Dual Mode NOx Sensor: Measuring Both the Accumulated Amount and Instantaneous Level at Low Concentrations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: The accumulating-type (or integrating-type) NOx sensor principle offers two operation modes to measure low levels of NOx: The direct signal gives the total amount dosed over a time interval and its derivative the instantaneous concentration. With a linear sensor response, no baseline drift, and both response times and recovery times in the range of the gas exchange time of the test bench (5 to 7 s), the integrating sensor is well suited to reliably detect low levels of NOx. Experimental results are presented demonstrating the sensor’s integrating properties for the total amount detection and its sensitivity to both NO and to NO2. We also show the correlation between the derivative of the sensor signal and the known gas concentration. The long-term detection of NOx in the sub-ppm range (e.g., for air quality measurements) is discussed. Additionally, a self-adaption of the measurement range taking advantage of the temperature dependency of the sensitivity is addressed.1. Introduction
Due to the tightening emission and safety regulations, the detection of harmful or toxic gases has become a large field of research in the past decades. Resistive or impedimetric solid state gas sensors that detect the actual concentration of a target gas are widely investigated. Usually, n-type semiconducting metal oxides are used that change their resistivity more or less selectively as a function of an analyte concentration. Numerous reviews on gas sensor devices of the so-called Taguchi type are available [1–4]. For NOx detection, the state-of-the-art metal oxide gas sensors are mostly based on WO3 [5], SnO2, TiO2 [6], or In2O3 [7]. The resistance of these sensors is continuously measured to obtain the actual concentration of the gaseous analyte, cNOx. Important measurement criteria for such sensors are—besides stability and cross-sensitivity to other varying gases—the sensitivity, the measurement range, and the sensor response time. In the automotive field, a concentration-detecting NOx sensor based on two pumping cells is applied to control the proper function of the NOx reduction catalysts and for On-board diagnostics (OBD). As reported in [8], the sensing performance of this Al2O3/ZrO2 based sensor in the second generation was highly improved by enhancements in the sensor design and control module, but the zero level value has still a uncertainty limiting the reliable detection in the low ppm range.
For the application in the automotive or industrial exhaust gas stream as well as for the air quality management, the mean values over a longer measurement period are of interest. For the latter, the requested NO2 thresholds are given e.g., from the EU immission legislation [9] and the German air quality standards (BImSchV [10]) as
the annual mean value (40 μg/m3),
the 1-hour value (200 μg/m3, not to be exceeded more than 18 times a calendar year) and
the alert value (400 μg/m3 measured over three consecutive hours).
The 1-hour limit of 200 μg/m3 corresponds to a mean NO2 concentration of 0.1 ppm. The detection of low concentrations of analyte gas over a long measurement period, like for instance for air quality monitoring purposes, is challenging with such types of concentration-detecting sensors described above. Noisy signals and the sometimes insufficient long-term stability of the sensor response at 0 ppm (baseline drift) [8,11,12] reduce the ability of these resistive or amperometric sensors to accurately determine the presence of low NOx levels. In addition, typical semiconductor gas sensors show an acceptable response rise time when the analyte, here NO or NO2, is added to a test gas. However, when the analyte is turned off, often only a slower recovery time (response fall time) is observed, even in the case of metal oxide sensitive layers with very high surface areas interacting with the gas phase [1,5,13,14]. Reasons for that may be a slow desorption of the strongly sorbed analyte NO2, and/or the non-linear sensor response of metal oxide gas sensors that also leads to a non-symmetry in the sensor’s response rise and recovery times. For all these reasons, it may be insufficient to determine the time averaged emission by an integration over time of the response of a concentration-detecting sensor.
To overcome this problem and to be able to detect reliably even small levels of an analyte gas over a longer period, the integrating (also called accumulating) sensing principle has been developed for NOx detection [15–17]. Analyte gas molecules that hit a sensitive layer are accumulated (i.e., sorbed in any way) in this layer. For a useful accumulative-type sensing material, at least one easy-to-detect physical material property should change linearly with the amount of sorbed analyte. For sufficient time intervals, the accuracy of an integrating sensor at low analyte concentration is increased compared to state-of-the-art sensors due to the accumulative properties of the sensitive layer. Even small analyte concentrations will accumulate and contribute to the sensor response. With such a sensor, the mathematical integration of a noisy concentration correlating sensor signal is not needed to determine the time-integrated amount. The ability of detecting the total amount of an analyte by chemical accumulation like a dosimeter or an active sampler, makes this sensor principle suitable for long-term monitoring purposes. In such applications, one is interested in the total amount of analyte that occurs per time period at the sensor (sometimes also called dose). However, also the peak concentrations are sometimes of interest.
It will be shown for an accumulation-type NOx sensor that both quantities, i.e., the dosed amount and instantaneous concentration, can be detected with just one sensor device. The response of such a senor correlates with the total amount of NOx gas, ANOx, dosed over a time interval and the concentration, cgas, at any given moment can be obtained from the derivative of this response with respect to time. Furthermore, we find that the sensor response is linear even in the very low ppm region and responds quickly to rapidly changing NOx concentrations. Both response and recovery times are about 5 to 7 s which is in the range of the gas exchange time of the test bench upstream of the sensitive device of 6 s. The measurement range can be self-adapted taking advantage of the temperature dependency of the sensitivity. This makes the sensor suitable for many applications. Cross-sensitivities are shortly addressed.
