Determination of Chloride Content in Cementitious Materials: From Fundamental Aspects to Application of Ag/AgCl Chloride Sensors


Abstract
1. Introduction
- In the first section, the main hydration products of cement paste in the bulk matrix and at the sensor–cement paste interface are described. The composition of the pore solution of cementitious materials, i.e., the various ions in contact with the sensor’s surface and hence of importance for sensors’ readings, is also assessed.
- Next, the classification of chloride ions in cementitious materials is reviewed and the importance of chloride sensors for local chloride measurements, specifically at the level of the reinforcement, is discussed.
- Section 4 presents the different techniques for the determination of chloride content in concrete and the advantages of chloride sensors in particular for free chloride determination.
- The next section bridges fundamental considerations and technical background, both related to the performance, characterization and application of Ag/AgCl sensors. The advantages and drawbacks of the chloride sensors are discussed. The thermodynamic behaviour of silver and the electrochemical kinetics of AgCl formation are discussed. Then, the relevance of these parameters to the reliability of the sensors’ response in cementitious materials is evaluated. The above framework outlines the structure of this paper, highlighting the principles, related mechanisms and determining factors for monitoring of chloride ions in cementitious materials.
2. Important Properties of Cementitious Materials in View of Chloride Measurements via Ag/AgCl Sensors
2.1. Hydration Products and Microstructure of Cement Paste
2.2. Ions in Pore Solution
2.2.1. Portland Cement
2.2.2. Blast Furnace Slag Cement
3. Chloride Ions in Concrete
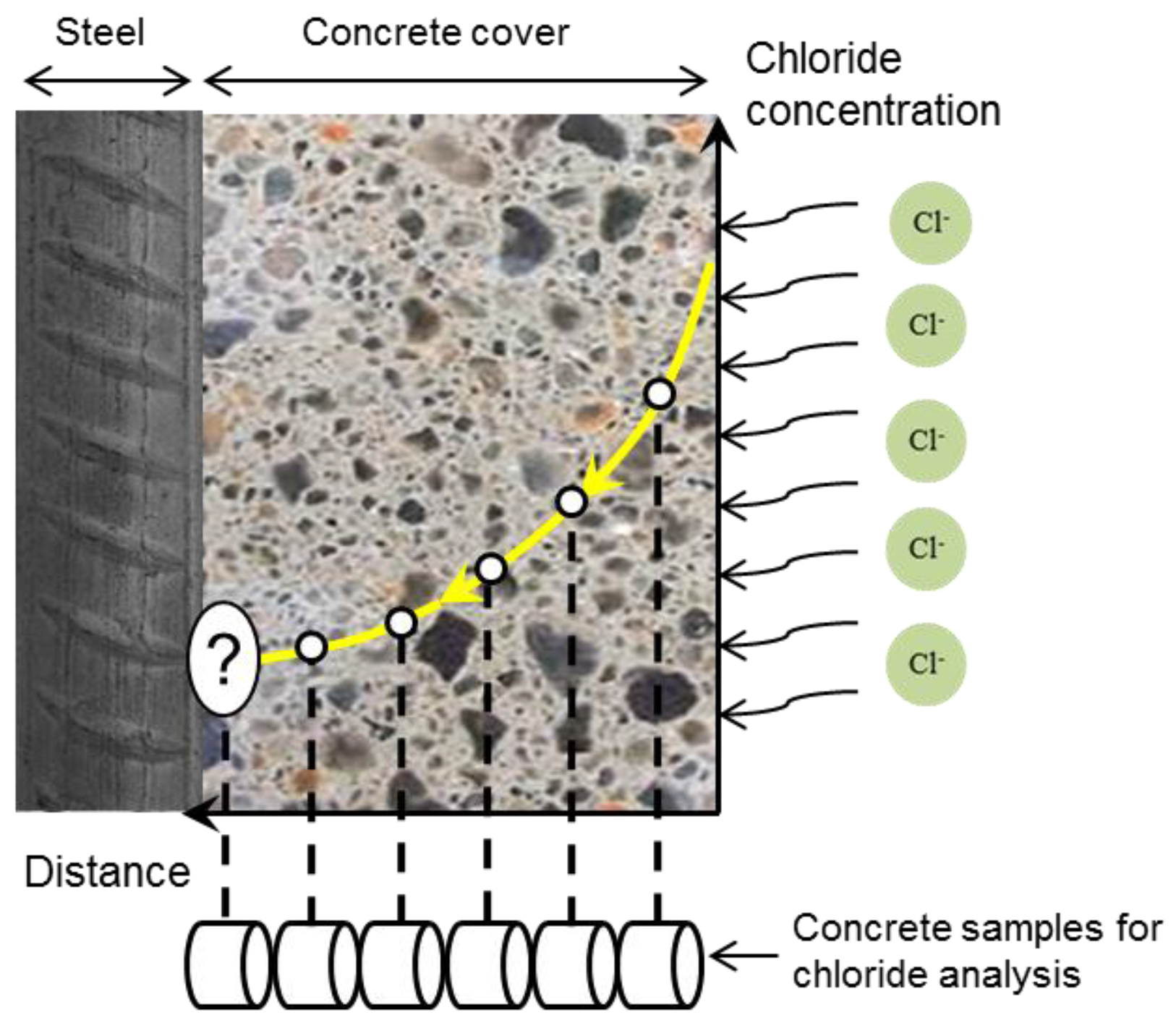
3.1. Classification of Chloride Ions
3.2. Free Chloride
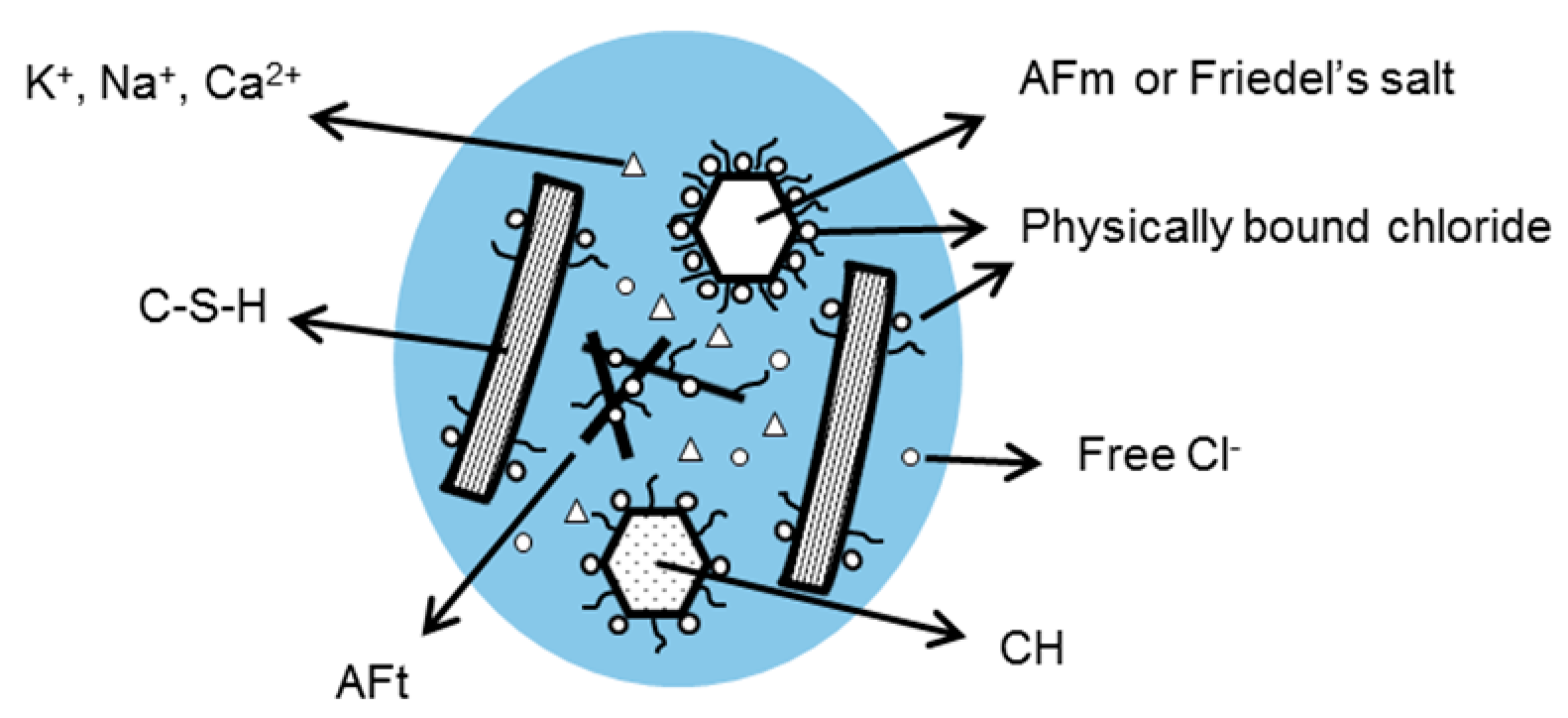
3.3. Bound Chloride
3.3.1. Chemical Binding of Chloride
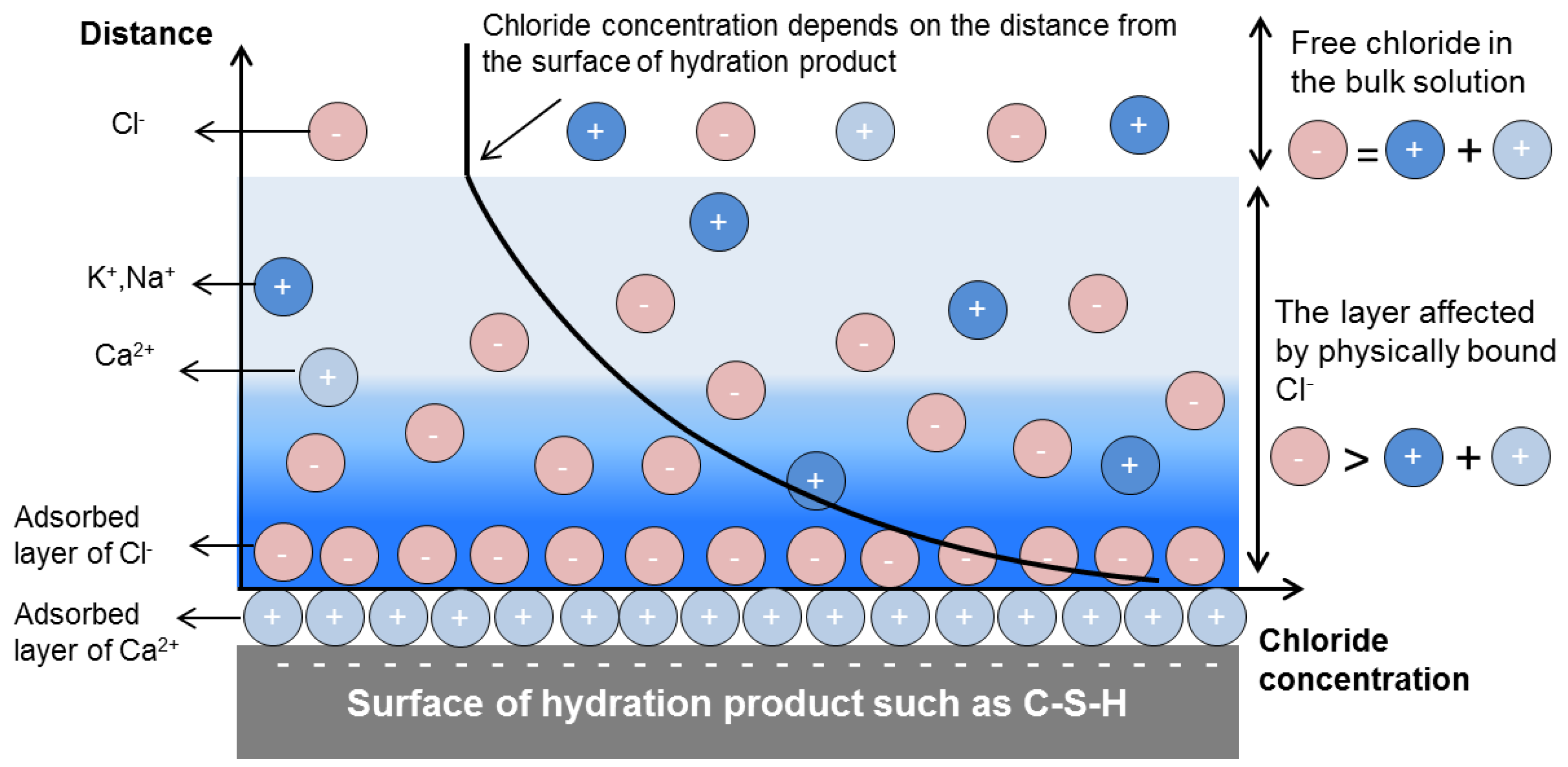
3.3.2. Physical Binding of Chloride
3.4. Chloride Threshold Value
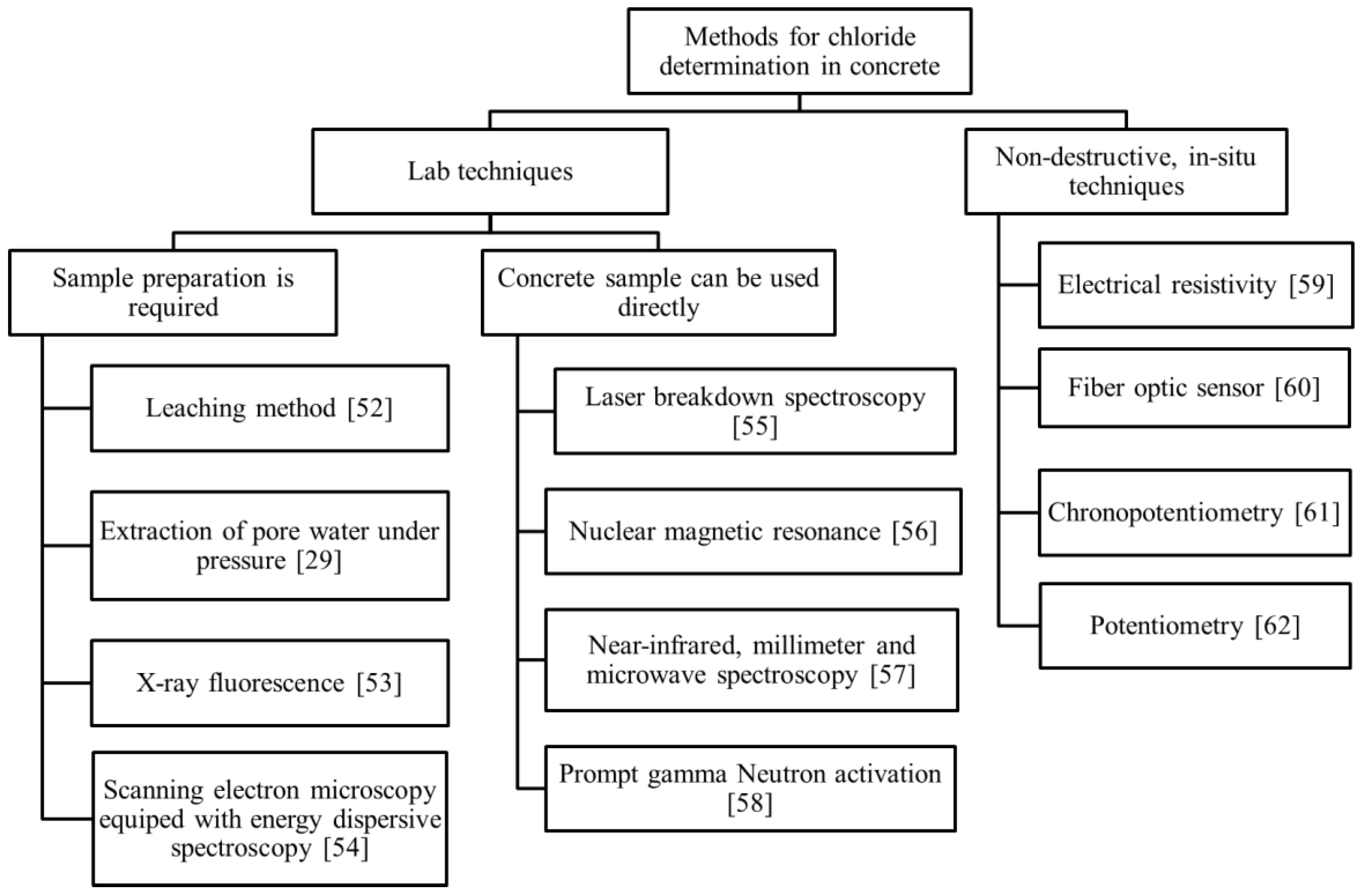
4. Techniques for Determination of the Chloride Content in Concrete
4.1. Lab Techniques
4.2. Non-Destructive In Situ Techniques
5. Working Principles of the Ag/AgCl Electrode
6. Discussion
6.1. Electrochemical Kinetics of AgCl Layer Formation
6.2. Thermodynamic Behaviour of Silver
6.3. Considerations with Regard to Chloride Ions’ Activity and pH
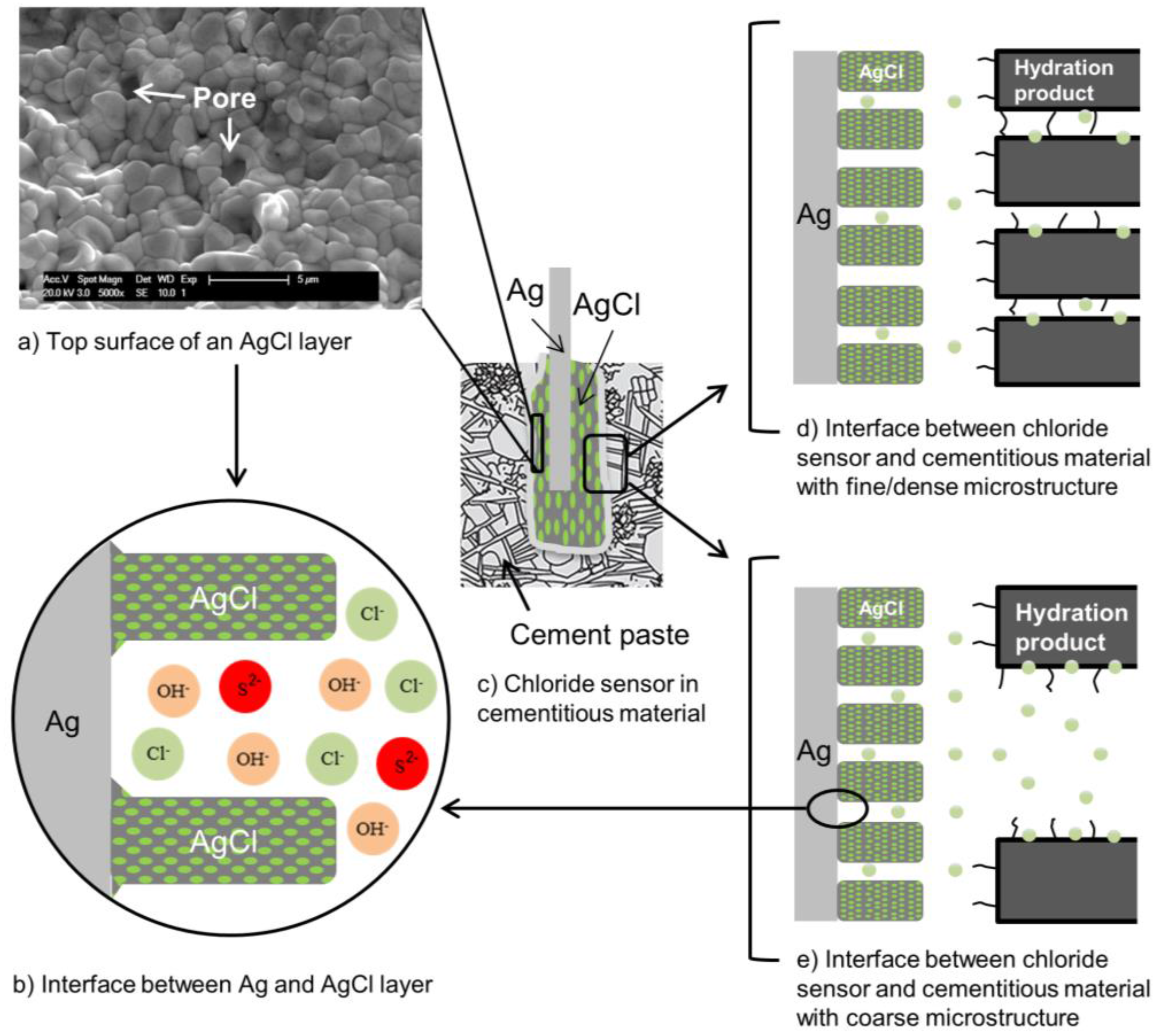
6.4. Reliability of Sensor Response in Cementitious Materials
7. Conclusions
- The interfacial properties between the silver substrate and the AgCl layer, i.e., morphology of the AgCl particles and the conductivity of the layer.
- The presence of interfering hydroxide and sulphide ions, i.e., the pore water composition.
- The relative distribution and compactness of cement hydration products at the sensor’s surface, i.e., the occurrence of “screening” effects.
Conflicts of Interest
References
