dPCR: A Technology Review


Abstract
:1. Introduction: Quantification of Nucleic Acids by Quantitative PCR and Digital PCR
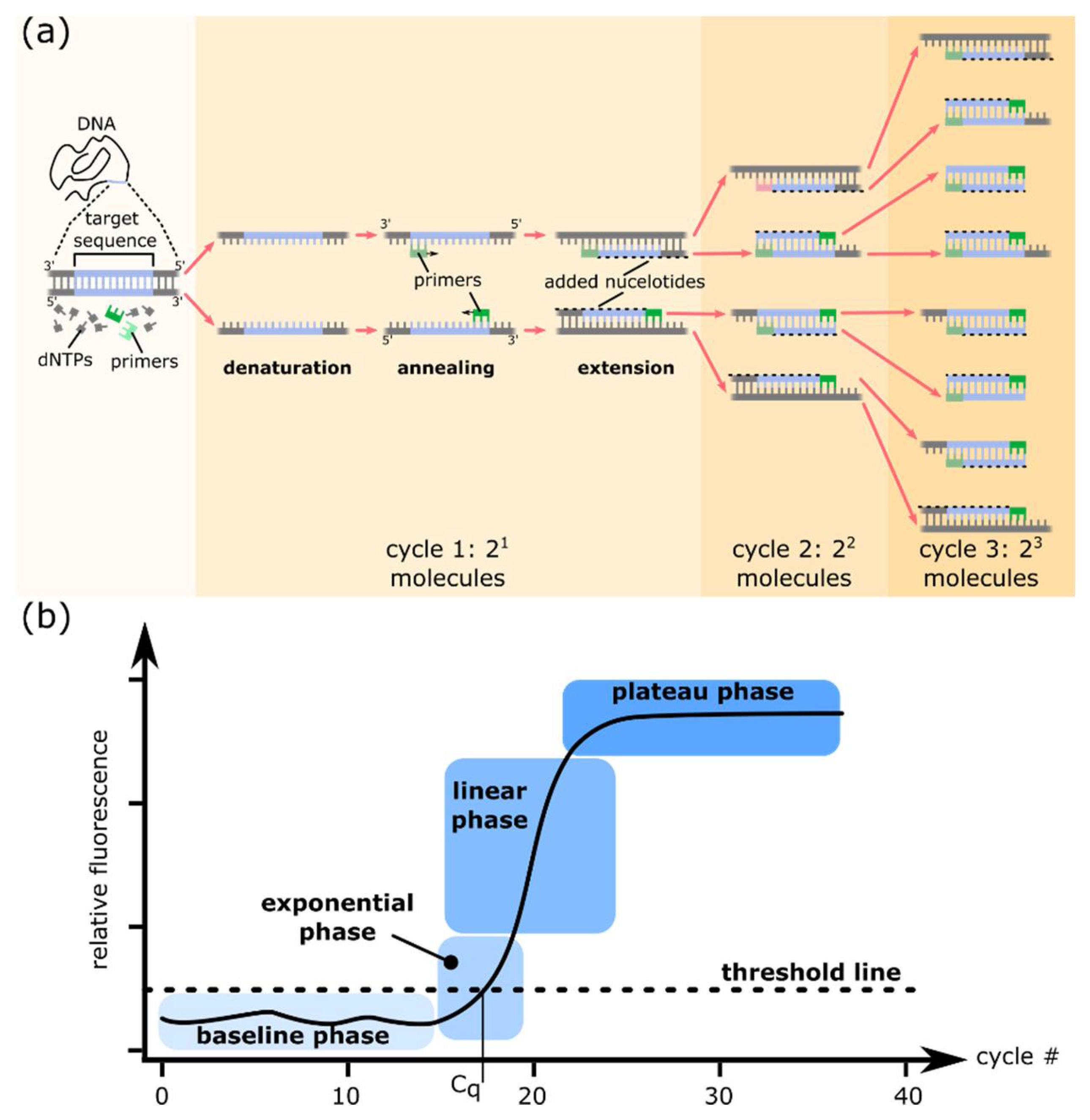
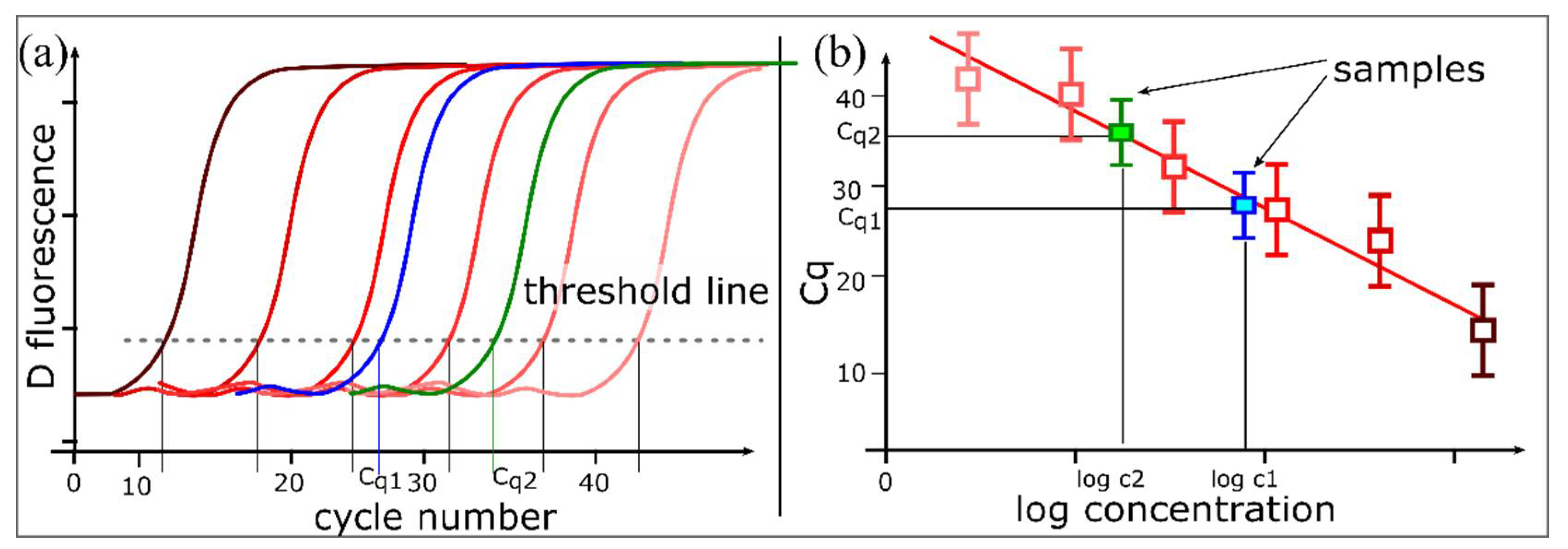
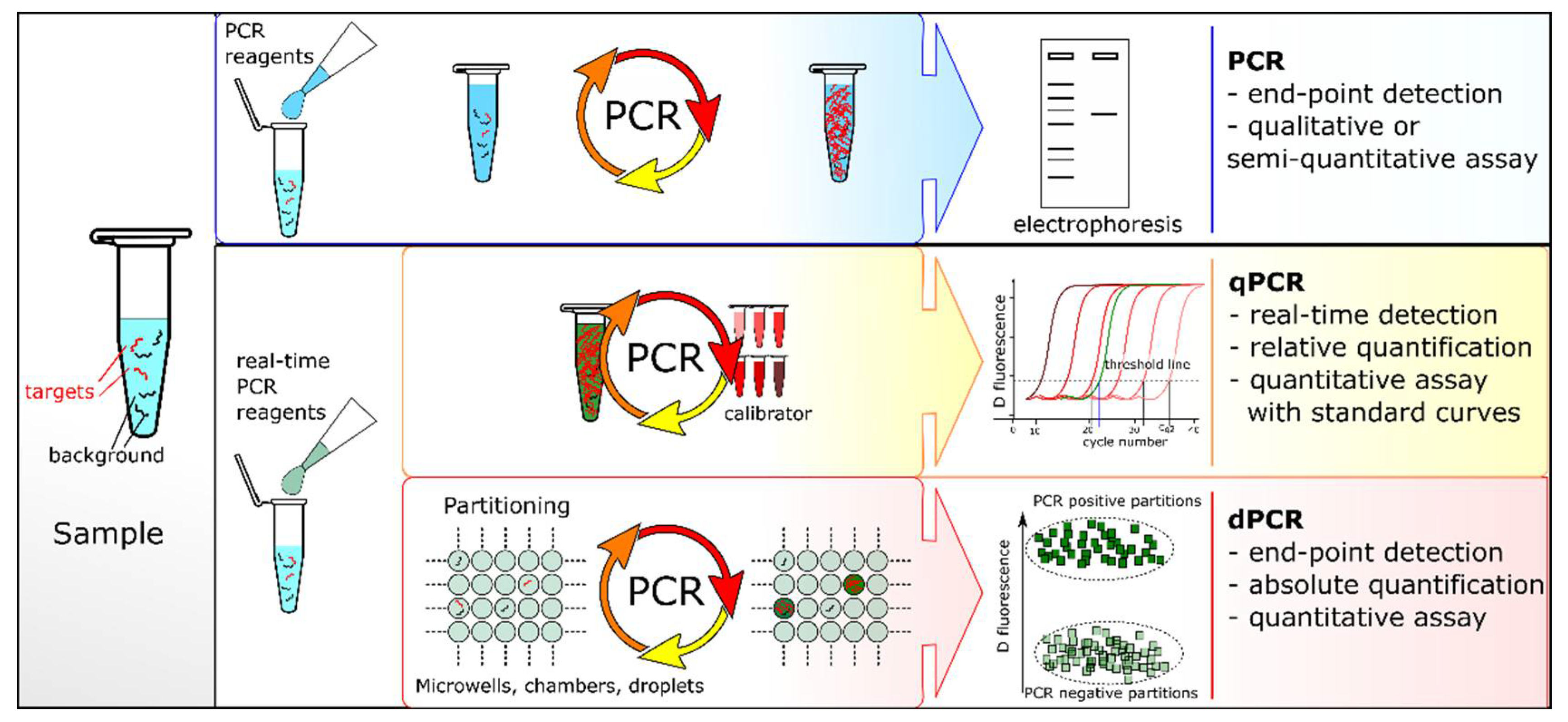
1.1. PCR and Quantitative PCR
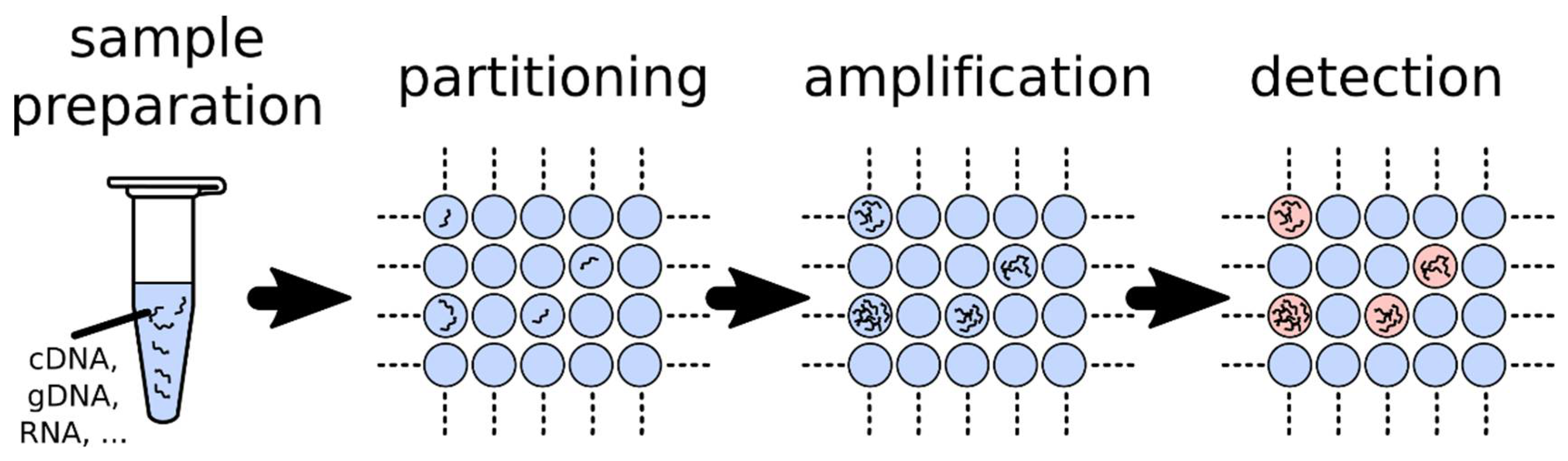
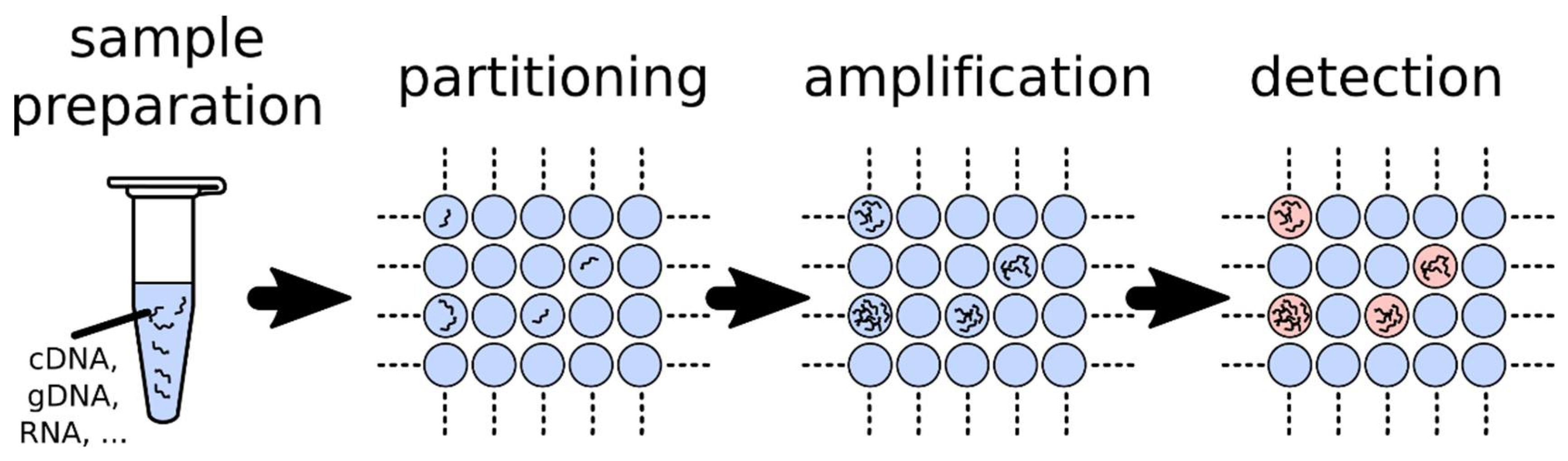
1.2. Fundamentals of dPCR
1.3. Fundamental Differences between dPCR and qPCR
2. Statistical Foundations of dPCR
2.1. Binomial Probability and Poisson Approximation
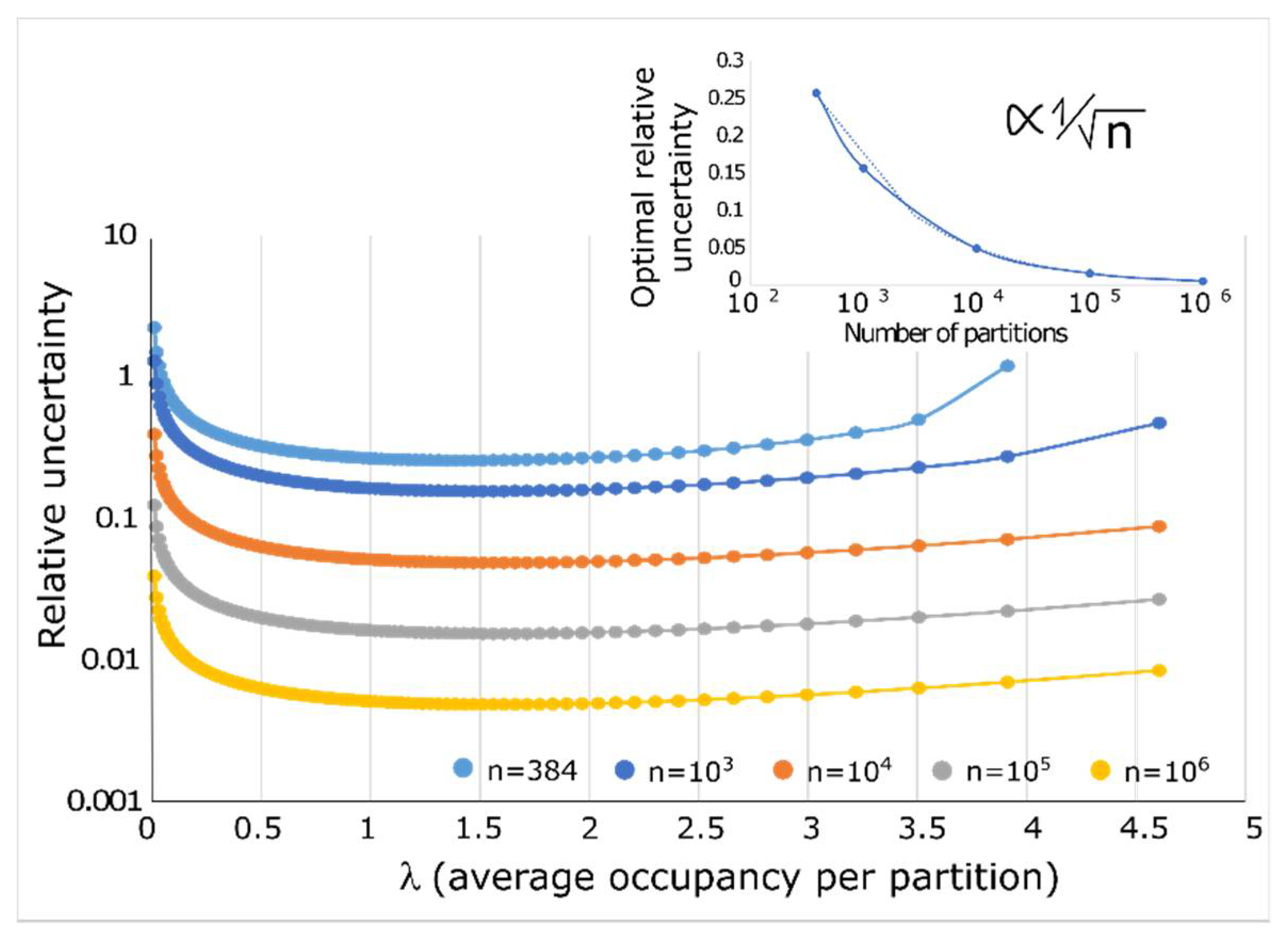
2.2. Quantification Accuracy
2.3. Most Probable Number (MPN)
2.4. Copy Number Variant (CNV) Applications
2.5. Absolute Limit of Quantification Due to Specimen Sampling
2.6. Hypothesis and Technological Implications
2.7. Conclusion of the Statistical Foundations of dPCR
3. Performance Metrics
3.1. Sensitivity of Detection
3.2. Dynamic Range of Detection
3.3. Practical Considerations in the Reliability of dPCR Measurements-False-Negative/Positive Signals
4. Miniaturization and Hyper-Compartmentalization
4.1. Introduction
4.2. Chamber Formats
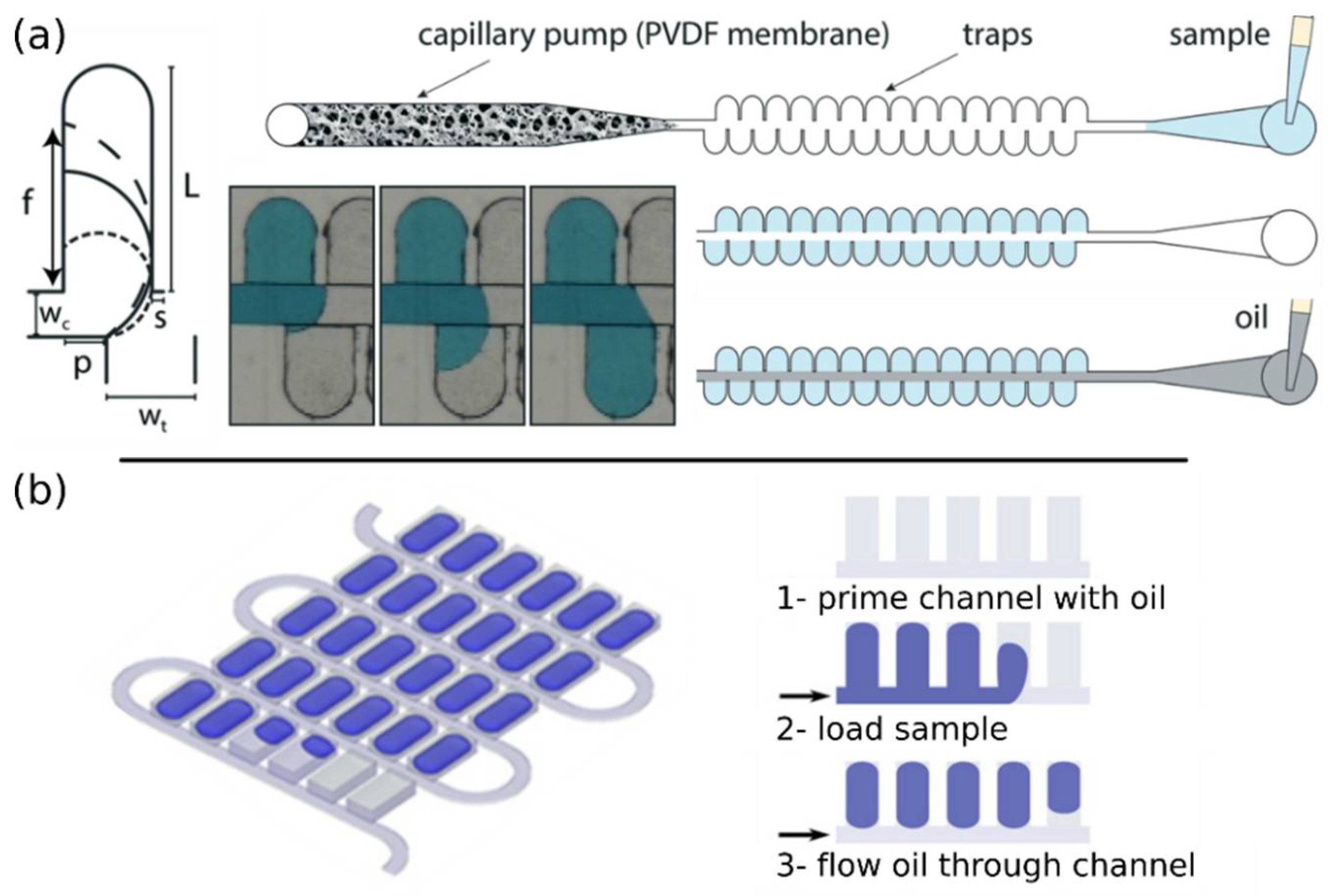
4.2.1. Active Partitioning Platforms
4.2.2. Passive Partitioning Platforms
4.2.3. Self-Digitization Platforms
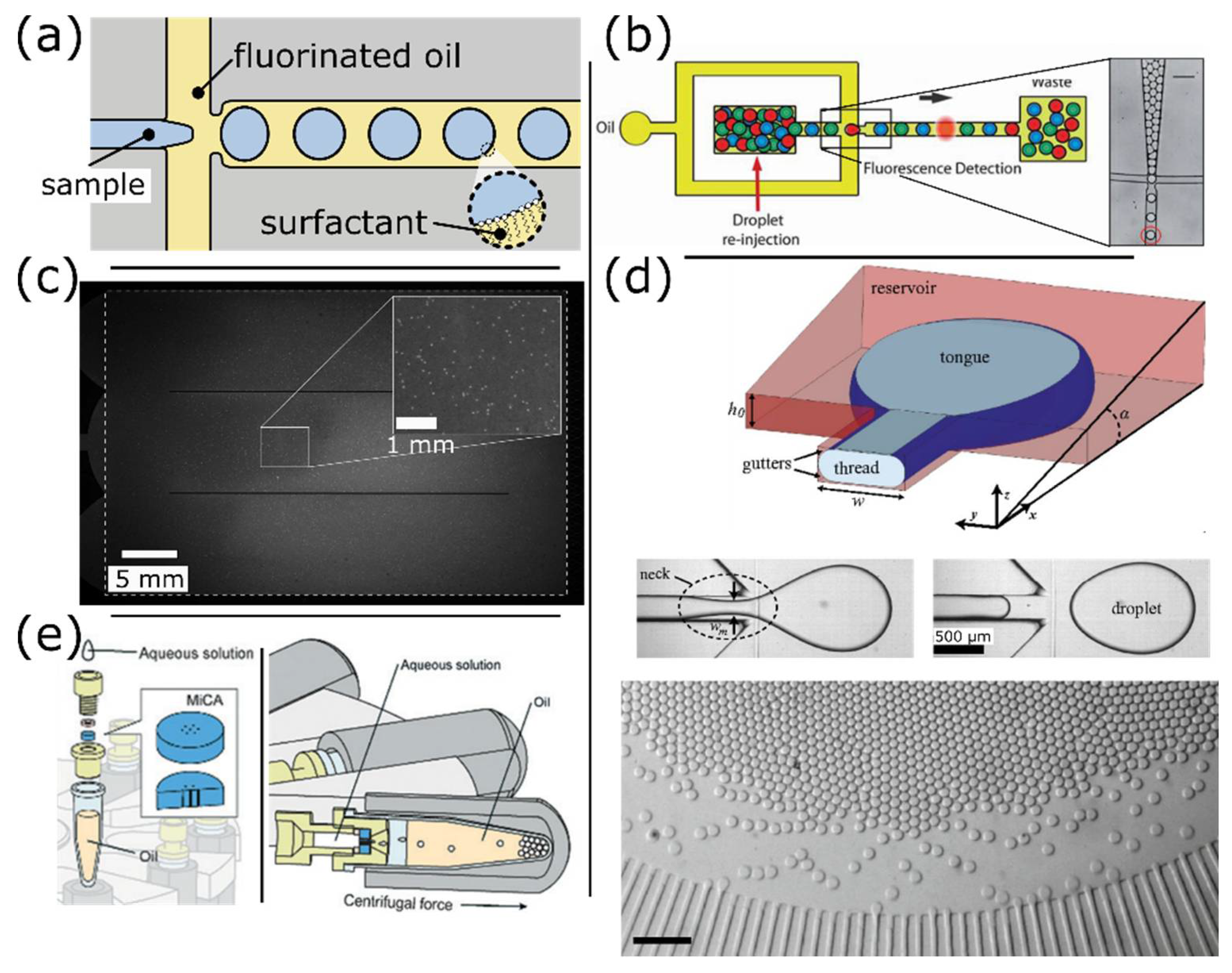
4.3. Droplet-Based Platforms
4.4. Conclusion of Hypercompartmentalization
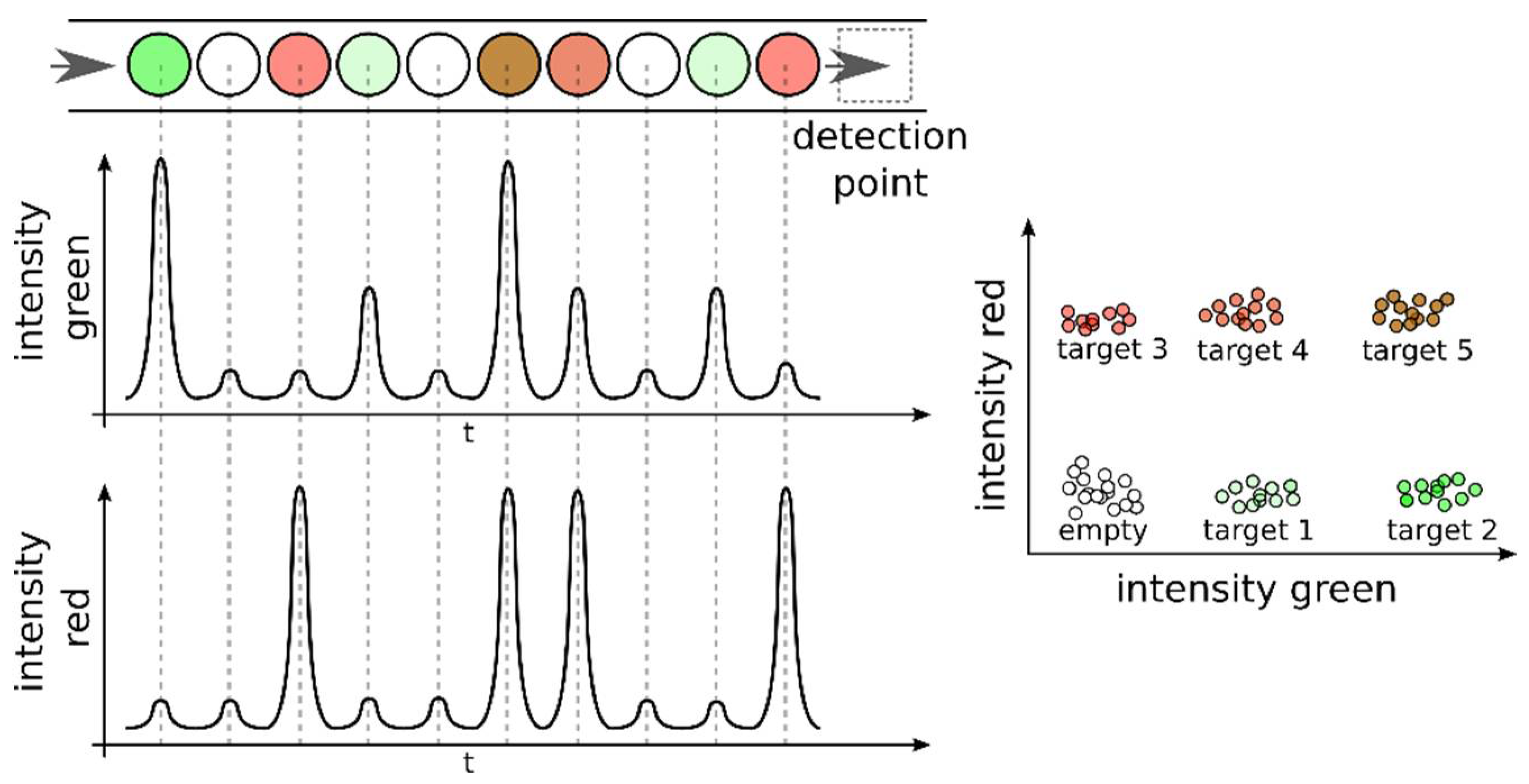
5. Detection Methods and Multiplexing Approaches in dPCR
6. Beyond PCR
6.1. Isothermal Amplification
6.2. Digital Isothermal Amplification Systems
7. Experimental Comparison of dPCR and qPCR
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R. Basic Principles of Real-Time Quantitative PCR. Expert Rev. Mol. Diagn. 2005, 5, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Svec, D.; Tichopad, A.; Novosadova, V.; Pfaffl, M.W.; Kubista, M. How Good Is a PCR Efficiency Estimate: Recommendations for Precise and Robust qPCR Efficiency Assessments. Biomol. Detect. Quantif. 2015, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G. The Need for Transparency and Good Practices in the qPCR Literature. Nat. Methods 2013, 10, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, J.R.; van Kempen, L.C.; Nagtegaal, I.D.; Bustin, S.A. Critical Appraisal of Quantitative PCR Results in Colorectal Cancer Research: Can We Rely on Published qPCR Results? Mol. Oncol. 2014, 8, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, R.T.; Hokanson, K.M.; Pounds, S.B.; Bankowski, M.J.; Belzer, S.W.; Carr, J.; Diorio, D.; Forman, M.S.; Joshi, Y.; Hillyard, D.; et al. Multicenter Comparison of Different Real-Time PCR Assays for Quantitative Detection of Epstein-Barr Virus. J. Clin. Microbiol. 2008, 46, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Grenier, S.; Nwachukwu, B.; Wei, C.; Lipton, J.H.; Suzanne, K.-R. Inter-Laboratory Comparison of Chronic Myeloid Leukemia Minimal Residual Disease Monitoring. J. Mol. Diagn. 2007, 9, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, O.; Lebedeva, I.; Brown, J.; Silver, J. Nanoliter Scale PCR with TaqMan Detection. Nucleic Acids Res. 1997, 25, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Sykes, P.J.; Neoh, S.H.; Brisco, M.J.; Hughes, E.; Condon, J.; Morley, A.A. Quantitation of Targets for PCR by Use of Limiting Dilution. Biotechniques 1992, 13, 444–449. [Google Scholar] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Dube, S.; Qin, J.; Ramakrishnan, R. Mathematical Analysis of Copy Number Variation in a DNA Sample Using Digital PCR on a Nanofluidic Device. PLoS ONE 2008, 3, e2876. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.S.; Cowen, S.; Foy, C.A.; Huggett, J.F. Methods for Applying Accurate Digital PCR Analysis on Low Copy DNA Samples. PLoS ONE 2013, 8, e58177. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The Digital MIQE Guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Wallis, S. Binomial Confidence Intervals and Contingency Tests: Mathematical Fundamentals and the Evaluation of Alternative Methods. J. Quant. Linguist. 2013, 20, 178–208. [Google Scholar] [CrossRef]
- Brown, L.D.; Cai, T.T.; Das Gupta, A. Interval Estimation for a Binomial Proportion. Stat. Sci. 2001, 16, 101–133. [Google Scholar]
