Amalgam Electrode-Based Electrochemical Detector for On-Site Direct Determination of Cadmium(II) and Lead(II) from Soils

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Material
2.2. Modification of Electrolytic Copper Wire to Serve as Working Electrode (Cu/Hg-WE)
2.3. Electrochemical Determination of Cadmium and Lead by the Amalgam Electrode
2.4. Electrochemical Determination of Cadmium and Lead by HMDE
2.5. Atomic Absorption Spectrometry (AAS)
2.6. Descriptive Statistics
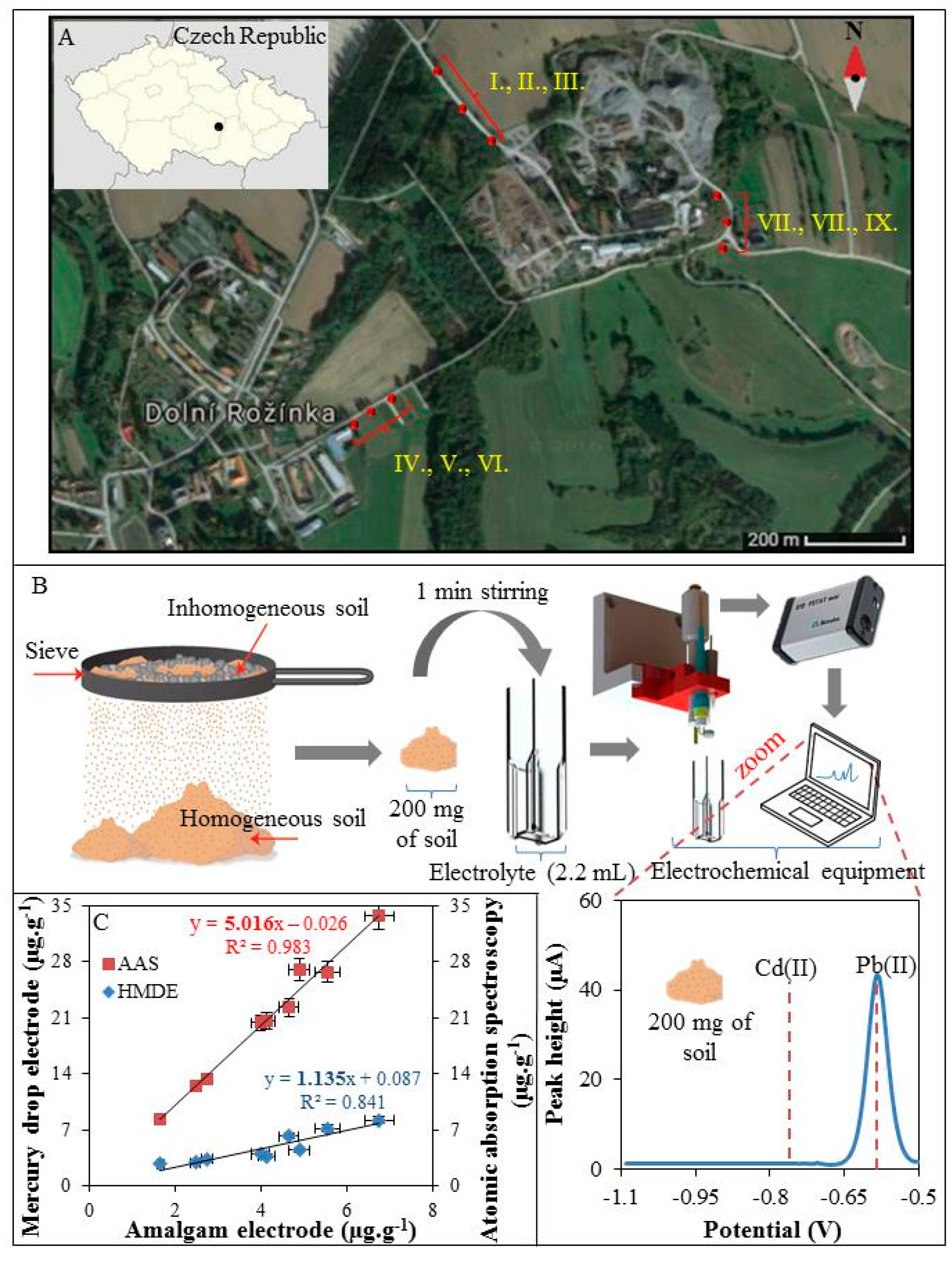
2.7. Description of the Studied Sites (Dolni Rozinka, Czech Republic) and Motivation for On-site Analysis
3. Results and Discussion
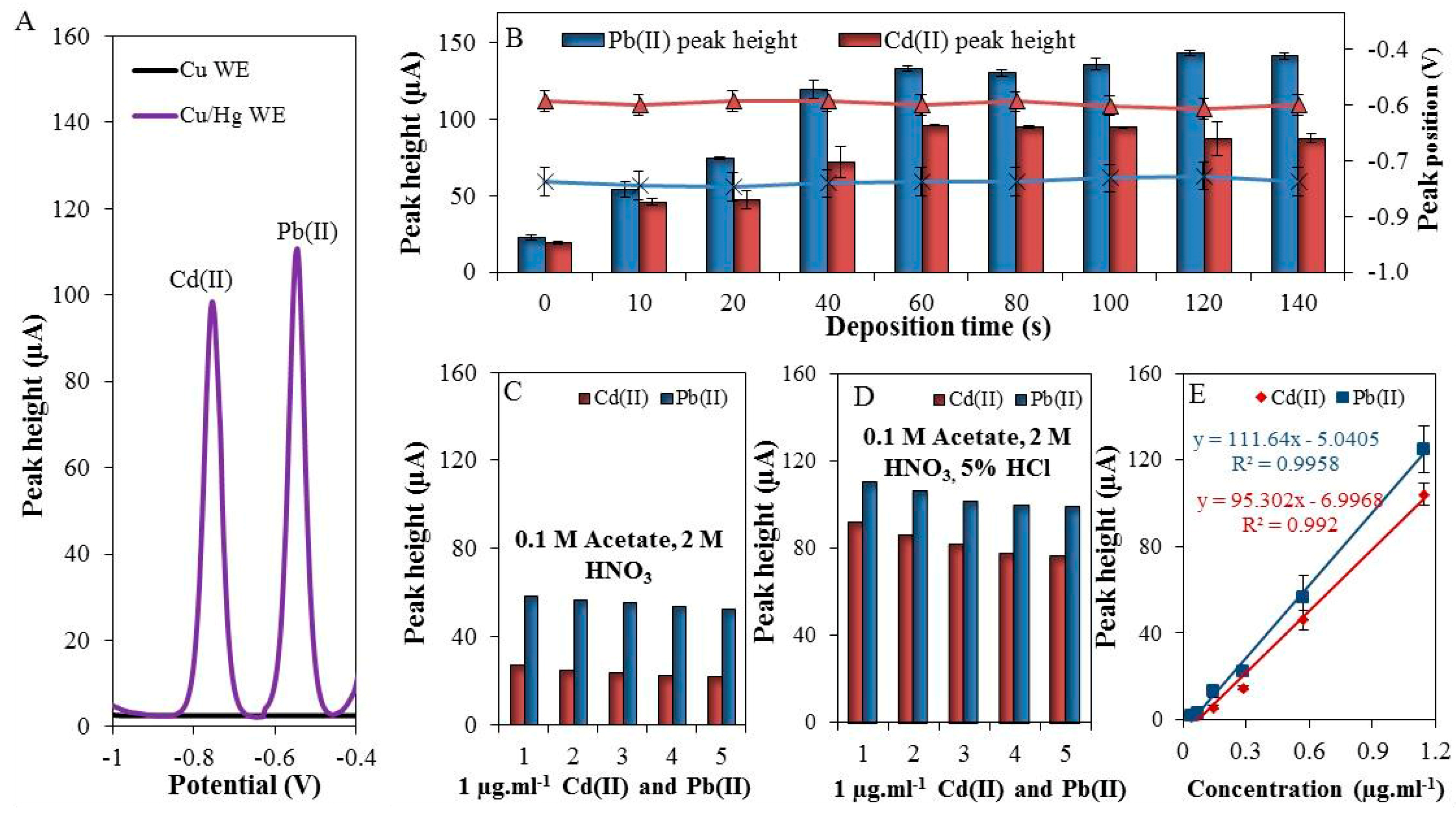
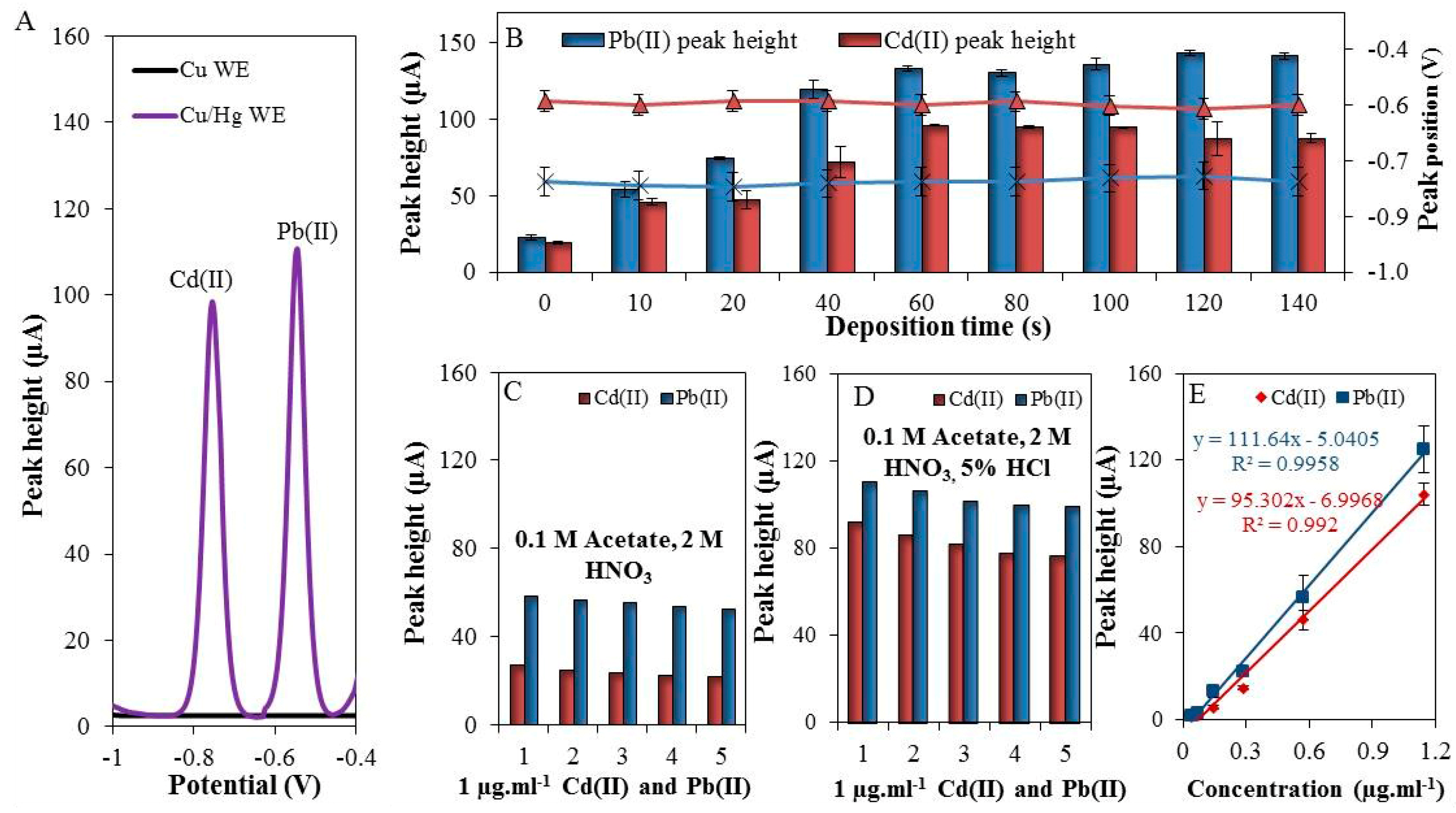
3.1. Preparation of Homemade Working Electrode
3.2. The Optimization of Pb(II) and Cd(II) Detection
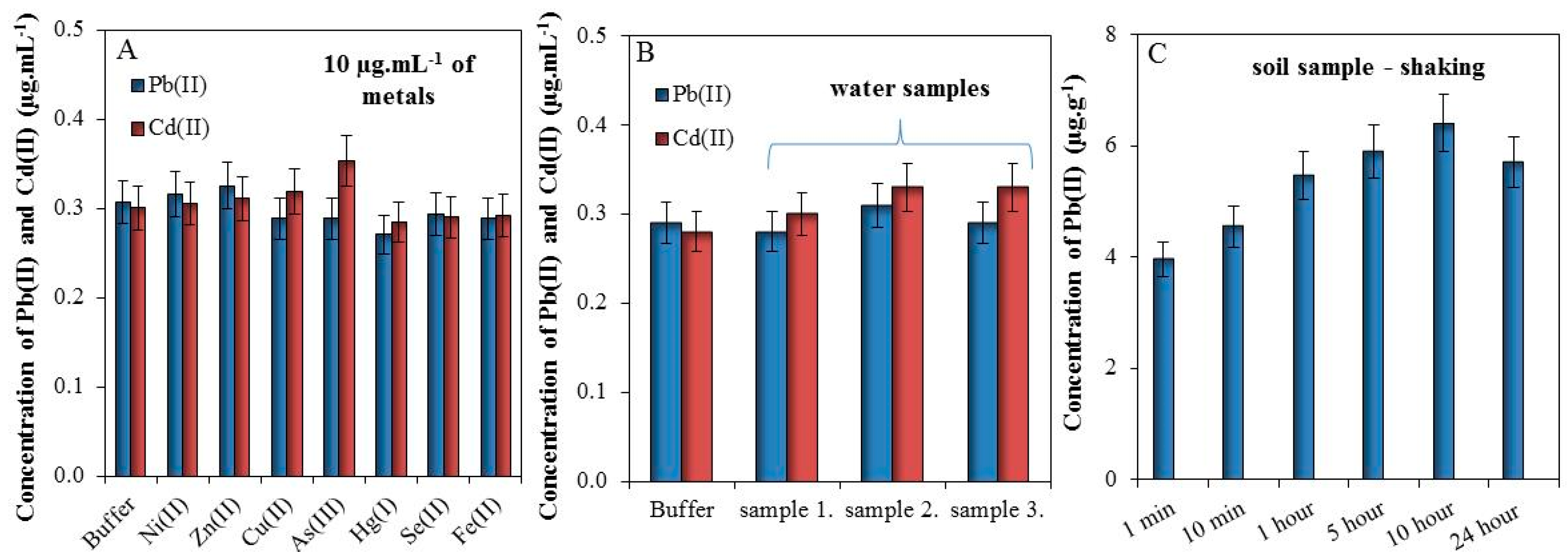
3.3. Study of Interfering Substances and Time of Extraction
3.4. On-Site Analysis of Soils Samples
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sun, H.F.; Li, Y.H.; Ji, Y.F.; Yang, L.S.; Wang, W.Y.; Li, H. Environmental contamination and health hazard of lead and cadmium around Chatian mercury mining deposit in western Hunan Province, China. Trans. Nonferr. Met. Soc. China 2010, 20, 308–314. [Google Scholar] [CrossRef]
- Petroczi, A.; Naughton, D.P. Mercury, cadmium and lead contamination in seafood: A comparative study to evaluate the usefulness of Target Hazard Quotients. Food Chem. Toxicol. 2009, 47, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Rahman, S.; Vance, G.F. Munn LC Geostatistical analyses of trace-elements in soils and plants. Soil Sci. 1995, 159, 383–390. [Google Scholar] [CrossRef]