2. Fundamentals
2.1. Fundamentals of the Accumulating Sensing Principle
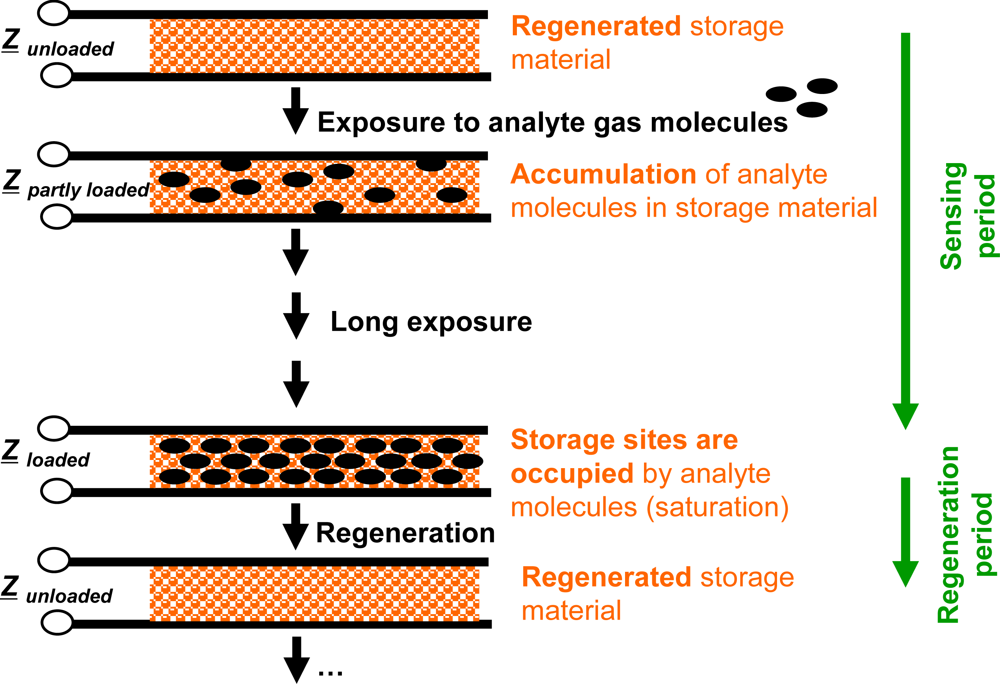
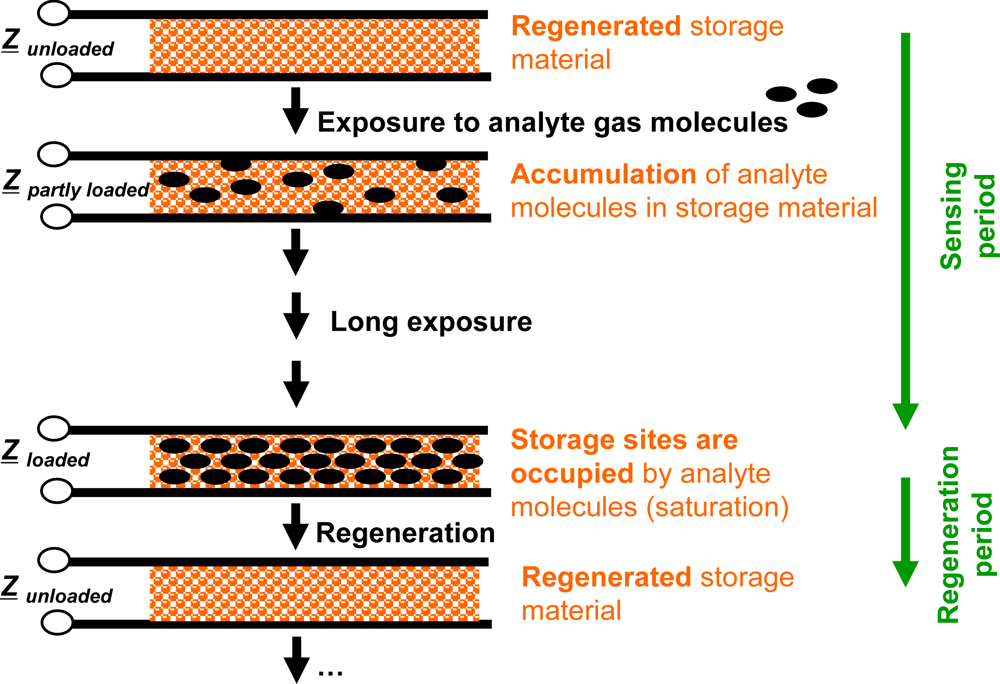
Compared to classical gas sensors, the accumulating gas sensing principle is different. Basic elements of accumulating type sensors are analyte gas storing materials applied as sensitive layers. Their material composition changes successively during gas exposure as the accumulation of the analyte molecules occurs in form of material transformations at the storage sites, i.e., in form of chemical reactions. Typically, such material transformations are reflected by the materials’ electrical properties [15,16,18–23], in particular by their complex impedances, Z, by the materials’ mass [24,25] or by their optical properties [26,27]. All these properties may serve as measurands. Therefore, the sensor response is expected to correlate with the total amount (dose), Agas, of analyte molecules being present in the gas stream over a time interval. Since the storage capacity is limited, the storage sites need to be regenerated as saturation effects deteriorate the integrating properties. These processes are illustrated in Figure 1 for the case of an impedimetric integrating-type gas sensor. Starting with the freshly regenerated material, the analyte gas molecules can be accumulated in the sensitive layer. After this accumulation phase, the storage sites are occupied and the operation mode needs to be shifted to the regenerating conditions to recover the original storage capacity. In the ideal case, long sensing periods alternate with short regeneration intervals.
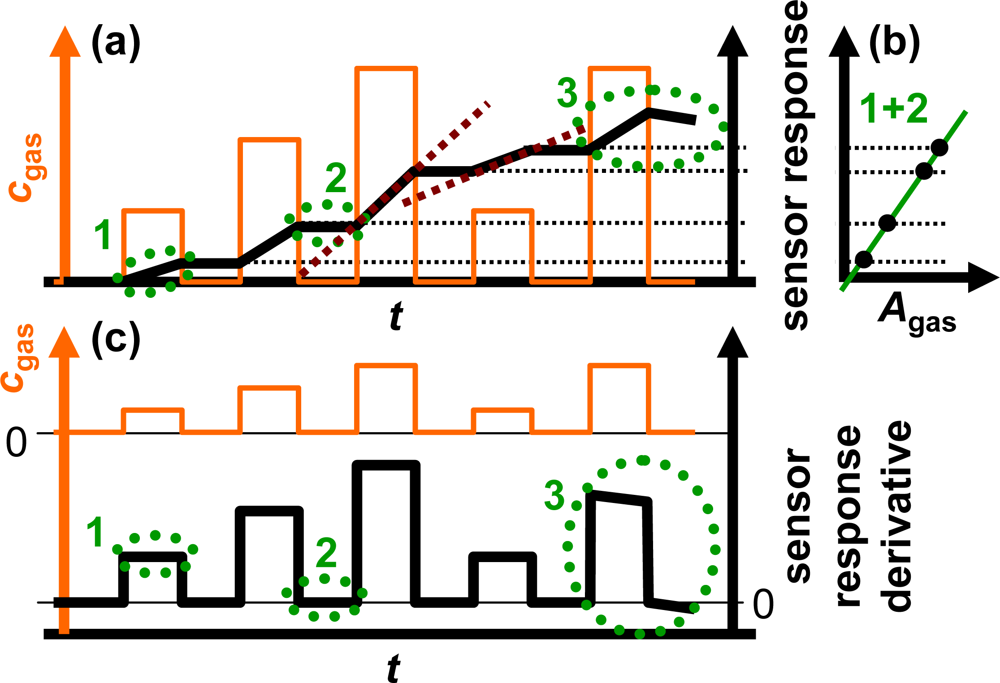
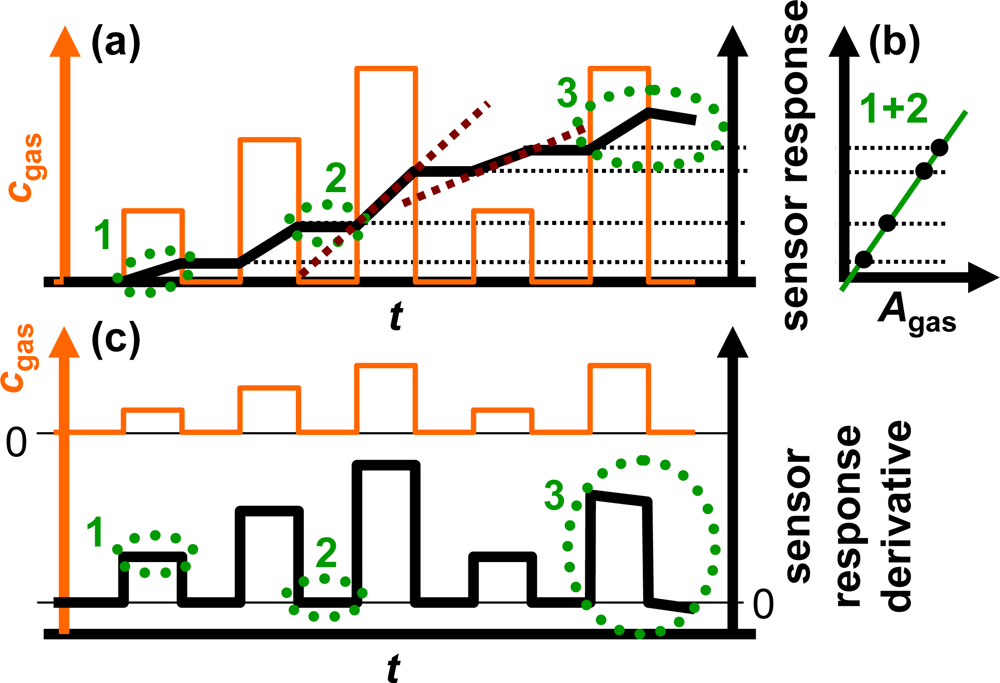
The sensor response of an ideal integrating sensor during cyclic analyte gas exposure with the analyte gas concentration cgas is illustrated in Figure 2(a). On the time scale, the sensor response increases in the presence of the analyte gas due to the accumulation of analyte molecules [linearity (1)] but remains constant in the absence of the analyte in the gas stream since the total amount of the stored analyte is not changing [holding capability (2)]. As an undesired effect, in the highly loaded state, the sensitivity may decrease and some of the formerly stored analyte molecules might desorb at 0 ppm analyte gas [saturation effects (3)]. The corresponding characteristic line in Figure 2(b) is obtained by correlating the sensor response with the amount of analyte gas, Agas. Agas here is defined as:
Another benefit of the integrating sensing principle is the fact that long-term drifts of the zero level signal (e.g., due to aging of the involved materials)—which often cause inaccuracies in state-of-the-art concentration detecting sensors [8]—are avoided by self-defining the zero value of the sensor response after each regeneration period.