- Molina, M. Zerstörungsfreie Erfassung der Gelösten Chloride im Beton. Ph.D. Thesis, ETH Zurich, Zürich, Switzerland, 1993. (In Germany). [Google Scholar]
- Bard, A.J.; Rubinstein, I. Electroanalytical Chemistry: A Series of Advances; Marcel Dekker, Inc.: New York, NY, USA, 1996; Volume 22. [Google Scholar]
- Hewlett, P.C. Lea’s Chemistry of Cement and Concrete, 4th ed.; Butterworth-Heinemann, Elsevier Ltd.: Oxford, UK, 2003. [Google Scholar]
- Lothenbach, B.; Le Saout, G.; Haha, M.B.; Figi, R.; Wieland, E. Hydration of a low-alkali CEM III/B–SiO2 cement (LAC). Cem. Concr. Res. 2012, 42, 410–423. [Google Scholar] [CrossRef]
- Mehta, P.K.; Monterio, P.J.M. Concrete, Microstructure, Properties and Materials, 3rd ed.; McGraw-Hill Companies: New York, NY, USA, 2006. [Google Scholar]
- Van Breugel, K. Simulation of Hydration and Formation of Structure in Hardening Cement-Based Materials. Ph.D. Thesis, Delft University of Technology, Delft, The Netherlands, 1991. [Google Scholar]
- Ollivier, J.P.; Maso, J.C.; Bourdette, B. Interfacial transition zone in concrete. Adv. Cem. Based Mater. 1995, 2, 30–38. [Google Scholar] [CrossRef]
- Swamy, R.N. The Alkali-Silica Reaction in Concrete; The Blackie Publishing Group: Glasgow, UK, 1992. [Google Scholar]
- Elakneswaran, Y.; Nawa, T.; Kurumisawa, K. Electrokinetic potential of hydrated cement in relation to adsorption of chlorides. Cem. Concr. Res. 2009, 39, 340–344. [Google Scholar] [CrossRef]
- Longuet, P.; Burglen, L.; Zelwer, A. La phase liquide du ciment hydrate. Rev. Mater. Constr. 1973, 676, 35–41. [Google Scholar]