- Nejdl, L.; Nguyen, H.V.; Richtera, L.; Krizkova, S.; Guran, R.; Masarik, M.; Hynek, D.; Heger, Z.; Lundberg, K.; Erikson, K.; et al. Label-free bead-based metallothionein electrochemical immunosensor. Electrophoresis 2015, 36, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Lamble, K.J.; Hill, S.J. Microwave digestion procedures for environmental matrices. Analyst 1998, 123. [Google Scholar] [CrossRef]
- Alves, G.M.S.; Magalhaes, J.; Salaun, P.; van den Berg, C.M.G.; Soares, H. Simultaneous electrochemical determination of arsenic, copper, lead and mercury in unpolluted fresh waters using a vibrating gold microwire electrode. Anal. Chim. Acta 2011, 703, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nejdl, L.; Ruttkay-Nedecky, B.; Kudr, J.; Kremplova, M.; Cernei, N.; Prasek, J.; Konecna, M.; Hubalek, J.; Zitka, O.; Kynicky, J.; et al. Behaviour of Zinc Complexes and Zinc Sulphide Nanoparticles Revealed by Using Screen Printed Electrodes and Spectrometry. Sensors 2013, 13, 14417–14437. [Google Scholar] [CrossRef] [PubMed]
- Hynek, D.; Krejcova, L.; Sochor, J.; Cernei, N.; Kynicky, J.; Adam, V.; Trnkova, L.; Hubalek, J.; Vrba, R.; Kizek, R. Study of Interactions between Cysteine and Cadmium(II) Ions using Automatic Pipetting System off-line Coupled with Electrochemical Analyser Dedicated United Nation Environment Program: Lead and Cadmium Initiatives. Int. J. Electrochem. Sci. 2012, 7, 1802–1819. [Google Scholar]
- Barcelo-Quintal, M.H.; Manzanilla-Cano, J.A.; Reyes-Salas, E.O.; Flores-Rodriguez, J. Implementation of a differential pulse anodic stripping voltammetry (DPASV) at a hanging mercury drop electrode (HMDE) procedure for the analysis of airborne heavy metals. Anal. Lett. 2001, 34, 2349–2360. [Google Scholar] [CrossRef]