- Shen, F.; Du, W.; Kreutz, J.E.; Fok, A.; Ismagilov, R.F. Digital PCR on a Slipchip. Lab Chip 2010, 10, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B. Probable Inference, the Law of Succession, and Statistical Inference. J. Am. Stat. Assoc. 1927, 22, 209–212. [Google Scholar] [CrossRef]
- Ziegler, N.R.; Halvorson, H.O. Application of Statistics to Problems in Bacteriology: IV. Experimental Comparison of the Dilution Method, the Plate Count, and the Direct Count for the Determination of Bacterial Populations. J. Bacteriol. 1935, 29, 609–634. [Google Scholar] [PubMed]
- Halvorson, H.O.; Ziegler, N.R. Application of Statistics to Problems in Bacteriology: I. A Means of Determining Bacterial Population by the Dilution Method. J. Bacteriol. 1933, 25, 101–121. [Google Scholar] [PubMed]
- Halvorson, H.O. Application of Statistics to Problems in Bacteriology: II. A Consideration of the Accuracy of Dilution Data Obtained by Using a Single Dilution. J. Bacteriol. 1933, 26, 331–339. [Google Scholar] [PubMed]
- Halvorson, H.O. Application of Statistics to Problems in Bacteriology: III. A Consideration of the Accuracy of Dilution Data Obtained by Using Several Dilutions. J. Bacteriol. 1933, 26, 559–567. [Google Scholar] [PubMed]
- Cochran, W.G. Estimation of Bacterial Densities by Means of the Most Probable Number. Biometrics 1950, 6, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Garthright, W.E.; Blodgett, R.J. FDA’S Preferred MPN Methods for Standard, Large or Unusual Tests, with a Spreadsheet. Food Microbiol. 2003, 20, 439–445. [Google Scholar] [CrossRef]
- Kiss, M.M.; Ortoleva-Donnelly, L.; Beer, N.R.; Warner, J.; Bailey, C.G.; Colston, B.W.; Rothberg, J.M.; Link, D.R.; Leamon, J.H. High-Throughput Quantitative Polymerase Chain Reaction in Picoliter Droplets. Anal. Chem. 2008, 80, 8975–8981. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, J.E.; Munson, T.; Huynh, T.; Shen, F.; Du, W.; Ismagilov, R.F. Theoretical Design and Analysis of Multivolume Digital Assays with Wide Dynamic Range Validated Experimentally with Microfluidic Digital PCR. Anal. Chem. 2011, 83, 8158–8168. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Sun, B.; Kreutz, J.E.; Davydova, E.K.; Du, W.; Reddy, P.L.; Joseph, L.J.; Ismagilov, R.F. Multiplexed Quantification of Nucleic Acids with Large Dynamic Range Using Multivolume Digital Rt-PCR on a Rotational Slipchip Tested with HIV and Hepatitis C Viral Load. J. Am. Chem. Soc. 2011, 133, 17705–17712. [Google Scholar] [CrossRef] [PubMed]
- Wald, A. Sequential Analysis; John Wiley & Sons, Inc.: New York, NY, USA, 1947. [Google Scholar]
- Shih, I.M.; Zhou, W.; Goodman, S.N.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Evidence That Genetic Instability Occurs at an Early Stage of Colorectal Tumorigenesis. Cancer Res. 2001, 61, 818–822. [Google Scholar] [PubMed]
- Zhou, W.; Galizia, G.; Goodman, S.N.; Romans, K.E.; Kinzler, K.W.; Vogelstein, B.; Choti, M.A.; Montgomery, E.A. Counting Alleles Reveals a Connection between Chromosome 18q Loss and Vascular Invasion. Nat. Biotechnol. 2001, 19, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.; Kurman, R.J.; Chang, H.W.; Cho, S.K.; Shih, I.M. Diverse Tumorigenic Pathways in Ovarian Serous Carcinoma. Lab. Investig. 2002, 82, 211A. [Google Scholar] [CrossRef]
- Chang, H.W.; Ali, S.Z.; Cho, S.K.R.; Kurman, R.J.; Shih, I.M. Detection of Allelic Imbalance in Ascitic Supernatant by Digital Single Nucleotide Polymorphism Analysis. Clin. Cancer Res. 2002, 8, 2580–2585. [Google Scholar] [PubMed]
- Lo, Y.M.; Lun, F.M.; Chan, K.C.; Tsui, N.B.; Chong, K.C.; Lau, T.K.; Leung, T.Y.; Zee, B.C.; Cantor, C.R.; Chiu, R.W. Digital PCR for the Molecular Detection of Fetal Chromosomal Aneuploidy. Proc. Natl. Acad. Sci. USA 2007, 104, 13116–13121. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.S.; Huggett, J.F.; Cowen, S.; Speirs, V.; Shaw, J.; Ellison, S.; Foy, C.A.; Scott, D.J. Comparison of Microfluidic Digital PCR and Conventional Quantitative PCR for Measuring Copy Number Variation. Nucleic Acids Res. 2012, 40, e82. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, T. Limits Ofanalysis. Anal. Chem. 1976, 48, 16A–18A. [Google Scholar] [CrossRef]