To obtain ideal integrating properties, it is essential that the slope of the sensor signal on the time scale depends linearly on the actual analyte concentration, cgas, since a doubled concentration results in a doubled amount per time unit. From Figure 2(a) it can be obtained that the slope of the sensor response of the ideal accumulating sensor increases linearly with the concentration of the analyte in the test gas as highlighted by the dashed red lines exemplarily for the third and fourth NOx step. This behavior can be used to determine the actual analyte gas concentration, cgas. As sketched in Figure 2(c), the timely derivative of the response of an ideally integrating sensor correlates with the actual concentration of the analyte gas, cgas, due to the linearity in the presence of the analyte (1) and the holding capabilities at 0 ppm (2). At high loading states, it is expected that saturation effects (3) lead to deviations from the ideal behavior.
Utilizing the sensor response of one single accumulating-type sensor and its derivative, one can obtain:
the total amount of the analyte molecules Agas (accumulative properties) from the sensor response and (Figure 2(b))
the time-resolved actual analyte gas concentration cgas (linearity) from the sensor response derivative (Figure 2(c)).
2.2. Materials Aspects
The appropriate selection of the material of the sensitive layer is essential for the integrating behavior of the accumulating-type sensor. In general, six main criteria for the material properties have been found to be important for the accumulating measurement principle:
Selective sorption: The analyte molecules are accumulated selectively in the sensitive layer, e.g., via a chemical storage process including material conversion. The amount of molecules stored per time unit should be proportional to the actual analyte concentration (linearity). The selectivity ensures negligible cross sensitivities to other gas components.
Strength of sorption: The sorption of the analyte molecules needs to be strongly enhanced compared to desorption, ensuring accumulation of the analyte molecules in the sensitive layer and no losses in the absence of analyte in the gas phase (holding capability).
A linear relationship between the sorbed analyte amount and the material property, e.g., the conductivity or the impedance, monitored as sensor response (characteristic line) is crucial.
The change of the material properties that serve as a measurand must be independent on the analyte loading level.
Regeneration: Only under defined conditions that are different from the operation conditions, the sorbed analyte molecules can be released from the storage sites to regenerate the material – otherwise it is a single-use sensor.
Measurement range: The storage capacity of the sensitive layer and the rate of sorption need to be adapted to the application requirements.
It is reported that (to some extent) several materials ranging from carbon nanotubes [20] to polymers [24,27], carbonates [25], zeolites [22,23,28] or materials used in passive samplers [29] show accumulative properties, even if it is often not explicitly denoted in the literature as accumulative. In [30], modified diamond surfaces adsorb at room temperature a very thin water film, in which water-soluble gases affect selectively the pH of the film. Since the gases get by far faster solved than dissolved, the pH signal, which can be conveniently measured, behaves like an accumulating sensor.
In our contribution, we focus on carbonate-based lean NOx trap (LNT) materials, known from automotive catalysts, that have been found to change their electrical properties during NOx exposure [18,31] and were therefore tested as NOx-sensitive materials working accumulatively like a dosimeter [14,15].
2.3. NOx Storage Layer for an Integrating NOx Sensor
Since the long-term reliable detection of low levels of NOx is challenging, the integrating NOx sensor based on a NOx storage layer was developed. Here we present experimental results for such a sensor demonstrating its ability to detect small concentrations of NO and NO2 (i.e., 200 ppb for air quality monitoring or 2 to 10 ppm for exhaust gas applications) at different temperatures.
Lean NOx trap (LNT) materials serve as sensitive layers of the presented integrating-type NOx sensor. LNTs were developed as automotive catalyst materials to reduce the NOx emissions in the exhaust of leanly operated engines. They are able to store NOx molecules chemically in the form of nitrates in atmospheres with excess oxygen and to release them in short rich intervals so that the NOx molecules can be reduced to harmless nitrogen [32,33]. The highest NOx conversion occurs in the temperature range from 350 to 400 °C due to kinetic limitations at lower temperatures [32,34,35] and a reduced thermodynamic stability of the formed nitrates leading to decomposition at the higher temperatures [32,33]. Generally, those catalyst formulations consist of [32–37]:
a porous oxide framework—typically stabilized alumina—providing a high inner surface area,
alkaline (earth) metal oxides or carbonates “MCO3” (e.g., of potassium or barium) being able to store NO2 molecules in the form of nitrites or nitrates “M(NO3)2”
oxygen storage components like zirconia stabilized ceria and
finely dispersed precious metal particles to catalyze oxidation and reduction reactions, e.g.,
The ability to oxidize NO to obtain NO2, that can be stored in a second step chemically by converting carbonates to nitrates and to release the formerly stored NOx in rich surroundings or at higher temperatures to recover the storage capacity, make LNTs good candidates as sensitive layers for accumulating total NOx sensors. Jung et al. [25] detected the NOx accumulation and release on BaCO3, the most popular NOx storage component, at 400 °C gravimetrically by the mass increase when converting the carbonate to nitrate. Since the complex impedance of the LNTs was found to be highly affected by the NOx loading level [21], the presented accumulating total NOx sensor has been developed to detect low levels of NO and NO2 by electrical means.