- Fernandez-Bobes, C.; Fernandez-Abedul, M.T.; Costa-Garcia, A. Anodic stripping of heavy metals using a hanging mercury drop electrode in a flow system. Electroanalysis 1998, 10, 701–706. [Google Scholar] [CrossRef]
- Fogg, A.G.; Ismail, R.; Yusoff, A.; Ahmad, R.; Banica, F.G. Cathodic stripping voltammetric determination at a hanging mercury drop electrode of the environmental heavy metal precipitant trimercapto-s-triazine (TMT). Talanta 1997, 44, 497–500. [Google Scholar] [CrossRef]
- Economou, A.; Fielden, P.R. Mercury film electrodes: Developments, trends and potentialities for electroanalysis. Analyst 2003, 128, 205–212. [Google Scholar] [CrossRef] [PubMed]
- McCreery, R.L. Advanced carbon electrode materials for molecular electrochemistry. Chem. Rev. 2008, 108, 2646–2687. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M. Recent Nanoarchitectures in Metal Nanoparticle-modified Electrodes for Electroanalysis. Anal. Sci. 2010, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.W.; Foord, J.S. Nanoparticle-Based Diamond Electrodes. In Novel Aspects of Diamond: From Growth to Applications; Yang, N., Ed.; Springer: Berlin, Germany, 2015; pp. 165–204. [Google Scholar]
- Amato, L.; Schulte, L.; Heiskanen, A.; Keller, S.S.; Ndoni, S.; Emnéus, J. Novel Nanostructured Electrodes Obtained by Pyrolysis of Composite Polymeric Materials. Electroanalysis 2015, 27, 1544–1549. [Google Scholar] [CrossRef]
- Ramachandran, R.; Chen, S.M.; Kumar, G.P.G.; Gajendran, P.; Devi, N.B. An Overview of Fabricating Nanostructured Electrode Materials for Biosensor Applications. Int. J. Electrochem. Sci. 2015, 10, 8607–8629. [Google Scholar]
