presence of glucose. These data suggest that when GGBP-M182C binds glucose, undergoes a conformational change that is able to give a rational placement of the fluorophores allowing glucose-dependent spatial realignment of the two probes.
In
Figure 4 is showed the real time measurements of glucose concentration by using GGBP-M182C/acrylodan. It appears that upon glucose binding the lifetime of GGBP-M182C/acrylodan decreased from 2.8 to 2.3 ns, which correlates well with the intensity changes observed for steady-state measurements.
4. Experimental Section
4.1 Construction of the M182C Mutant of GGBP
The mglb gene that encodes for wild-type GGBP and its natural promoter were isolated from the E. coli K-12 genome by Polymerase Chain Reaction (PCR). The gene-promoter DNA fragment was cloned into the SacI/PstI restriction sites of the plasmid pTz18U. The resulting recombinant plasmid, was used as template for the construction of the GGBP-M182C mutant. Site-directed mutagenesis was accomplished using Overlap-Extention PCR. The forward primer mglB-FW 5′-AGGAATTCGAGCTCACTTCATTAACTCTAC-3′ including the natural promoter of GGBP was designed to introduce a SacI site (underlined). The reverse primer mglB-RV 5′-AACAGCTGTTATTTCTTGCTGAATTCAAGC-3′, covering the stop codon of GGBP, was used to introduce a PstI site (underlined). Two internal primers were used for point mutation to replace the methionine at position 182 with a cysteine; the forward primer (mglB-M182C-forward) had the following sequence: 5′-TAGATACCGCATGTTGGGACACCGCTCAGGCA-3′ and reverse primer (mglB-M182C-reverse) had a sequence as follow: 5′-AGcGGtGtcccaACATGCGGTATCTAACTGTAAC-3′; the mutated codon is indicated as bold faced letters. PCR cycle condition were 95°C for 5 min followed by 30 cycles of 95°C for 1 min, 50°C for 1 min, 72°C for 2 min. The amplified 1100 bp DNA fragment was ligated into the SacI/PstI site of the high copy number plasmid pTZ18U. The DNA sequencing data (PRIMM-SeqCore, Naples-Italy) verified that no mutation occurred except for of the desired point mutation. Transformation and subsequent expression of the resulting GGBP-M182C gene was performed in E. coli strain NM303.
4.2 Isolation of GGBP-M182C
The mono-cysteine mutant of GGBP was overproduced in
E. coli NM303 (F1
mgl 503
lacZ lacY1
recA1), a mutant strain that does not produce GGBP. The cultures consisted of 0.5% inoculum, 50 μg/ml ampicillin in 2 L Luria-Bertani medium (10 g/liter bacto-tryptone, 5 g/liter bacto-yeast extract, 10 g/liter NaCl, pH 7.2) and 1.0 mM D-fucose incubated at 37°C for 16-18 hours. Cells were harvested and GGBP-M182C was extracted by osmotic shock as Neu and Heppel protocol [
13]. The crude extract was dialyzed against 10 mM Tris–HCl pH 8.0 at 4°C for 16-18 hours. The GGBP-M182C was purified using a DEAE anion-exchange column (Bio-Rad). GGBP-M182C was eluted with a 10 mM Tris–HCl, pH 8.0, gradient from 0 to 0.5 M NaCl. Fractions were analyzed for the presence of GGBP-M182C by SDS–PAGE, and fractions containing GGBP-M182C were pooled and dialyzed against 10 mM Tris–HCl pH 7.2 at 4°C for 16-18 hours.
4.3 Steady-State Fluorescence Spectroscopy
Fluorescence data were measured on samples of GGBP-M182C in 10 mM Tris-HCl pH 7.2 with the protein concentration of 0.05 mg/ml and in the presence of increasing amount of glucose. Steady state fluorescence measurements were performed on the K2 fluorometer (ISS, Champaign, IL, USA) equipped with the two-cell temperature controlled sample holder. Acrylodan fluorescence was excited at 340 nm with the slit width of 1 nm.
4.4 Fluorescence Labelling of GGBP-M182C
A threefold excess of acrylodan (6-acryloyl-2-dimethylaminonaphthalene) in DMSO (N,N-dimethylformamide) was added drop-wise to a solution of 5.0 mg/ml of GGBP-M182C in 10 mM Tris-HCl pH 7.2, and then the solution was left to react for 3 hours at room temperature. The resulting labeled protein was separated from the free dye by Sephadex G-25 column.
The GBP-M182C-Acrylodan obtained was reacted with a threefold excess rhodamine (Tetrametylrhodamine Isothiocyanate) in DMSO for 30 minutes at 37°C. The resulting protein labeled with both fluorophores was separated from the free dye by Sephadex G-25 column.
4.5 Frequency-domain Fluorescence Spectroscopy
Frequency domain measurements were performed on the K2 fluorometer (ISS, Champaign, IL, USA) equipped with 2-cell temperature controlled sample holder. The excitation source was lamp at 350 nm. The GGBP-M182C/Acrylodan emission was selected by a Schott 450 nm long pass filter.
Fluorescence intensity decays were analyzed in terms of the multi-exponential model
where αi are the pre-exponential factors, τi the decay times, and ∑αi = 1.0. The fractional intensity fi of each decay component were calculated from the formula
and the mean lifetimes as τmean = ∑ fi τi.

{kind=link}
{kind=link}
{kind=link}
{kind=link}