3. Experimental Section
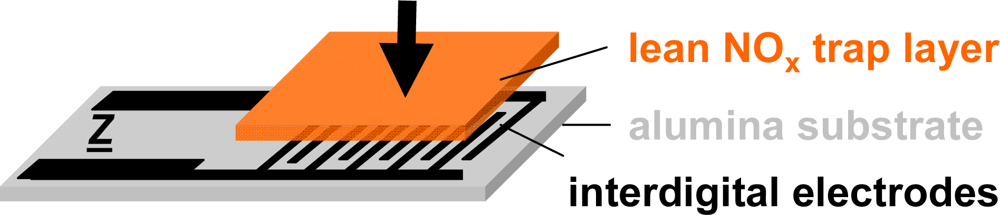
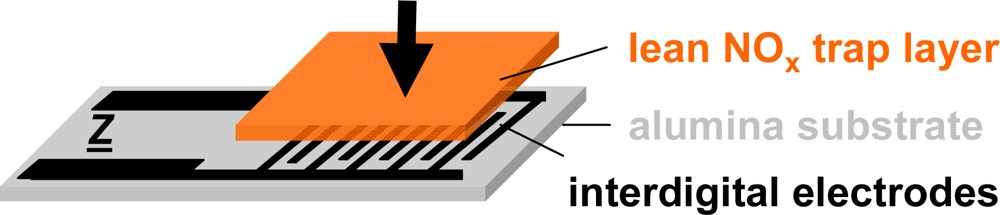
The setup of the presented resistive type integrating NOx sensor is shown in Figure 3. The sensor devices consist of a 96% pure alumina substrate (630 μm thick) with screen-printed interdigital electrodes (IDE) made from gold (DuPont) or platinum (Heraeus)—the electrode material was found to have no influence on the sensor performance—with an electrode width and spacing of 100 or 50 μm each. The electrode areas (5 × 6 mm) are covered with layers of a potassium-based LNT-material in various thicknesses ranging from about 30–100 μm. The raw material was provided by Johnson Matthey and the composition is described in [38]. The sensors were operated in a quartz tube (25 mm in diameter) located in a heated furnace at 380 °C (furnace temperature) and supplied with various synthetic gas mixtures of 2 L/min. The gas exchange time of the test bench upstream of the sensor (dosing unit, quartz tube) was estimated to be in the range of 6 s.
To avoid polarization effects, the electrical properties were monitored by impedance spectroscopy in the frequency range from 1 Hz to 1 MHz at 1 V (rms). Nyquist spectra in the regenerated and in the NOx-loaded state proved that the electrical properties can be described almost perfectly by an RC parallel equivalent circuit. The capacitance C was found to be only slightly affected by NOx, whereas the resistance R depended highly on the NOx loading state. To investigate the time-dependent behavior, the complex impedance, Z, was recorded every 1 or 2 seconds at 1 kHz and the resistance R was calculated from the impedance by:
The time derivative of the measured resistance values is correlated with the actual NOx concentration during NOx exposure. The derivative |dR/dt| is calculated by the absolute value of dividing the resistance change of two subsequent data points by the time (t) difference between them, which is usually 1.6 to 1.7 s:
The response rise time t90 is determined from the measurement curve of |dR/dt| taking the time difference between the beginning of the increase of the signal due to NOx and reaching 90% of the end value of |dR/dt| corresponding to the actual concentration. The recovery time is defined as the duration until |dR/dt| reaches the zero value. For each measurement, the mean values of t90 and the recovery time concerning the NOx steps besides of the first ones are given.
The integrating behavior has been demonstrated with various sensor devices having slightly different porous sensitive layer geometries and morphologies. These devices, each providing probably a different amount of accessible storage sites, resulted in different absolute resistance values and sensitivities.
As test gas base composition, a humidified gas mixture (50% N2 in a water bubbler at room temperature) of 10% O2 and 5% CO2 in N2 was used. The gas composition was mixed using mass flow controllers. A three-way valve was applied to the NO and NO2 line. The valve enabled to drain NOx to the vent prior to admixing it to the main gas flow. This avoided pressure pulses resulting in an uncontrolled overdosing of NOx at the beginning of the NOx dosing intervals. The NO and NO2 concentrations downstream of the sensor position were verified under steady state conditions with a chemiluminescence detector (Eco Physics CLD 700 EL ht).
4. Results and Discussion