- Hao, C.; Shen, Y.R.; Shen, J.X.; Xu, K.Y.; Wang, X.H.; Zhao, Y.; Ge, C. A glassy carbon electrode modified with bismuth oxide nanoparticles and chitosan as a sensor for Pb(II) and Cd(II). Microchim. Acta 2016, 183, 1823–1830. [Google Scholar] [CrossRef]
- Yang, D.; Wang, L.; Chen, Z.L.; Megharaj, M.; Naidu, R. Anodic stripping voltammetric determination of traces of Pb(II) and Cd(II) using a glassy carbon electrode modified with bismuth nanoparticles. Microchim. Acta 2014, 181, 1199–1206. [Google Scholar] [CrossRef]
- Do Nascimento, M.E.; Martelli, P.B.; Furtado, C.A.; Santos, A.P.; de Oliveira, L.F.C.; de Fátima Gorgulho, H. Determination of lead(II) in aqueous solution using carbon nanotubes paste electrodes modified with Amberlite IR-120. Microchim. Acta 2011, 173, 485–493. [Google Scholar] [CrossRef]
- Vanderlinden, W.E.; Dieker, J.W. Glassy-carbon as electrode material in electroanalytical chemistry. Anal. Chim. Acta 1980, 119, 1–24. [Google Scholar] [CrossRef]
- Svancara, I.; Vytras, K.; Barek, J.; Zima, J. Carbon paste electrodes in modern electroanalysis. Crit. Rev. Anal. Chem. 2001, 31, 311–345. [Google Scholar] [CrossRef]
- Shaidarova, L.G.; Budnikov, G.K. Chemically modified electrodes based on noble metals, polymer films, or their composites in organic voltammetry. J. Anal. Chem. 2008, 63, 922–942. [Google Scholar] [CrossRef]
- Tallman, D.E.; Petersen, S.L. Composite electrodes for electroanalysis—Principles and applications. Electroanalysis 1990, 2, 499–510. [Google Scholar] [CrossRef]
- Green, R.A.; Baek, S.; Poole-Warren, L.A.; Martens, P.J. Conducting polymer-hydrogels for medical electrode applications. Sci. Technol. Adv. Mater. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.T.; Shao, G.J.; Ji, M.T.; Ma, Z.P. Research Progress in Nano-Structured MnO2 as Electrode Materials for Supercapacitors. Asian J. Chem. 2013, 25, 7065–7070. [Google Scholar]