- Barneyback, R.S.; Diamond, S. Expression and analysis of pore fluids from hardened cement pastes and mortars. Cem. Concr. Res. 1981, 11, 279–285. [Google Scholar] [CrossRef]
- Page, C.L.; Vennesland, Ø. Pore solution composition and chloride binding capacity of silica-fume cement pastes. Matér. Constr. 1983, 16, 19–25. [Google Scholar] [CrossRef]
- Lothenbach, B.; Winnefeld, F. Thermodynamic modelling of the hydration of Portland cement. Cem. Concr. Res. 2006, 36, 209–226. [Google Scholar] [CrossRef]
- Diamond, S. ASR—Another Look at Mechanisms. In Proceedings of the Eighth International Conference on Alkali-Aggregate Reaction in Concrete, Kyoto, Japan, 17–20 July 1989; pp. 83–94. [Google Scholar]
- Baron, J.; Ollivier, J.P. La durabilité des Bétons; Presse de l’École Nationale des Ponts et chaussées: Paris, France, 1992. (In French) [Google Scholar]
- Garcia, V.; Francois, R.; Carcasses, M.; Gegout, P. Potential measurement to determine the chloride threshold concentration that initiates corrosion of reinforcing steel bar in slag concretes. Mater. Struct. 2014, 47, 1483–1499. [Google Scholar] [CrossRef]
- Glass, G.K.; Wang, Y.; Buenfeld, N.R. An investigation of experimental methods used to determine free and total chloride contents. Cem. Concr. Res. 1996, 26, 1443–1449. [Google Scholar] [CrossRef]
- Analysis of total chloride content in concrete. Available online: https://www.rilem.net/images/publis/1340.pdf (Accessed on 28 October 2017).