In the following six sections, the sensor response and its derivative towards various NOx programs are presented, demonstrating the accumulating properties of this resistive-type integrating sensor under various conditions and the dual mode signal functionalities. The presented NOx runs address the detection of very low levels of NO like 2 ppm for just 25 s during cyclic exposure, the influence of varying NO concentrations, and the comparison of the sensitivity to NO and NO2. Additionally shown is a 50 min run with very low levels of NO2 (concentration in the sub-ppm range) that was not interrupted by regeneration periods proofing the suitability for air quality measurements. Each plot is arranged as in the scheme in Figure 2. It consists: (a) of the sensor response |ΔR|/R0 during cyclic NOx dosing; (b) of the corresponding characteristic line correlating |ΔR|/R0 with the total amount of NOx, ANOx, and (c) of the curve of the resistance derivative |dR/dt| mirroring the actual NOx concentration, cNOx. As a reference, cNOx calculated with data of the mass flow controller outputs is shown as well. The demonstration of the performance of the integrating NOx sensor and its benefits is complemented in a fifth section with results on the temperature dependency of the sensor response, which can be employed to self-adapt the measurement range to the requirements. Since the selectivity of gas sensors to the analyte strongly affects accuracy, cross-sensitivities to other present gases will be discussed in the last section.
4.1. Detection of Low Levels of NO
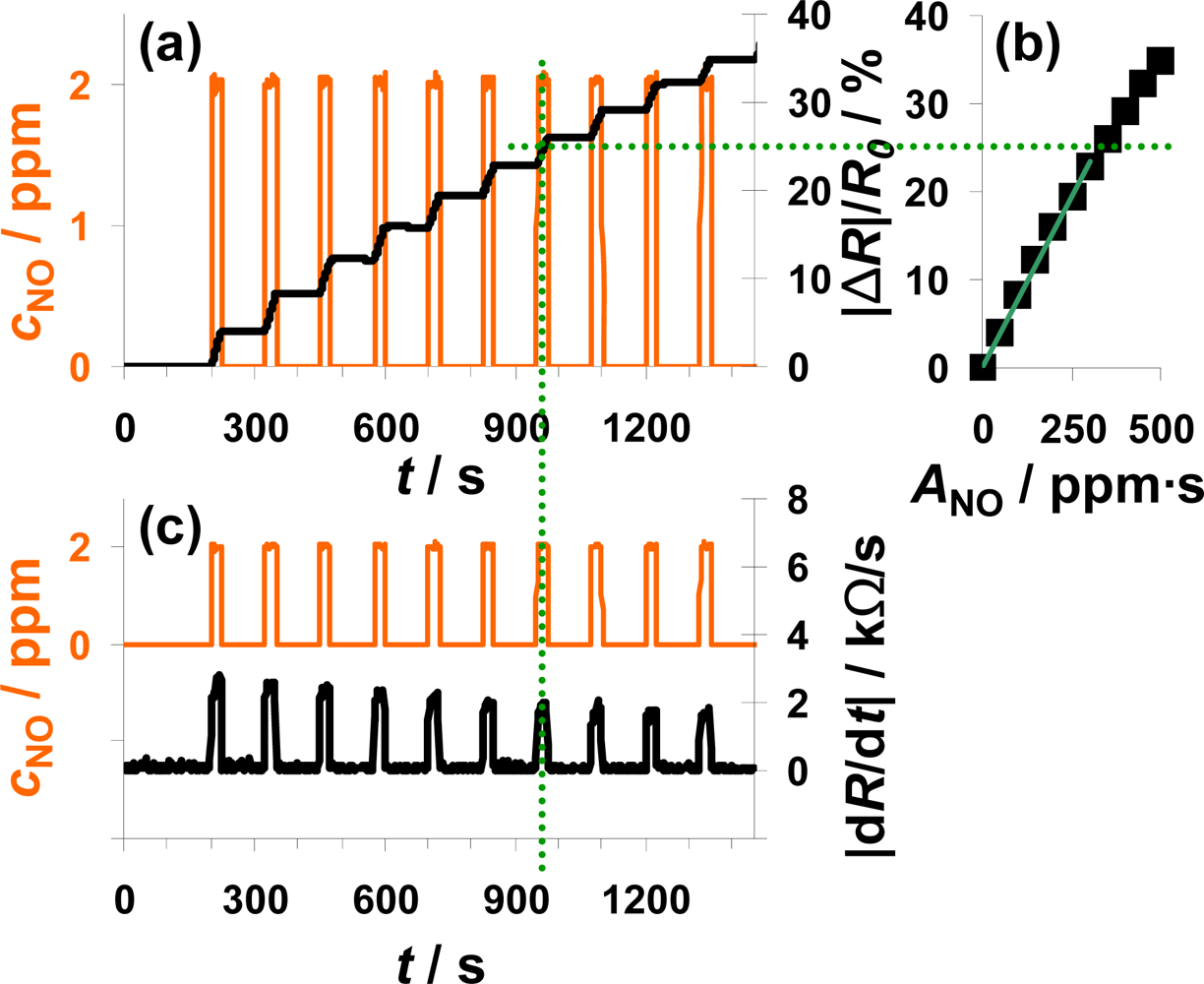
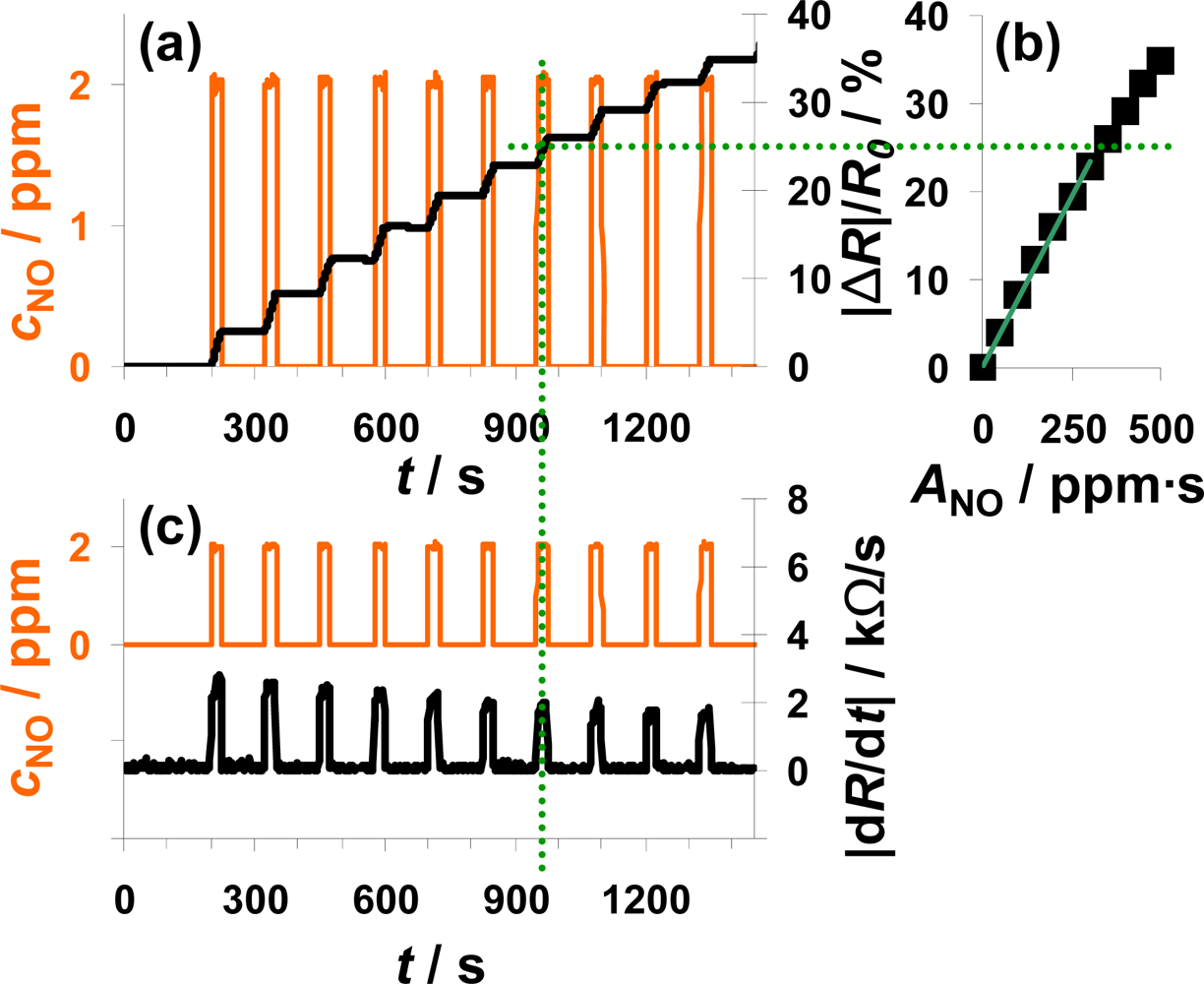
In Figure 4, the sensor response towards a pulsed addition of 2 ppm NO to the lean gas mixture is shown. Starting at 200 s, 2 ppm NO for 25 s alternate with 0 ppm NO for 100 s. The orange line (left axis) is the NO concentration as calculated from the mass flow controller outputs. The constant resistance value before the first NO step was used as R0 for the calculation of the sensor response |ΔR|/R0 according to Equation (5). As can be seen in Figure 4(a), |ΔR|/R0 increases in the presence of 2 ppm NO due to an increased conductivity of the NOx trap layer (R < R0). The slope appears to be almost constant during each NO pulse period. In the absence of NO, |ΔR|/R0 remains constant. Both, the linear increase of the sensor response in the presence of NO and the constant sensor response at 0 ppm NO confirm the integrating behavior of the sensor.
The characteristic line of the integrating NOx sensor correlates the sensor response |ΔR|/R0 with the total amount of NOx, ANOx, calculated from Equation (1). The characteristic line was extracted from the measurement data after each NO step (Figure 4(a)) and plotted in Figure 4(b). The correlation between |ΔR|/R0 and ANOx is linear up to a sensor response of about 25% and the sensitivity, S, (Equation (6)) in this measurement range is about 0.078%/ppm·s for this 100 μm Pt-IDE sensor. At higher loading states, the characteristic line gets nonlinear and the sensitivity decreases (denoted by the dotted line). As described above, the slope of the sensor response of an integrating gas sensor correlates with the actual concentration, cgas, at a constant gas flow rate. To prove this correlation, the curves of the absolute value of the derivative of the resistance |dR/dt| and of the added NO concentration, cNO, are compared in Figure 4(c). The resistance changes by about 2.5 kΩ/s in the presence of 2 ppm NO in the lowly loaded state. This value decreases slightly with time due to saturation effects. In the case of 0 ppm NO, the derivative is zero (holding capabilities) independent on the baseline-resistance.