- Xu, J.H.; Wang, Y.Z.; Hu, S.S. Nanocomposites of graphene and graphene oxides: Synthesis, molecular functionalization and application in electrochemical sensors and biosensors. A review. Microchim. Acta 2017, 184, 1–44. [Google Scholar] [CrossRef]
- Dos Santos, V.B.; Fava, E.L.; Curi, N.S.D.; Faria, R.C.; Guerreiro, T.B.; Fatibello-Filho, O. An electrochemical analyzer for in situ flow determination of Pb(II) and Cd(II) in lake water with on-line data transmission and a global positioning system. Anal. Methods 2015, 7, 3105–3112. [Google Scholar] [CrossRef]
- Nejdl, L.; Kudr, J.; Cihalova, K.; Chudobova, D.; Zurek, M.; Žalud, L.; Kopecný, L.; Burian, F.; Ruttkay-Nedecký, B.; Prášek, J.; et al. Remote-controlled robotic platform Orpheus as a new tool for detection of bacteria in the environment. Electrophoresis 2014, 35, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.; Garcia, M.B.G.; Santos, D.H.; Fanjul-Bolado, P.; Ribotti, A.; McCaul, M.; Diamond, D.; Magni, P. Screen-printed electrodes for environmental monitoring of heavy metal ions: A review. Microchim. Acta 2016, 183, 503–517. [Google Scholar] [CrossRef]
- Li, M.; Li, Y.T.; Li, D.W.; Long, Y.T. Recent developments and applications of screen-printed electrodes in environmental assays—A review. Anal. Chim. Acta 2012, 734, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Yosypchuk, B.; Novotny, L. Nontoxic electrodes of solid amalgams. Crit. Rev. Anal. Chem. 2002, 32, 141–151. [Google Scholar] [CrossRef]