- Debski, P.R.; Garstecki, P. Designing and Interpretation of Digital Assays: Concentration of Target in the Sample and in the Source of Sample. Biomol. Detect. Quantif. 2016, 10, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Herrmann, J.; Armishaw, P.; Corbisier, P.; Emslie, K.R. Single Molecule Detection in Nanofluidic Digital Array Enables Accurate Measurement of DNA Copy Number. Anal. Bioanal. Chem. 2009, 394, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Heyries, K.A.; Tropini, C.; Vaninsberghe, M.; Doolin, C.; Petriv, O.I.; Singhal, A.; Leung, K.; Hughesman, C.B.; Hansen, C.L. Megapixel Digital PCR. Nat. Methods 2011, 8, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Kallioniemi, O.P.; Kallioniemi, A.; Kurisu, W.; Thor, A.; Chen, L.C.; Smith, H.S.; Waldman, F.M.; Pinkel, D.; Gray, J.W. ERBB2 Amplification in Breast Cancer Analyzed by Fluorescence in Situ Hybridization. Proc. Natl. Acad. Sci. USA 1992, 89, 5321–5325. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.H.; Jiang, W.; Jayaraman, J.; Trowsdale, J.; Holland, M.J.; Traherne, J.A. Killer-Cell Immunoglobulin-Like Receptor Gene Linkage and Copy Number Variation Analysis by Droplet Digital PCR. Genome Med. 2014, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a Droplet Digital Polymerase Chain Reaction Format for DNA Copy Number Quantification. Anal. Chem. 2011, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Cowen, S.; Foy, C.A. Considerations for Digital PCR as an Accurate Molecular Diagnostic Tool. Clin. Chem. 2015, 61, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Meng, Y.; Sui, Z.; Wang, J.; Wu, L.; Fu, B. Comparison of Four Digital PCR Platforms for Accurate Quantification of DNA Copy Number of a Certified Plasmid DNA Reference Material. Sci. Rep. 2015, 5, 13174. [Google Scholar] [CrossRef] [PubMed]
- Hatch, A.C.; Fisher, J.S.; Tovar, A.R.; Hsieh, A.T.; Lin, R.; Pentoney, S.L.; Yang, D.L.; Lee, A.P. 1-Million Droplet Array with Wide-Field Fluorescence Imaging for Digital PCR. Lab Chip 2011, 11, 3838–3845. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR Versus qPCR for Gene Expression Analysis with Low Abundant Targets: From Variable Nonsense to Publication Quality Data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Jenkins, G.; Zhang, W.; Zhang, M.; Guan, Z.; Yang, C.J. Single-Molecule Emulsion PCR in Microfluidic Droplets. Anal. Bioanal. Chem. 2012, 403, 2127–2143. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; McLaughlin, J.L.H.; Emslie, K.R. Effect of Sustained Elevated Temperature Prior to Amplification on Template Copy Number Estimation Using Digital Polymerase Chain Reaction. Analyst 2011, 136, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, K.R.; Burke, D.G.; Emslie, K.R. Quantitative Polymerase Chain Reaction: A Framework for Improving the Quality of Results and Estimating Uncertainty of Measurement. Anal. Methods 2011, 3, 2201–2211. [Google Scholar] [CrossRef]
- Sanders, R.; Huggett, J.F.; Bushell, C.A.; Cowen, S.; Scott, D.J.; Foy, C.A. Evaluation of Digital PCR for Absolute DNA Quantification. Anal. Chem. 2011, 83, 6474–6484. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Lee, S.; Goodman, S.; Zhang, Z.; Chan, D.W.; Kurman, R.J.; Shih, I.M. Assessment of Plasma DNA Levels, Allelic Imbalance, and CA-125 as Diagnostic Tests for Cancer. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1198S. [Google Scholar] [CrossRef]
- Hong, J.W.; Quake, S.R. Integrated Nanoliter Systems. Nat. Biotechnol. 2003, 21, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.A.; Whitesides, G.M. Rapid Prototyping of Microfluidic Systems in Poly(Dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Li, L.; Nichols, K.P.; Ismagilov, R.F. Slipchip. Lab Chip 2009, 9, 2286–2292. [Google Scholar] [CrossRef] [PubMed]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xue, G.; Yeung, E.S. Selective Detection of Individual DNA Molecules by Capillary Polymerase Chain Reaction. Anal. Chem. 2001, 73, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Jarvius, J.; Melin, J.; Goransson, J.; Stenberg, J.; Fredriksson, S.; Gonzalez-Rey, C.; Bertilsson, S.; Nilsson, M. Digital Quantification Using Amplified Single-Molecule Detection. Nat. Methods 2006, 3, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.A.; Chou, H.P.; Thorsen, T.; Scherer, A.; Quake, S.R. Monolithic Microfabricated Valves and Pumps by Multilayer Soft Lithography. Science 2000, 288, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.; Hurley, J.; Garcia, J.; Yoder, K.; Katz, A.; Roberts, D.; Cho, J.; Kanigan, T.; Ilyin, S.E.; Horowitz, D.; et al. Nanoliter High Throughput Quantitative PCR. Nucleic Acids Res. 2006, 34, e123. [Google Scholar] [CrossRef] [PubMed]