The measurement results shown in Figure 4 clearly prove that the described NOx sensor based on a LNT-sensitive layer works like an integrating sensor. In addition, the measurement results clarify that it is well adapted to detect even very small NOx levels like 2 ppm NO for just 25 s with a high sensitivity (here: 0.078%/ppm·s NO). In contrast to classical gas sensors, the response time and the recovery time are identical and very fast in the range of 4 to 5 s. Since this is approximately the gas exchange time of the test setup, it is an indication that the sensor might respond even faster. The correlation between the signal derivative and the instantaneous analyte concentration is obvious. It should be noted here that the use of the time derivate of the sensor response to increase the speed of its response is not novel. It has been successfully applied for instance for the detection of ozone with phthalocyanine thin films [39]. However, in these cases, the sensor materials do not show an integrating behavior, but they do respond slowly to the changing analyte concentration. In other words, they do not collect the offered analyte molecules and do therefore also desorb the analytes after turning off the analyte.
4.2. Effect of Different NO Concentrations
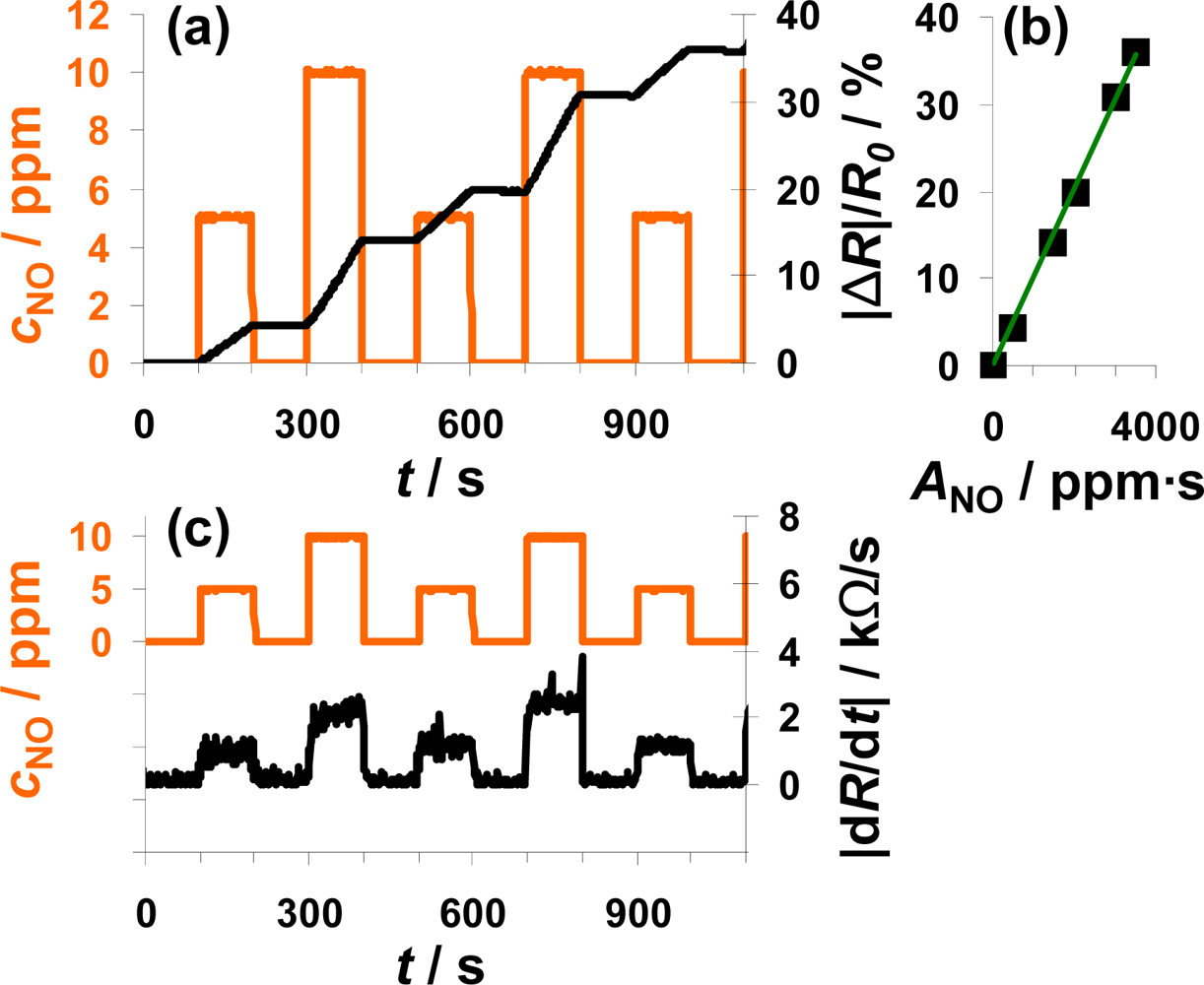
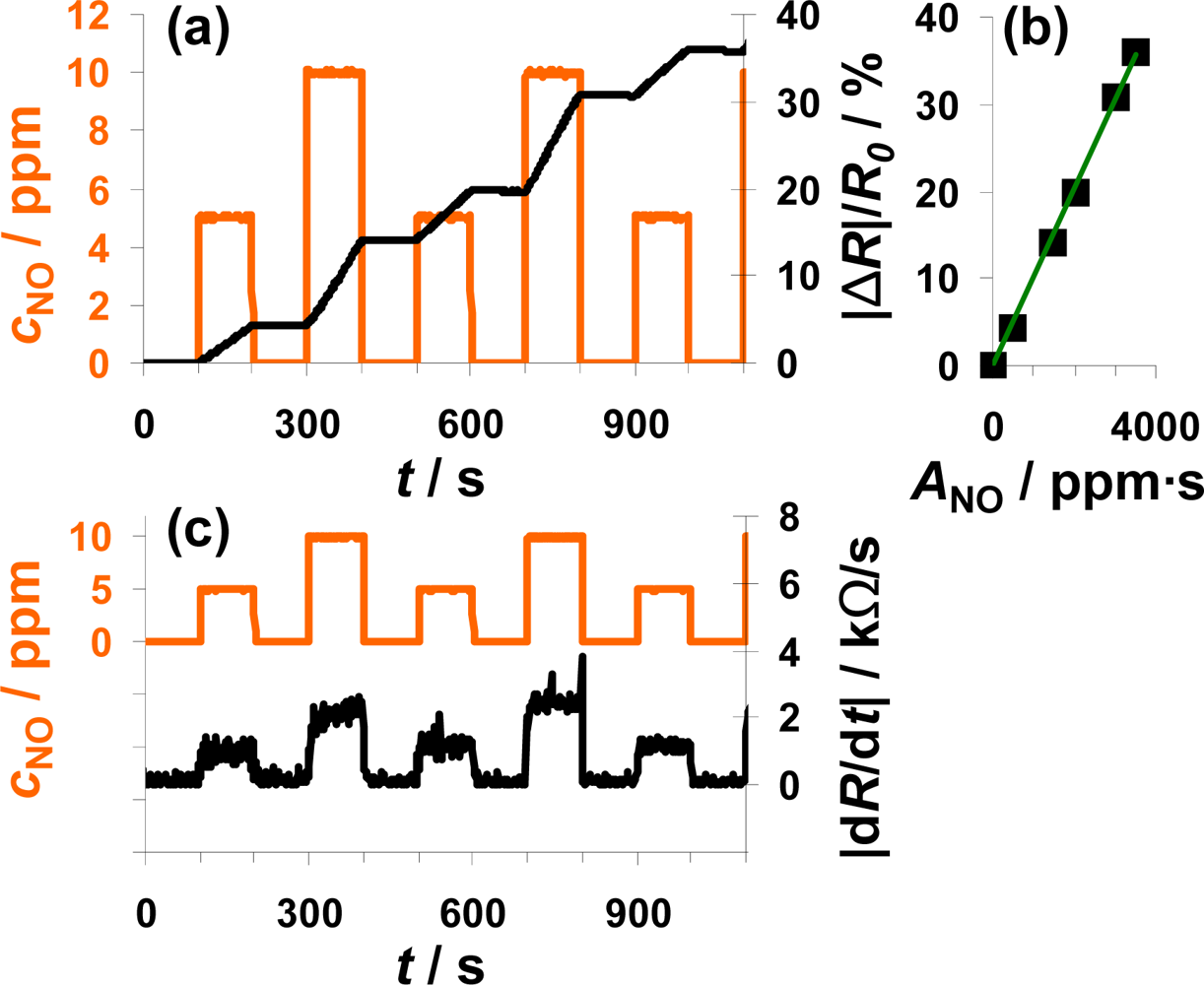
Another interesting point is to investigate whether the sensor also works as an integrator if the NO concentration varies. The sensor response to alternating 0, 5, and 10 ppm NO for 100 s each is plotted in Figure 5. Again, the sensor response increases during NO exposure but remains constant in the absence of the analyte (Figure 5(a)). In the case of 10 ppm NO, |ΔR|/R0 increases faster than in the presence of 5 ppm—the slope seems to be doubled. By calculating ANO from Equation (1), the doubled concentration is taken into account. Looking at the characteristic line in Figure 5(b), it becomes obvious that the measurement points form a straight line if plotted versus ANO. This means that the characteristic line in Figure 5(b) is independent on the actual concentration and that the sensor works as an NO-accumulating device, detecting the total amount of NO in the low ppm range. The sensitivity of this sensor device (100 μm Pt-IDE) is 0.010%/ppm·s NO. The sensitivity obtained in this measurement is much smaller compared to the sensitivity reported in the previous section (0.078%/ppm·s NO) in Figure 4. The measurement results presented were obtained with different