- ASTM C1152/C1152M–04(2012)e1. Standard Test Method for Acid-Soluble Chloride in Mortar and Concrete; ASTM International: Philadelphia, PA, USA, 2003. [Google Scholar]
- Nordtest. Concrete, Hardened: Chloride Content by Volhard Titration; NT Build 208; Nordtest: Espoo, Finland, 1996. [Google Scholar]
- Dhir, R.K.; Jones, M.R.; Ahmed, H.E.H. Determination of total and soluble chlorides in concrete. Cem. Concr. Res. 1990, 20, 579–590. [Google Scholar] [CrossRef]
- Arya, C. Assessment of Chlorides in Concrete. Ph.D. Thesis, University of London, London, UK, 1988. [Google Scholar]
- Broomfield, J.P. Corrosion of Steel in Concrete, 2nd ed.; Taylor & Francis: London, UK, 2007. [Google Scholar]
- Birnin-Yauri, U.A.; Glasser, F.P. Friedel’s salt, Ca2Al(OH)6(Cl,OH)·2H2O: Its solid solutions and their role in chloride binding. Cem. Concr. Res. 1998, 28, 1713–1723. [Google Scholar] [CrossRef]
- Maslehuddin, M.; Page, C.L. Rasheeduzzafar Temperature effect on the pore solution chemistry in contaminated cements. Mag. Concr. Res. 1997, 49, 5–14. [Google Scholar] [CrossRef]
- Zibara, H. Binding of External Chloride by Cement Pastes. Ph.D. Thesis, Department of Building Materials, University of Toronto, Toronto, ON, Canada, 2001. [Google Scholar]
- Chatterji, S.; Kawamura, M. Electrical double layer, ion transport and reactions in hardened cement paste. Cem. Concr. Res. 1992, 22, 774–782. [Google Scholar] [CrossRef]
- Elakneswaran, Y. Multi-Ionic Transport in Cementitious Materials with Ion-Cement Hydrates Interactions. Ph.D. Thesis, Hokkaido University, Solid Waste, Resources and Geoenviromental Engineering, Sapporo, Japan, 2009. [Google Scholar]
- Tritthart, J. Chloride binding in Cement II. The influence of the hydroxide concentration in the pore solution of hardened cement paste on chloride binding. Cem. Concr. Res. 1989, 19, 683–691. [Google Scholar] [CrossRef]
- Birdi, K.A.S. Handbook of Surface and Colloid Chemistry; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Friedmann, H.; Amiri, O.; Ait-Mokhtar, A. Physical modeling of the electrical double layer effects on multispecies ions transport in cement-based materials. Cem. Concr. Res. 2008, 38, 1394–1400. [Google Scholar] [CrossRef]
- Maage, M.; Helland, S.; Poulsen, E.; Vennesland, Ø.; Carlsen, J.E. Service life prediction of existing concrete structures exposed to marine environment. ACI Mater. J. 1996, 93, 602–608. [Google Scholar]
- Hausmann, D.A. Steel corrosion in concrete. How does it occur? Mater. Prot. 1967, 6, 19–23. [Google Scholar]
- Melchers, R.E.; Li, C.Q. Reinforcement corrosion initiation and activation times in concrete structures exposed to severe marine environments. Cem. Concr. Res. 2009, 39, 1068–1076. [Google Scholar] [CrossRef]
- Melchers, R.E. Developing realistic deterioration models. In Life-Cycle of Engineering Systems: Emphasis on Sustainable Civil Infrastructure; CRC Press: Boca Raton, FL, USA, 2016; pp. 28–32. [Google Scholar]
- Alonso, C.; Andrade, C.; Castellote, M.; Castro, P. Chloride threshold values to depassivate reinforcing bars embedded in a standardized OPC mortar. Cem. Concr. Res. 2000, 30, 1047–1055. [Google Scholar] [CrossRef]
- Alonso, C.; Castellote, M.; Andrade, C. Chloride threshold dependence of pitting potential of reinforcements. Electrochim. Acta 2002, 47, 3469–3481. [Google Scholar] [CrossRef]
- Oh, B.H.; Jang, S.Y.; Shin, Y.S. Experimental investigation of the threshold chloride concentration for corrosion initiation in reinforced concrete structures. Mag. Concr. Res. 2003, 55, 117–124. [Google Scholar] [CrossRef]
- Anon. Condition Investigation Manual for Concrete Facade Panels: BY 42; Concrete Association of Finland: Helsinki, Finland, 2002; p. 178. (In Finnish) [Google Scholar]
- Stratfull, R.F.; Jurkovich, W.J.; Spellman, D.L. Corrosion Testing of Bridge Decks; Highway Research Record 539; Transportation Research Board: Washington, DC, USA, 1975. [Google Scholar]
- Ferreira, R.M. Probability-Based Durability Analysis of Concrete Structures in Marine Environment. Ph.D. Thesis, University of Minho, Braga, Portugal, 2004. [Google Scholar]
- Maes, M.; Gruyaert, E.; de Belie, N. Resistance of concrete with blast-furnace slag against chlorides. investigated by comparing chloride profiles after migration and diffusion. Mater. Struct. 2013, 46, 89–103. [Google Scholar] [CrossRef]
- Schiessl, P.; Raupach, M. Corrosion of Reinforcement in Concrete; Elsevier Applied Science: London, UK, 1990. [Google Scholar]
- Daily, S.F. Understanding Corrosion and Cathodic Protection of Reinforced Concrete Structures; Corrpro Companies, Incorporated: Medina, OH, USA, 1999. [Google Scholar]