- Mikkelsen, O.; Schroder, K.H. Amalgam electrodes for electroanalysis. Electroanalysis 2003, 15, 679–687. [Google Scholar] [CrossRef]
- Yosypchuk, B.; Novotny, L. Copper solid amalgam electrodes. Electroanalysis 2003, 15, 121–125. [Google Scholar] [CrossRef]
- Yosypchuk, B.; Barek, J. Analytical Applications of Solid and Paste Amalgam Electrodes. Crit. Rev. Anal. Chem. 2009, 39, 189–203. [Google Scholar] [CrossRef]
- Jelen, F.; Yosypchuk, B.; Kourilova, A.; Novotny, L.; Palecek, E. Label-free determination of picogram quantities of DNA by stripping voltammetry with solid copper amalgam or mercury electrodes in the presence of copper. Anal. Chem. 2002, 74, 4788–4793. [Google Scholar] [CrossRef] [PubMed]
- Novakova, K.; Navratil, T.; Dytrtova, J.J.; Chylkova, J. Use of Copper Solid Amalgam Electrode for Determination of Triazolic Fungicide Tebuconazole. In Xxxii Moderni Elektrochemicke Metody; J. Heyrovsky Institute of Physical Chemistry AS ČR: Prague, Czech Republic, 2012; pp. 87–90. [Google Scholar]
- De Souza, D.; de Toledo, R.A.; Mazo, L.H.; Machado, S.A.S. Utilization of a copper solid amalgam electrode for the analytical determination of atrazine. Electroanalysis 2005, 17, 2090–2094. [Google Scholar] [CrossRef]
- Yosypchuk, B.; Sestakova, I.; Novotny, L. Voltammetric determination of phytochelatins using copper solid amalgam electrode. Talanta 2003, 59, 1253–1258. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, H.; Liu, G. Electrochemical Determination of Trace Cadmium in Soil by a Bismuth Film/Graphene-β-cyclodextrin-Nafion Composite Modified Electrode. Int. J. Electrochem. Sci. 2016, 11, 1840–1851. [Google Scholar]