sensor samples that were coated by screen-printing or dipping in an aqueous solution of the LNT-material resulting in sensitive layers with varies thicknesses and morphologies. In further tests, it was found that the sensitivity to NOx varies with the thickness of the sensitive layer, which can be explained by the dependency of the number of accessible storage sites on the volume and the morphology of the sensitive layer influencing the diffusion of NOx into the storage material and therefore the rate of storage. In Figure 5(c), |dR/dt| is plotted in parallel to cNO and follows it. In the presence of 5 ppm NO, the resistance changes by about 1.25 kΩ/s, in 10 ppm it is about 2.5 kΩ/s, which demonstrates once more the linearity. Again, the sensor signal responds and recovers very fast (5 to 6 s) with only a slight dependency on the actual NO concentration.
From a chemical reaction point of view, the results indicate that in the low loading state and in the case of low concentrations, the sensitive layer is able to incorporate always a constant fraction of the incoming NO molecules, i.e., the reaction rate of the rate determining reaction (either Equation (2) or Equation (3)) is constant. These results show further that the amount of NO flowing in a tube can be measured independently of the actual NO concentration in the low ppm-range and that the actual concentration can be determined by looking on the derivative of the sensor response.
4.3. Sensitivity to NO and NO2
To obtain a total NOx sensor, it is essential that the sensor responds to NO the same way as it does to NO2. This means for an integrating NOx sensor that the linearity as well as the holding capabilities should be equal for both NO and NO2.
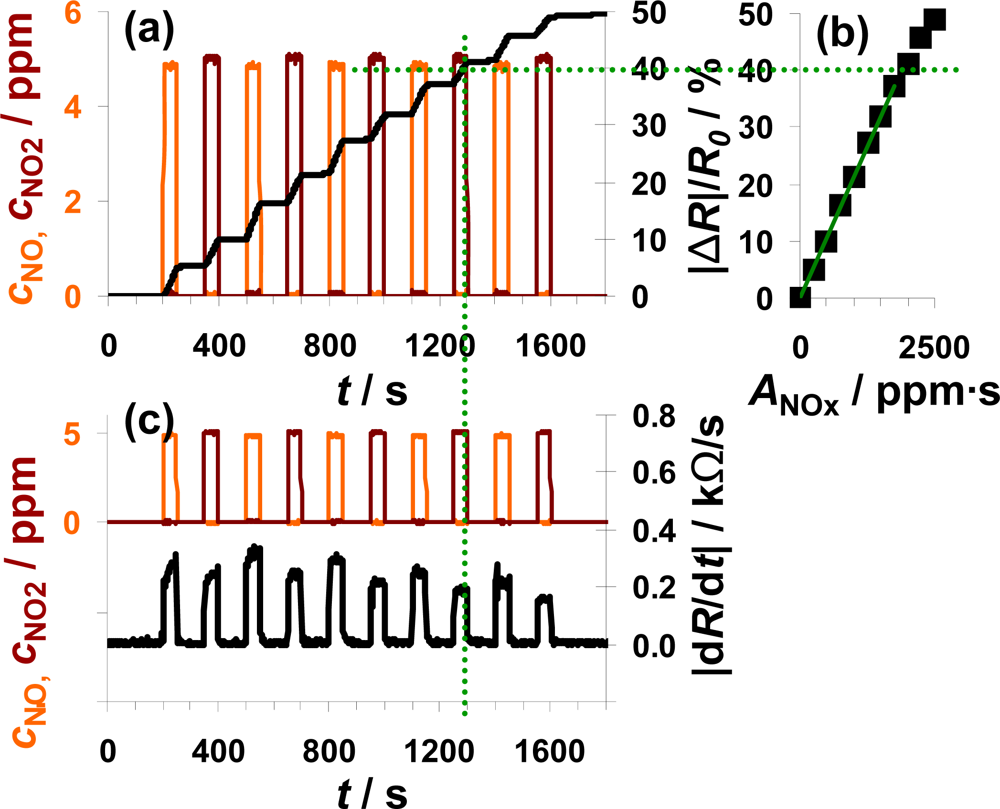
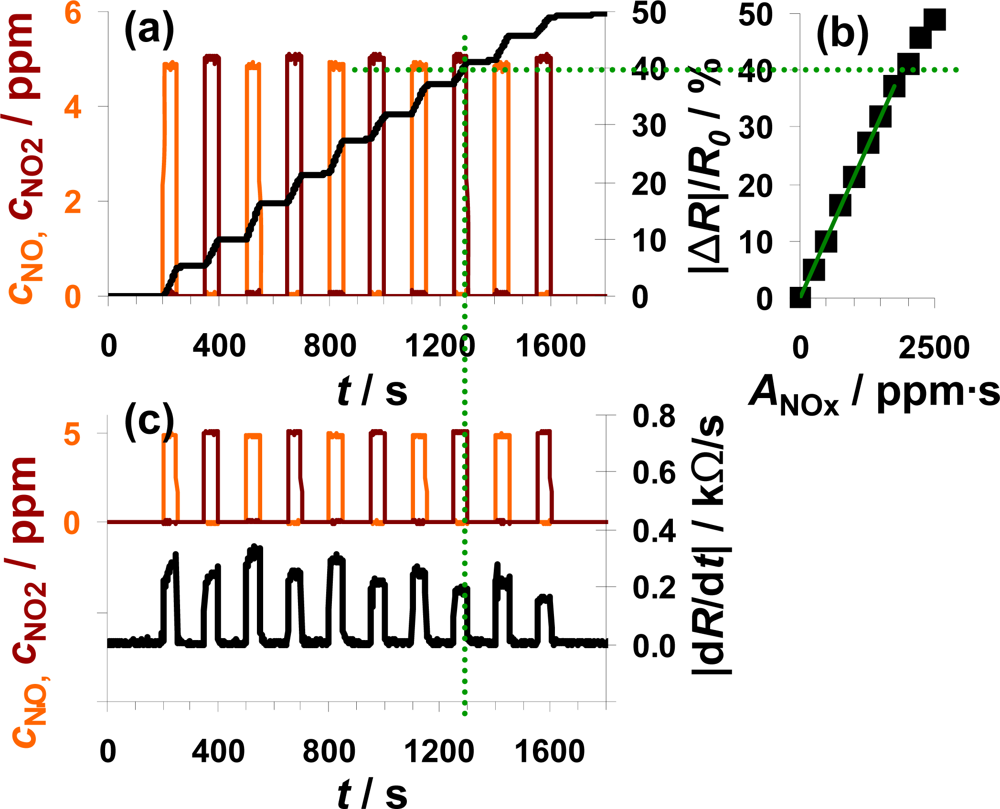
NO and NO2 were added alternately to the lean base gas (Figure 6). Each step consisted of 5 ppm NO or 5 ppm NO2 lasting 50 s which were separated by 100 s intervals of 0 ppm NOx. Figure 6(a) shows the sensor response as a function of time together with the dosed analyte gas concentrations. NOx dosing started with NO (orange line) which was later followed by a NO2 pulse (dark red line), with this cycle repeated five times. The sensor responds to NO in an integrating manner as already discussed in Figures 4 and 5. In the presence of NO2, |ΔR|/R0 increases with a constant slope and after switching off the NO2 flow, |ΔR|/R0 remains constant. Comparing the course of |ΔR|/R0 during NO and NO2 exposure, the sensor works