- Jackman, R.J.; Duffy, D.C.; Ostuni, E.; Willmore, N.D.; Whitesides, G.M. Fabricating Large Arrays of Microwells with Arbitrary Dimensions and Filling Them Using Discontinuous Dewetting. Anal. Chem. 1998, 70, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Akagi, Y.; Rao, S.R.; Morita, Y.; Tamiya, E. Optimization of Fluorescent Cell-Based Assays for High-Throughput Analysis Using Microchamber Array Chip Formats. Sci. Technol. Adv. Mater. 2004, 5, 343–349. [Google Scholar] [CrossRef]
- Cohen, D.E.; Schneider, T.; Wang, M.; Chiu, D.T. Self-Digitization of Sample Volumes. Anal. Chem. 2010, 82, 5707–5717. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, Y.; Kerman, K.; Kobayashi, M.; Yamamura, S.; Morita, Y.; Takamura, Y.; Tamiya, E. On-Chip Nanoliter-Volume Multiplex Taqman Polymerase Chain Reaction from a Single Copy Based on Counting Fluorescence Released Microchambers. Anal. Chem. 2004, 76, 6434–6439. [Google Scholar] [CrossRef] [PubMed]
- Kinpara, T.; Mizuno, R.; Murakami, Y.; Kobayashi, M.; Yamaura, S.; Hasan, Q.; Morita, Y.; Nakano, H.; Yamane, T.; Tamiya, E. A Picoliter Chamber Array for Cell-Free Protein Synthesis. J. Biochem. 2004, 136, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Wiktor, P.; Brunner, A.; Kahn, P.; Qiu, J.; Magee, M.; Bian, X.; Karthikeyan, K.; LaBaer, J. Microreactor Array Device. Sci. Rep. 2015, 5, 8736. [Google Scholar] [CrossRef] [PubMed]
- Dimov, I.K.; Lu, R.; Lee, E.P.; Seita, J.; Sahoo, D.; Park, S.; Weissman, I.L.; Lee, L.P. Discriminating Cellular Heterogeneity Using Microwell-Based RNA Cytometry. Nat. Commun. 2014, 5, 3451. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.A.; Hong, J.W.; Quake, S.R.; Leadbetter, J.R. Microfluidic Digital PCR Enables Multigene Analysis of Individual Environmental Bacteria. Science 2006, 314, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Davydova, E.K.; Du, W.; Kreutz, J.E.; Piepenburg, O.; Ismagilov, R.F. Digital Isothermal Quantification of Nucleic Acids via Simultaneous Chemical Initiation of Recombinase Polymerase Amplification Reactions on Slipchip. Anal. Chem. 2011, 83, 3533–3540. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, E.; Chen, C.S.; Ingber, D.E.; Whitesides, G.M. Selective Deposition of Proteins and Cells in Arrays of Microwells. Langmuir 2001, 17, 2828–2834. [Google Scholar] [CrossRef]
- Reddy, S.; Schunk, P.R.; Bonnecaze, R.T. Dynamics of Low Capillary Number Interfaces Moving through Sharp Features. Phys. Fluids 2005, 17, 122104. [Google Scholar] [CrossRef]
- Sposito, A.J.; DeVoe, D.L. Staggered Trap Arrays for Robust Microfluidic Sample Digitization. Lab Chip 2017, 17, 4105–4112. [Google Scholar] [CrossRef] [PubMed]
- Vulto, P.; Podszun, S.; Meyer, P.; Hermann, C.; Manz, A.; Urban, G.A. Phaseguides: A Paradigm Shift in Microfluidic Priming and Emptying. Lab Chip 2011, 11, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gao, Y.; Yu, B.; Ren, H.; Qiu, L.; Han, S.; Jin, W.; Jin, Q.; Mu, Y. Self-Priming Compartmentalization Digital Lamp for Point-of-Care. Lab Chip 2012, 12, 4755–4763. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Song, Q.; Xu, Y.; Zhu, Q.; Yu, B.; Jin, W.; Jin, Q.; Mu, Y. A Localized Temporary Negative Pressure Assisted Microfluidic Device for Detecting Keratin 19 in A549 Lung Carcinoma Cells with Digital PCR. Anal. Methods 2015, 7, 2006–2011. [Google Scholar] [CrossRef]
- Ma, Y.-D.; Chang, W.-H.; Luo, K.; Wang, C.-H.; Liu, S.-Y.; Yen, W.-H.; Lee, G.-B. Digital Quantification of DNA via Isothermal Amplification on a Self-Driven Microfluidic Chip Featuring Hydrophilic Film-Coated Polydimethylsiloxane. Biosens. Bioelectron. 2018, 99, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Gansen, A.; Herrick, A.M.; Dimov, I.K.; Lee, L.P.; Chiu, D.T. Digital Lamp in a Sample Self-Digitization (SD) Chip. Lab Chip 2012, 12, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, S.O.; Wittwer, C.T.; Gao, C.; Gale, B.K. Spinning Disk Platform for Microfluidic Digital Polymerase Chain Reaction. Anal. Chem. 2010, 82, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; Gansen, A.; Paguirigan, A.L.; Kreutz, J.E.; Radich, J.P.; Chiu, D.T. Self-Digitization Microfluidic Chip for Absolute Quantification of Mrna in Single Cells. Anal. Chem. 2014, 86, 12308–12314. [Google Scholar] [CrossRef] [PubMed]