- Zhao, G.; Wang, H.; Liu, G.; Wang, Z.Q.; Cheng, J. Simultaneous determination of trace Cd(II) and Pb(II) based on Bi/Nafion/reduced graphene oxide-gold nanoparticle nanocomposite film-modified glassy carbon electrode by one-step electrodeposition. Ionics 2017, 23, 767–777. [Google Scholar] [CrossRef]
- Aragay, G.; Puig-Font, A.; Cadevall, M.; Merkoci, A. Surface Characterizations of Mercury-Based Electrodes with the Resulting Micro and Nano Amalgam Wires and Spheres Formations May Reveal Both Gained Sensitivity and Faced Nonstability in Heavy Metal Detection. J. Phys. Chem. C 2010, 114, 9049–9055. [Google Scholar] [CrossRef]
- Golimowski, J.; Golimowska, K. UV-photooxidation as pretreatment step in inorganic analysis of environmental samples. Anal. Chim. Acta 1996, 325, 111–133. [Google Scholar] [CrossRef]
- Nascimento, P.C.; Del-Fabro, L.D.; Bohrer, D.; De Carvalho, L.M.; Rosa, M.B.; Noremberg, S.M. Al(III) and Fe(III) Balance in Hemodialysis Treatment Assessed via Fluid Analysis by Adsorptive Stripping Voltammetry and UV Sample Digestion. Electroanalysis 2008, 20, 1078–1084. [Google Scholar] [CrossRef]
- Baccaro, A.L.B.; Gutz, I.G.R. Novel photoelectrocatalytic approach aiming at the digestion of water samples, estimation of organic matter content and stripping analysis of metals in a special UV-LED irradiated cell with a TiO2-modified gold electrode. Electrochem. Commun. 2013, 31, 28–30. [Google Scholar] [CrossRef]
- Li, X.Z.; Li, F.B. Study of Au/Au3+-TiO2 photocatalysts toward visible photooxidation for water and wastewater treatment. Environ. Sci. Technol. 2001, 35, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Pekakis, P.A.; Xekoukoulotakis, N.P.; Mantzavinos, D. Treatment of textile dyehouse wastewater by TiO2 photocatalysis. Water Res. 2006, 40, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Wuana, R.A.; Okieimen, F.E. Heavy Metals in Contaminated Soils: A Review of Sources, Chemistry, Risks and Best Available Strategies for Remediation. ISRN Ecol. 2011, 2011, 1–20. [Google Scholar] [CrossRef]