- Pettersson, K. Corrosion Threshold Value and Corrosion Rate in Reinforced Concrete; CBI Report 2; Swedish Cement and Concrete Research Institute: Stockholm, Sweden, 1992. [Google Scholar]
- Elsener, B.; Zimmermann, L.; Fluckiger, D.; Burchler, D.; Bohni, H. Chloride penetration- non-destructive determination of the free chloride content in mortar and concrete. In Proceedings of the RILEM International Workshop on Chloride Penetration into Concrete, Paris, France, 15–18 October 1995. [Google Scholar]
- Zimmermann, L.; Elsener, B.; Bohni, H. Critical factors for the initiation of rebar corrosion. In Proceedings of the EUROCORR ’99, European Federation of Corrosion, Aachen, Germany, 30 August–2 September 1999. [Google Scholar]
- Lambert, P.; Page, C.L.; Vassie, P.R. Investigations of reinforcement corrosion. 2. Electrochemical monitoring of steel in chloride-contaminated concrete. Mater. Struct. 1991, 24, 351–358. [Google Scholar] [CrossRef]
- Tuutti, K. Corrosion of Steel in Concrete; Swedish Cement and Concrete Research Institute: Stockholm, Sweden, 1982. [Google Scholar]
- Mohammed, T.U.; Hamada, H. Relationship between free chloride and total chloride contents in concrete. Cem. Concr. Res. 2003, 33, 1487–1490. [Google Scholar] [CrossRef]
- Silva, N.; Tang, L.; Lindqvist, J.E.; Boubitsas, D. Chloride profiles along the concrete-steel interface. Int. J. Struct. Eng. 2013, 4, 100–112. [Google Scholar] [CrossRef]
- Arya, C.; Buenfeld, N.R.; Newman, J.B. Assessment of simple methods of determining the free chloride ion content of cement paste. Cem. Concr. Res. 1987, 17, 907–918. [Google Scholar] [CrossRef]
- Proverbio, E.; Carassiti, F. Evaluation of chloride content in concrete by X-ray fluorescence. Cem. Concr. Res. 1997, 27, 1213–1223. [Google Scholar] [CrossRef]
- Skoglund, P. Chloride Transport and Reinforcement Corrosion in the Vicinity of the Transition Zone between Substrate and Repair Concrete. Licentiate Thesis, KEYI, Stockholm, Sweden, 2006. [Google Scholar]
- Wilsch, G.; Weritz, F.; Schaurich, D.; Wiggenhauser, H. Determination of chloride content in concrete structures with laser-induced breakdown spectroscopy. Constr. Build. Mater. 2005, 19, 724–730. [Google Scholar] [CrossRef]
- De J Cano, F.; Bremner, T.W.; McGregor, R.P.; Balcom, B.J. Magnetic resonance imaging of 1H, 23Na, and 35Cl penetration in Portland cement mortar. Cem. Concr. Res. 2002, 32, 1067–1070. [Google Scholar] [CrossRef]
- Tripathi, S.R.; Inoue, H.; Hasegawa, T.; Kawase, K. Non-destructive inspection of chloride ion in concrete structures using attenuated total reflection of millimeter waves. J. Infrared Millim. Terahertz Waves 2013, 34, 181–186. [Google Scholar] [CrossRef]
- Livingston, R.A.; Al-Sheikhly, M.; Mohamed, A.B. Numerical simulation of the PGNA signal from chlorine diffusion gradients in concrete. Appl. Radiat. Isot. 2010, 68, 679–682. [Google Scholar] [CrossRef] [PubMed]
- McPolin, D.; Basheer, P.A.M.; Long, A.E.; Grattan, K.T.V.; Sun, T. Obtaining progressive chloride profiles in cementitious materials. Constr. Build. Mater. 2005, 19, 666–673. [Google Scholar] [CrossRef]
- Laferriere, F.; Inaudi, D.; Kronenberg, P.; Smith, I.F. A new system for early chloride detection in concrete. Smart Mater. Struct. 2008, 17, 045017. [Google Scholar] [CrossRef]
- Abbas, Y. In-Situ Measurement of Chloride ion Concentration in Concrete. Ph.D. Thesis, University of Twente, Enschede, The Netherlands, 2015. [Google Scholar]
- Atkins, C.P.; Scantlebury, J.D.; Nedwell, P.J.; Blatch, S.P. Monitoring chloride concentrations in hardened cement pastes using ion selective electrodes. Cem. Concr. Res. 1996, 26, 319–324. [Google Scholar] [CrossRef]
- ASTM C114–15. Standard Test Methods for Chemical Analysis of Hydraulic Cement; American Society for Testing and Materials (ASTM): Philadelphia, PA, USA, 2000. [Google Scholar]
- Maierhofer, C.; Reinhardt, H.W.; Dobmann, G. (Eds.) Non-Destructive Evaluation of Reinforced Concrete Structures: Non-Destructive Testing Methods; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Torres-Luque, M.; Bastidas-Arteaga, E.; Schoefs, F.; Sanchez-Silva, M.; Osma, J.F. Non-destructive methods for measuring chloride ingress into concrete: State-of-the-art and future challenges. Constr. Build. Mater. 2014, 68, 68–81. [Google Scholar] [CrossRef]
- Haque, M.N.; Kayali, O.A. Free and water soluble chloride in concrete. Cem. Concr. Res. 1995, 25, 531–542. [Google Scholar] [CrossRef]
- ASTM C1218/C1218M–17. ASTM C1218. Standard Test Method for Water-Soluble Chloride in Mortar and Concrete; American Society for Testing and Materials (ASTM): Philadelphia, PA, USA, 2015. [Google Scholar]
- Analysis of water soluble chloride content in concrete. Available online: https://www.rilem.net/images/publis/1341.pdf (Accessed on 28 October 2017).