- Tice, J.D.; Song, H.; Lyon, A.D.; Ismagilov, R.F. Formation of Droplets and Mixing in Multiphase Microfluidics at Low Values of the Reynolds and the Capillary Numbers. Langmuir 2003, 19, 9127–9133. [Google Scholar] [CrossRef]
- Boukellal, H.; Selimovic, S.; Jia, Y.; Cristobal, G.; Fraden, S. Simple, Robust Storage of Drops and Fluids in a Microfluidic Device. Lab Chip 2009, 9, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Link, D.R.; Anna, S.L.; Weitz, D.A.; Stone, H.A. Geometrically Mediated Breakup of Drops in Microfluidic Devices. Phys. Rev. Lett. 2004, 92, 054503. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-C.; Fisher, J.S.; Lee, A.I.; Cristini, V.; Lee, A.P. Design of Microfluidic Channel Geometries for the Control of Droplet Volume, Chemical Concentration, and Sorting. Lab Chip 2004, 4, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Yen, G.S.; Thompson, A.M.; Burnham, D.R.; Chiu, D.T. Self-Digitization of Samples into a High-Density Microfluidic Bottom-Well Array. Anal. Chem. 2013, 85, 10417–10423. [Google Scholar] [CrossRef] [PubMed]
- Holtze, C.; Rowat, A.C.; Agresti, J.J.; Hutchison, J.B.; Angile, F.E.; Schmitz, C.H.; Koster, S.; Duan, H.; Humphry, K.J.; Scanga, R.A.; et al. Biocompatible Surfactants for Water-in-Fluorocarbon Emulsions. Lab Chip 2008, 8, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; He, Y.; Kenneth, W.K.; Vogelstein, B.; Dressman, D. Beaming: Single-Molecule PCR on Microparticles in Water-in-Oil Emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Diehl, F.; Dressman, D.; Vogelstein, B.; Kinzler, K.W. Beaming up for Detection and Quantification of Rare Sequence Variants. Nat. Methods 2006, 3, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming Single DNA Molecules into Fluorescent Magnetic Particles for Detection and Enumeration of Genetic Variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef] [PubMed]
- Taly, V.; Pekin, D.; El Abed, A.; Laurent-Puig, P. Detecting Biomarkers with Microdroplet Technology. Trends Mol. Med. 2012, 18, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Dangla, R.; Kayi, S.C.; Baroud, C.N. Droplet Microfluidics Driven by Gradients of Confinement. Proc. Natl. Acad. Sci. USA 2013, 110, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liao, P.; Zhang, F.; Jiang, M.; Zhu, Y.; Huang, Y. Centrifugal Micro-Channel Array Droplet Generation for Highly Parallel Digital PCR. Lab Chip 2017, 17, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, T.; Roberts, R.W.; Arnold, F.H.; Quake, S.R. Dynamic Pattern Formation in a Vesicle-Generating Microfluidic Device. Phys. Rev. Lett. 2001, 86, 4163–4166. [Google Scholar] [CrossRef] [PubMed]
- Anna, S.L.; Bontoux, N.; Stone, H.A. Formation of Dispersions Using “Flow Focusing” in Microchannels. Appl. Phys. Lett. 2003, 82, 364–366. [Google Scholar] [CrossRef]
- Sugiura, S.; Nakajima, M.; Iwamoto, S.; Seki, M. Interfacial Tension Driven Monodispersed Droplet Formation from Microfabricated Channel Array. Langmuir 2001, 17, 5562–5566. [Google Scholar] [CrossRef]
- Courtois, F.; Olguin, L.F.; Whyte, G.; Theberge, A.B.; Huck, W.T.S.; Hollfelder, F.; Abell, C. Controlling the Retention of Small Molecules in Emulsion Microdroplets for Use in Cell-Based Assays. Anal. Chem. 2009, 81, 3008–3016. [Google Scholar] [CrossRef] [PubMed]
- Beverung, C.J.; Radke, C.J.; Blanch, H.W. Protein Adsorption at the Oil/Water Interface: Characterization of Adsorption Kinetics by Dynamic Interfacial Tension Measurements. Biophys. Chem. 1999, 81, 59–80. [Google Scholar] [CrossRef]