- Perez-Sirvent, C.; Martinez-Sanchez, M.J.; Garcia-Lorenzo, M.L.; Molina, J.; Tudela, M.L. Geochemical background levels of zinc, cadmium and mercury in anthropically influenced soils located in a semi-arid zone (SE, Spain). Geoderma 2009, 148, 307–317. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Electrode Area (mm2) | Amalgamation Time (s) | Deposition Time (s) | Regression Equation | Linear Dynamic Range (ng·mL−1) | R2 | LOD (ng·mL−1) | RSD (%) |
|---|---|---|---|---|---|---|---|---|
| Cd(II) | 15 | 180 | 60 | y = 95.302x − 6.9968 | 200–1143 | 0.996 | 80 | 8 |
| Pb(II) | 15 | 180 | 60 | y = 111.64x − 5.0405 | 200–1143 | 0.993 | 50 | 7.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nejdl, L.; Kynicky, J.; Brtnicky, M.; Vaculovicova, M.; Adam, V. Amalgam Electrode-Based Electrochemical Detector for On-Site Direct Determination of Cadmium(II) and Lead(II) from Soils. Sensors 2017, 17, 1835. https://doi.org/10.3390/s17081835
Nejdl L, Kynicky J, Brtnicky M, Vaculovicova M, Adam V. Amalgam Electrode-Based Electrochemical Detector for On-Site Direct Determination of Cadmium(II) and Lead(II) from Soils. Sensors. 2017; 17(8):1835. https://doi.org/10.3390/s17081835
Chicago/Turabian StyleNejdl, Lukas, Jindrich Kynicky, Martin Brtnicky, Marketa Vaculovicova, and Vojtech Adam. 2017. "Amalgam Electrode-Based Electrochemical Detector for On-Site Direct Determination of Cadmium(II) and Lead(II) from Soils" Sensors 17, no. 8: 1835. https://doi.org/10.3390/s17081835