- Arya, C.; Newman, J.B. An Assessment of Four Methods of Determining the Free Chloride Content of Concrete. Mater. Struct. 1990, 23, 319–330. [Google Scholar] [CrossRef]
- Jin, M.; Jiang, L.; Zhu, Q. Monitoring chloride ion penetration in concrete with different mineral admixtures based on embedded chloride ion selective electrodes. Constr. Build. Mater. 2017, 143, 1–15. [Google Scholar] [CrossRef]
- Climent-Llorca, M.A.; Viqueira-Perez, E.; Lopez-Atalaya, M.M. Embeddable Ag/AgCl sensors for in-situ monitoring chloride contents in concrete. Cem. Concr. Res. 1996, 26, 1157–1161. [Google Scholar] [CrossRef]
- De Vera, G.; Climent, M.A.; Anton, C.; Hidalgo, A.; Andrade, C. Determination of the selectivity coefficient of a chloride ion selective electrode in alkaline media simulating the cement paste pore solution. J. Electroanal. Chem. 2010, 639, 43–49. [Google Scholar] [CrossRef]
- Climent, M.A.; Anton, C.; de Vera, G.; Hidalgo, A.; Andrade, C. The interference of OH-ions in the potentiometric determination of free Cl-in cement paste pore solution. In Proceedings of the International RILEM Conference on Advances in Construction Materials through Science and Engineering, Hong Kong, China, 5–7 September 2011; RILEM Publications: Bagneux, France, 2011. [Google Scholar]
- Page, C.L. Mechanism of corrosion protection in reinforced concrete marine structures. Nature 1975, 258, 514–515. [Google Scholar] [CrossRef]
- Sandberg, P. Chloride initiated reinforcement corrosion in marine concrete. In Report TVBM 1015; Division of Building Materials, LTH, Lund University: Lund, Sweden, 1998. [Google Scholar]
- Ghods, P. Multi-Scale Investigation of the Formation and Breakdown of Passive Films on Carbon Steel Rebar in Concrete. Ph.D. Thesis, Carleton University, Ottawa, ON, Canada, 2010. [Google Scholar]
- Page, C.; Havdahl, J. Electrochemical monitoring of corrosion of steel in microsilica cement pastes. Mater. Struct. 1985, 18, 41–47. [Google Scholar] [CrossRef]
- Carmody, W.R. A study of the silver chloride electrode. J. Am. Chem. Soc. 1929, 51, 2901–2904. [Google Scholar] [CrossRef]
- Ha, H.; Payer, J. The effect of silver chloride formation on the kinetics of silver dissolution in chloride solution. Electrochim. Acta 2011, 56, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Janata, J. Principles of Chemical Sensors; Springer Book: New York, NY, USA, 2009. [Google Scholar]
- Polk, B.J.; Stelzenmuller, A.; Mijares, G.; MacCrehan, W.; Gaitan, M. Ag/AgCl microelectrodes with improved stability for microfluidics. Sens. Actuators B Chem. 2006, 114, 239–247. [Google Scholar] [CrossRef]
- Dobos, D. Electrochemical Data: A Handbook for Electrochemists in Industry and Universities; Elsevier Science and Technology: Amsterdam, The Netherlands, 1975. [Google Scholar]
- De Vera, G.; Hidalgo, A.; Climent, M.A.; Andrade, C.; Alonso, C. Chloride-Ion Activities in Simplified Synthetic Concrete Pore Solutions: The Effect of the Accompanying Ions. J. Am. Ceram. Soc. 2000, 83, 640–644. [Google Scholar] [CrossRef]
- Hidalgo, A.; Vera, G.; Climent, M.A.; Andrade, C.; Alonso, C. Measurements of chloride activity coefficients in real Portland cement paste pore solutions. J. Am. Ceram. Soc. 2001, 84, 3008–3012. [Google Scholar] [CrossRef]
- Jaya, S.; Rao, T.P.; Rao, G.P. Mono- and multilayer formation studied of silver chloride on silver electrodes from chloride-containing solutions. J. Appl. Electrochem. 1987, 17, 635–640. [Google Scholar] [CrossRef]
- Jovic, B.M.; Jovic, V.D.; Drazic, D.M. Kinetics of chloride ion adsorption and the mechanism of AgCl layer formation on the (111), (100) and (110) faces of silver. J. Electroanal. Chem. 1995, 399, 197. [Google Scholar] [CrossRef]
- Katan, T.; Szpak, S.; Douglas, N.B. Silver/silver chloride electrodes: Surface morphology on charging and discharging. J. Electrochem. Soc. 1974, 121, 757–764. [Google Scholar] [CrossRef]
- Jin, X.; Lu, J.; Liu, P.; Tong, H. The electrochemical formation and reduction of a thick AgCl deposition layer on a silver substrate. J. Electroanal. Chem. 2003, 542, 85–96. [Google Scholar] [CrossRef]
- Lal, H.; Thirsk, H.R.; Jones, W.F.K.W. A study of the behavior of polarized electrodes, Part I—The silver/silver halide system. Trans. Faraday Soc. 1951, 47, 70–77. [Google Scholar] [CrossRef]
- Birss, V.I.; Smith, C.K. The anodic behavior of silver in chloride solution—I. The formation and reduction of thin silver chloride films. Electrochim. Acta 1987, 32, 259–268. [Google Scholar] [CrossRef]
- Stoica, D.; Brewer, P.J.; Brown, R.J.; Fisicaro, P. Influence of fabrication procedure on the electrochemical performance of Ag/AgCl reference electrodes. Electrochim. Acta 2011, 56, 10009–10015. [Google Scholar] [CrossRef]
- Beck, T.R.; Rice, D.E. Conductivity of anodic silver chloride during formation. J. Electrochem. Soc. 1984, 131, 89–93. [Google Scholar] [CrossRef]
- Elsener, B.; Zimmermann, L.; Bohni, H. Non-destructive determination of the free chloride content in cement based materials. Mater. Corros. 2003, 54, 440–446. [Google Scholar] [CrossRef]