- Beer, N.R.; Wheeler, E.K.; Lee-Houghton, L.; Watkins, N.; Nasarabadi, S.; Hebert, N.; Leung, P.; Arnold, D.W.; Bailey, C.G.; Colston, B.W. On-Chip Single-Copy Real-Time Reverse-Transcription PCR in Isolated Picoliter Droplets. Anal. Chem. 2008, 80, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Kumaresan, P.; Yang, C.J.; Cronier, S.A.; Blazej, R.G.; Mathies, R.A. High-Throughput Single Copy DNA Amplification and Cell Analysis in Engineered Nanoliter Droplets. Anal. Chem. 2008, 80, 3522–3529. [Google Scholar] [CrossRef] [PubMed]
- Schaerli, Y.; Wootton, R.C.; Robinson, T.; Stein, V.; Dunsby, C.; Neil, M.A.A.; French, P.M.W.; de Mello, A.J.; Abell, C.; Hollfelder, F. Continuous-Flow Polymerase Chain Reaction of Single-Copy DNA in Microfluidic Microdroplets. Anal. Chem. 2008, 81, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Mazutis, L.; Araghi, A.F.; Miller, O.J.; Baret, J.C.; Frenz, L.; Janoshazi, A.; Taly, V.; Miller, B.J.; Hutchison, J.B.; Link, D.; et al. Droplet-Based Microfluidic Systems for High-Throughput Single DNA Molecule Isothermal Amplification and Analysis. Anal. Chem. 2009, 81, 4813–4821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jenkins, G.; Zou, Y.; Zhu, Z.; Yang, C.J. Massively Parallel Single-Molecule and Single-Cell Emulsion Reverse Transcription Polymerase Chain Reaction Using Agarose Droplet Microfluidics. Anal. Chem. 2012, 84, 3599–3606. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, A.; Devenish, S.R.A.; Kintses, B.; Colin, P.; Fischlechner, M.; Hollfelder, F. One in a Million: Flow Cytometric Sorting of Single Cell-Lysate Assays in Monodisperse Picolitre Double Emulsion Droplets for Directed Evolution. Anal. Chem. 2014, 86, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.K.; Ali, M.M.; Zhang, K.; Huang, S.S.; Peterson, E.; Digman, M.A.; Gratton, E.; Zhao, W. Rapid Detection of Single Bacteria in Unprocessed Blood Using Integrated Comprehensive Droplet Digital Detection. Nat. Commun. 2014, 5, 5427. [Google Scholar] [CrossRef] [PubMed]
- Schuler, F.; Schwemmer, F.; Trotter, M.; Wadle, S.; Zengerle, R.; von Stetten, F.; Paust, N. Centrifugal Step Emulsification Applied for Absolute Quantification of Nucleic Acids by Digital Droplet Rpa. Lab Chip 2015, 15, 2759–2766. [Google Scholar] [CrossRef] [PubMed]
- Madic, J.; Zocevic, A.; Senlis, V.; Fradet, E.; Andre, B.; Muller, S.; Dangla, R.; Droniou, M.E. Three-Color Crystal Digital PCR. Biomol. Detect. Quantif. 2016, 10, 34–46. [Google Scholar] [CrossRef] [PubMed]
- McDermott, G.P.; Do, D.; Litterst, C.M.; Maar, D.; Hindson, C.M.; Steenblock, E.R.; Legler, T.C.; Jouvenot, Y.; Marrs, S.H.; Bemis, A.; et al. Multiplexed Target Detection Using DNA-Binding Dye Chemistry in Droplet Digital PCR. Anal. Chem. 2013, 85, 11619–11627. [Google Scholar] [CrossRef] [PubMed]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex Picodroplet Digital PCR to Detect Kras Mutations in Circulating DNA from the Plasma of Colorectal Cancer Patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Bhattacharya, S.; Kotsopoulos, S.; Olson, J.; Taly, V.; Griffiths, A.D.; Link, D.R.; Larson, J.W. Multiplex Digital PCR: Breaking the One Target Per Color Barrier of Quantitative PCR. Lab Chip 2011, 11, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Navarro, E.; Serrano-Heras, G.; Castaño, M.J.; Solera, J. Real-Time Pcr Detection Chemistry. Clin. Chim. Acta 2015, 439, 231–250. [Google Scholar] [CrossRef] [PubMed]
- Sint, D.; Raso, L.; Traugott, M. Advances in Multiplex PCR: Balancing Primer Efficiencies and Improving Detection Success. Methods Ecol. Evol. 2012, 3, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.S.; Huggett, J.F.; Tzonev, S. Fundamentals of Multiplexing with Digital PCR. Biomol. Detect. Quantif. 2016, 10, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Lau, B.T.; Wood-Bouwens, C.; Ji, H.P. Robust Multiplexed Clustering and Denoising of Digital PCR Assays by Data Gridding. Anal. Chem. 2017, 89, 11913–11917. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Novak, T.; Garson, J.A.; Green, C.; Morris-Jones, S.D.; Miller, R.F.; Zumla, A. Differential Susceptibility of PCR Reactions to Inhibitors: An Important and Unrecognised Phenomenon. BMC Res. Notes 2008, 1, 70. [Google Scholar] [CrossRef] [PubMed]
- Lizardi, P.M.; Huang, X.; Zhu, Z.; Bray-Ward, P.; Thomas, D.C.; Ward, D.C. Mutation Detection and Single-Molecule Counting Using Isothermal Rolling-Circle Amplification. Nat. Genet. 1998, 19, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Compton, J. Nucleic Acid Sequence-Based Amplification. Nature 1991, 350, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef] [PubMed]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive Human Genome Amplification Using Multiple Displacement Amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Xu, Y.; Kong, H. Helicase-Dependent Isothermal DNA Amplification. Embo Rep. 2004, 5, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.T.; Little, M.C.; Nadeau, J.G.; Shank, D.D. Isothermal in Vitro Amplification of DNA by a Restriction Enzyme/DNA Polymerase System. Proc. Natl. Acad. Sci. USA 1992, 89, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Li, Z.; Liu, C.; Cheng, Y. Ultrasensitive Detection of Micrornas by Exponential Isothermal Amplification. Angew. Chem. Int. Ed. 2010, 49, 5498–5501. [Google Scholar] [CrossRef] [PubMed]
- Van Ness, J.; van Ness, L.K.; Galas, D.J. Isothermal Reactions for the Amplification of Oligonucleotides. Proc. Natl. Acad. Sci. USA 2003, 100, 4504–4509. [Google Scholar] [CrossRef] [PubMed]
- Schoepp, N.G.; Schlappi, T.S.; Curtis, M.S.; Butkovich, S.S.; Miller, S.; Humphries, R.M.; Ismagilov, R.F. Rapid Pathogen-Specific Phenotypic Antibiotic Susceptibility Testing Using Digital Lamp Quantification in Clinical Samples. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C.; Quake, S.R. Digital MDA for Enumeration of Total Nucleic Acid Contamination. Nucleic Acids Res. 2011, 39, e19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kang, D.K.; Ali, M.M.; Liu, L.; Labanieh, L.; Lu, M.; Riazifar, H.; Nguyen, T.N.; Zell, J.A.; Digman, M.A.; et al. Digital Quantification of miRNA Directly in Plasma Using Integrated Comprehensive Droplet Digital Detection. Lab Chip 2015, 15, 4217–4226. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Shen, F.; McCalla, S.E.; Kreutz, J.E.; Karymov, M.A.; Ismagilov, R.F. Mechanistic Evaluation of the Pros and Cons of Digital RT-LAMP for HIV-1 Viral Load Quantification on a Microfluidic Device and Improved Efficiency Via a Two-Step Digital Protocol. Anal. Chem. 2013, 85, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Schuler, F.; Siber, C.; Hin, S.; Wadle, S.; Paust, N.; Zengerle, R.; von Stetten, F. Digital Droplet Lamp as a Microfluidic App on Standard Laboratory Devices. Anal. Methods 2016, 8, 2750–2755. [Google Scholar] [CrossRef]
- Nixon, G.; Garson, J.A.; Grant, P.; Nastouli, E.; Foy, C.A.; Huggett, J.F. Comparative Study of Sensitivity, Linearity, and Resistance to Inhibition of Digital and Nondigital Polymerase Chain Reaction and Loop Mediated Isothermal Amplification Assays for Quantification of Human Cytomegalovirus. Anal. Chem. 2014, 86, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Dingle, T.C.; Sedlak, R.H.; Cook, L.; Jerome, K.R. Tolerance of Droplet-Digital PCR vs. Real-Time Quantitative PCR to Inhibitory Substances. Clin. Chem. 2013, 59, 1670–16702. [Google Scholar] [CrossRef] [PubMed]
- Hayden, R.T.; Gu, Z.; Ingersoll, J.; Abdul-Ali, D.; Shi, L.; Pounds, S.; Caliendo, A.M. Comparison of Droplet Digital PCR to Real-Time PCR for Quantitative Detection of Cytomegalovirus. J. Clin. Microbiol. 2013, 51, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Rački, N.; Dreo, T.; Gutierrez-Aguirre, I.; Blejec, A.; Ravnikar, M. Reverse Transcriptase Droplet Digital PCR Shows High Resilience to PCR Inhibitors from Plant, Soil and Water Samples. Plant Methods 2014, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xia, Q.; Yin, Y.; Wang, Z. Comparison of Droplet Digital PCR and Quantitative PCR Assays for Quantitative Detection of Xanthomonas citri Subsp. Citri. PLoS ONE 2016, 11, e0159004. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Inagaki, F. Molecular Quantification of Environmental DNA Using Microfluidics and Digital PCR. Syst. Appl. Microbiol. 