- Duffo, G.S.; Farina, S.B.; Giordano, C.M. Characterization of solid embeddable reference electrodes for corrosion monitoring in reinforced concrete structures. Electrochim. Acta 2009, 54, 2010–2020. [Google Scholar] [CrossRef]
- Montemor, M.F.; Alves, J.H.; Simoes, A.M.; Fernandes, J.C.S.; Lourenço, Z.; Costa, A.J.S.; Appleton, A.J.; Ferreira, M.G.S. Multiprobe chloride sensor for in situ monitoring of reinforced concrete structures. Cem. Concr. Compos. 2006, 28, 233–236. [Google Scholar] [CrossRef]
- Bard, A.J.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution; Marcel Dekker: New York, NY, USA, 1985; pp. 294–312. [Google Scholar]
- Giles, R.D. The anodic behaviour of silver single crystal electrodes in concentrated chloride solutions. J. Electroanal. Chem. Interfacial Electrochem. 1970, 27, 11–19. [Google Scholar] [CrossRef]
- Winston, R. Uhlig’s Corrosion Handbook; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Welham, N.J.; Kelsall, G.H.; Diaz, M.A. Thermodynamics of Ag-Cl-H2O, Ag-Br-H2O and Ag-I-H2O systems at 298 K. J. Electroanal. Chem. 1993, 361, 39–41. [Google Scholar] [CrossRef]
- Angst, U.; Elsener, B.; Larsen, C.K.; Vennesland, O. Potentiometric determination of the chloride ion activity in cement based materials. J. Appl. Electrochem. 2010, 40, 561–573. [Google Scholar] [CrossRef]
- Birss, V.I.; Wright, G.A. The kinetics of the anodic formation and reduction of phase silver sulfide films on silver in aqueous sulfide solutions. Electrochim. Acta 1981, 26, 1809–1817. [Google Scholar] [CrossRef]
- Graedel, T.E. Corrosion mechanisms for silver exposed to the atmosphere. J. Electrochem. Soc. 1992, 139, 1963–1970. [Google Scholar] [CrossRef]
- Femenias, Y.S.; Angst, U.; Caruso, F.; Elsener, B. Ag/AgCl ion-selective electrodes in neutral and alkaline environments containing interfering ions. Mater. Struct. 2016, 49, 2637–2651. [Google Scholar] [CrossRef]
- Moody, G.J.; Rigdon, L.P.; Meisenheimer, R.G.; Frazer, J.W. Selectivity parameters of homogeneous solid-state chloride ion-selective electrodes and the surface, morphology of silver chloride-silver sulphide discs under simulated interference conditions. Analyst 1981, 106, 547–556. [Google Scholar] [CrossRef]
- Suzuki, H.; Hiratsuka, A.; Sasaki, S.; Karube, I. Problems associated with the thin-film Ag/AgCl reference electrode and a novel structure with improved durability. Sens. Actuators B Chem. 1998, 46, 104–113. [Google Scholar] [CrossRef]
- Altunbulduk, T.; Kocker, H.M.; Frenzel, W. Studies on the elimination of sulfide interference in the potentiometric determination of chloride using ion selective electrodes in a flow injection system. Fresenius’ J. Anal. Chem. 1995, 351, 593–598. [Google Scholar] [CrossRef]
- Raynauld, J.P.; Laviolette, J.R. The silver-silver chloride electrode: A possible generator of offset voltages and currents. J. Neurosci. Methods 1987, 19, 249–255. [Google Scholar] [CrossRef]
- Janz, G.J.; Ives, D.J. Silver, silver chloride electrodes. Ann. N. Y. Acad. Sci. 1968, 148, 210–221. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (mmol/L) | ||||||
|---|---|---|---|---|---|---|
| Na+ | K+ | Ca2+ | Cl− | SO42− | Ionic strength (mmol/L) | |
| Hydrated cement paste | 88.9 | 74.8 | 4.8 | 1.4 | 0.2 | 178.3 |
| Aggressive Species | Expressed as | Reported Chloride Threshold Value | Reference |
|---|---|---|---|
| Total chloride (including free chloride) | % by weight of binder | (1.24–3.08), (1–8.34), (0.68–0.97) | [38,39,40] |
| % by weight of concrete | (0.03–0.07), (0.03–0.2), (0.06–0.2) (0.06–0.37) | [41,42,43,44] | |
| Free chloride | % by weight of binder | (0.39–1.16), (1-4), (0.5–2) | [33,38,39] |
| % by weight of concrete 1 | 0.026 | [45] | |
| mol/L | (0.36–3.22), (0.44–0.65), (0.045–0.55) | [46,47,48] | |
| [Cl−]/[OH−] | (1.17–3.98), (1.7–20), (3–20) | [38,39,49] |
| Species | Gibbs Free Energy (KJ/mol) | Species | Gibbs Free Energy (KJ/mol) |
|---|---|---|---|
| Ag+ | 77 | Ag(OH)2− | −260 |
| Cl− | −131 | AgCl (c) | −110 |
| Ag2O (c) | −11 | AgCl (aq) | −54 |
| AgO (c) | 15 | AgCl2− | −216 |
| AgOH | −80 | AgCl43− | −478 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pargar, F.; Koleva, D.A.; Van Breugel, K. Determination of Chloride Content in Cementitious Materials: From Fundamental Aspects to Application of Ag/AgCl Chloride Sensors. Sensors 2017, 17, 2482. https://doi.org/10.3390/s17112482
Pargar F, Koleva DA, Van Breugel K. Determination of Chloride Content in Cementitious Materials: From Fundamental Aspects to Application of Ag/AgCl Chloride Sensors. Sensors. 2017; 17(11):2482. https://doi.org/10.3390/s17112482
Chicago/Turabian StylePargar, Farhad, Dessi A. Koleva, and Klaas Van Breugel. 2017. "Determination of Chloride Content in Cementitious Materials: From Fundamental Aspects to Application of Ag/AgCl Chloride Sensors" Sensors 17, no. 11: 2482. https://doi.org/10.3390/s17112482
APA StylePargar, F., Koleva, D. A., & Van Breugel, K. (2017). Determination of Chloride Content in Cementitious Materials: From Fundamental Aspects to Application of Ag/AgCl Chloride Sensors. Sensors, 17(11), 2482. https://doi.org/10.3390/s17112482