2012, 35, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, R.H.; Kuypers, J.; Jerome, K.R. A Multiplexed Droplet Digital PCR Assay Performs Better Than qPCR on Inhibition Prone Samples. Diagn. Microbiol. Infect. Dis. 2014, 80, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Carbonneau, J.; Shelton, D.N.; Boivin, G. Optimization of Droplet Digital PCR from RNA and DNA Extracts with Direct Comparison to Rt-qPCR: Clinical Implications for Quantification of Oseltamivir-Resistant Subpopulations. J. Virol. Methods 2015, 224, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Fontanelli, G.; Baratè, C.; Ciabatti, E.; Guerrini, F.; Grassi, S.; del Re, M.; Morganti, R.; Petrini, I.; Arici, R.; Barsotti, S.; et al. Real-Time PCR and Droplet Digital PCR: Two Techniques for Detection of the JAK2V617F Mutation in Philadelphia-Negative Chronic Myeloproliferative Neoplasms. Int. J. Lab. Hematol. 2015, 37, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, A. Quantitative Cell-Free Circulating BRAFV600E Mutation Analysis by Use of Droplet Digital PCR in the Follow-up of Patients with Melanoma Being Treated with BRAF Inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Strain, M.C.; Lada, S.M.; Luong, T.; Rought, S.E.; Gianella, S.; Terry, V.H.; Spina, C.A.; Woelk, C.H.; Richman, D.D. Highly Precise Measurement of HIV DNA by Droplet Digital PCR. PLoS ONE 2013, 8, e55943. [Google Scholar] [CrossRef] [PubMed]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute Quantification by Droplet Digital PCR Versus Analog Real-Time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Alikian, M.; Whale, A.S.; Akiki, S.; Piechocki, K.; Torrado, C.; Myint, T.; Cowen, S.; Griffiths, M.; Reid, A.G.; Apperley, J.; et al. Rt-qPCR and Rt-Digital PCR: A Comparison of Different Platforms for the Evaluation of Residual Disease in Chronic Myeloid Leukemia. Clin. Chem. 2017, 63, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Kiselinova, M.; Pasternak, A.O.; de Spiegelaere, W.; Vogelaers, D.; Berkhout, B.; Vandekerckhove, L. Comparison of Droplet Digital PCR and Seminested Real-Time PCR for Quantification of Cell-Associated HIV-1 RNA. PLoS ONE 2014, 9, e85999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich, T.J.; Gallien, S.; Li, J.Z.; Pereyra, F.; Kuritzkes, D.R. Low-Level Detection and Quantitation of Cellular HIV-1 DNA and 2-LTR Circles Using Droplet Digital PCR. J. Virol. Methods 2012, 186, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Goh, H.-G.; Lin, M.; Fukushima, T.; Saglio, G.; Kim, D.; Choi, S.; Kim, S.; Lee, J.; Lee, Y.; Oh, S.; et al. Sensitive Quantitation of Minimal Residual Disease in Chronic Myeloid Leukemia Using Nanofluidic Digital Polymerase Chain Reaction Assay. Leuk. Lymphoma 2011, 52, 896–904. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metrics | Accuracy | Sensitivity | Upper Limit |
|---|---|---|---|
| Design parameter | Number of partitions Variation in partition volume | Total assay volume or -Number of partitions × volume of partition | Volume of partition |
| Comments | Relative accuracy scales like Variation in partition volume dominates inaccuracy at very high number of partitions | 1/(Total assay volume) | Benefits from small partition volume |
| Partitioning Method | Number of Partitions | Volume of Partitions | Principles | Reference |
|---|---|---|---|---|
| microfluidic valving | 104 | 10 nL | relies on the elasticity of the material | [56] |
| SlipChip | 104 | 10 nL | slipping for partitioning | [16,52,66] |
| open arrays of microwells | 105 | 10 pL | both active and passive strategies demonstrated | [61,62,63,64,67] |
| microfluidic chambers | 106 | 10 pL | pinning of the oil interface to isolate chambers | [37] |
| self-digitization | 104 | 10 nL | plug splitting within a network of chambers pre-wet with immiscible oil | [76,81] |
| self-filling | 103 | 10 nL | control of the pinned interfaces for controlled filling | [69] |
| spinning disk | 103 | 10 nL | [75] | |
| droplet generator | 104–106 | 101–102 pL | droplets used as partitions | [40,43,87,88,101,102,103,104,105] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quan, P.-L.; Sauzade, M.; Brouzes, E. dPCR: A Technology Review. Sensors 2018, 18, 1271. https://doi.org/10.3390/s18041271
Quan P-L, Sauzade M, Brouzes E. dPCR: A Technology Review. Sensors. 2018; 18(4):1271. https://doi.org/10.3390/s18041271
Chicago/Turabian StyleQuan, Phenix-Lan, Martin Sauzade, and Eric Brouzes. 2018. "dPCR: A Technology Review" Sensors 18, no. 4: 1271. https://doi.org/10.3390/s18041271
APA StyleQuan, P.-L., Sauzade, M., & Brouzes, E. (2018). dPCR: A Technology Review. Sensors, 18(4), 1271. https://doi.org/10.3390/s18041271