accumulative for NOx: both linearity and holding abilities seem not to be affected whether NO or NO2 is added to the gas. This observation is verified by plotting the measurement data as a characteristic line similar to that shown in the previous two figures. The correlation between |ΔR|/R0 and ANOx, with ANOx being the sum of ANO and ANO2, gives a linear line up to a sensor response of about 40% with a sensitivity of 0.021%/ppm·s (50 μm Au-IDE). This demonstrates graphically the relative independency of the sensor response on the NO/NO2-ratio. Like the sensitivity, the difference in the range of linearity varies between the used samples. Having a closer look on the timely derivative in Figure 6(c), the correlation between |dR/dt| and the NOx composition can be seen in much more detail. In the presence of 5 ppm NO, the resistance decreases about 0.30 kΩ/s compared to about 0.25 kΩ/s at 5 ppm NO2. Again, we also see in Figure 6(c) the trend of a smaller sensor response with increasing NOx loading, when the saturation becomes noticeable. As in the previous measurements, the sensor response time is in the range of the gas exchange time of the system (6 s) and is not influenced by the composition of the dosed NOx. Unlike before, the curve of |dR/dt| recovers after a NO2 step in 12 s whereas it takes 17 s after NO exposure.
The data in Figure 6 demonstrate the ability of the presented integrating sensor to detect low levels of NO and NO2 with almost the same sensitivity. The integrating properties seem not to be affected by the NOx composition and the linearity of the sensor signal as well as the holding capabilities are given for the both analyte gases NO and NO2.
According to [40], the storage of NO occurs via the nitrite and the nitrate route, both involving NO oxidation. Since the NO/NO2-ratio in automotive exhausts is mainly influenced by temperature, precious metal particles are added to the catalyst formulations to establish the equilibrium reaction of NO and NO2 (Equation (3)). If the catalytic activity of the catalyst coating is sufficiently high, the efficiency of the LNT should not be affected by the NO/NO2 ratio in the feed gas. The measurement results in Figure 6 demonstrate that the sensor response of the proposed integrating sensor is not affected by the NOx composition and indicate that the oxidizing properties of the sensitive layer are sufficient to accumulate upcoming NO with the same rate as NO2.
4.4. Long-Term Monitoring of Low Levels of NO2
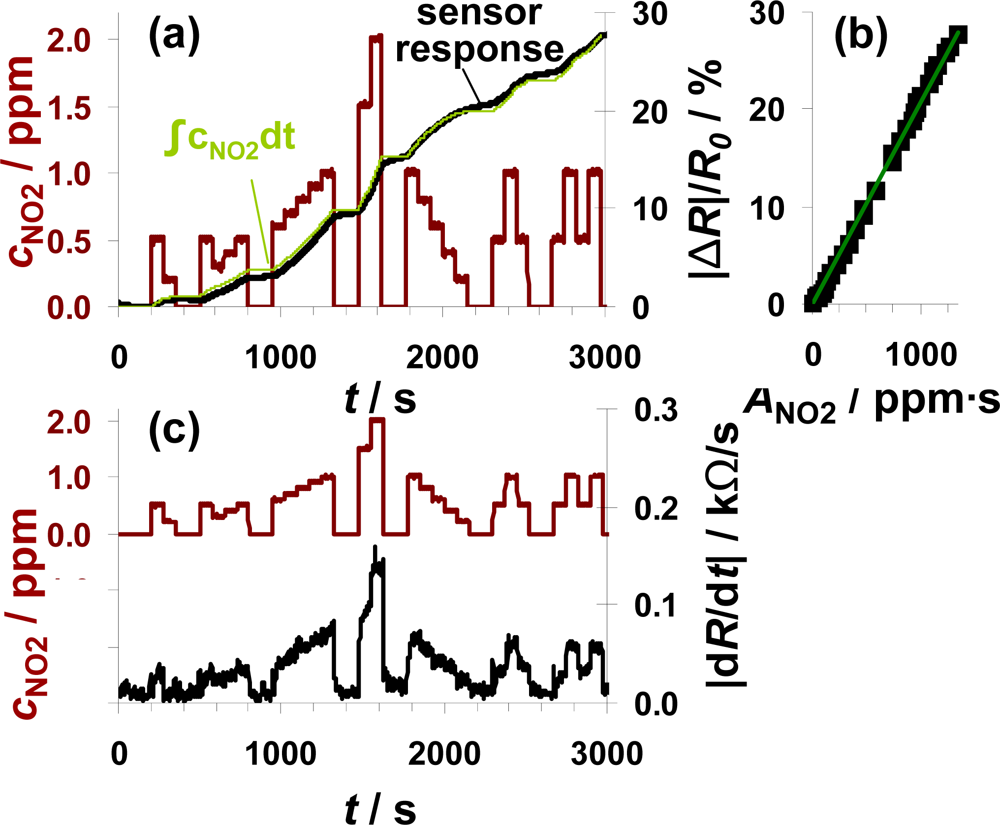
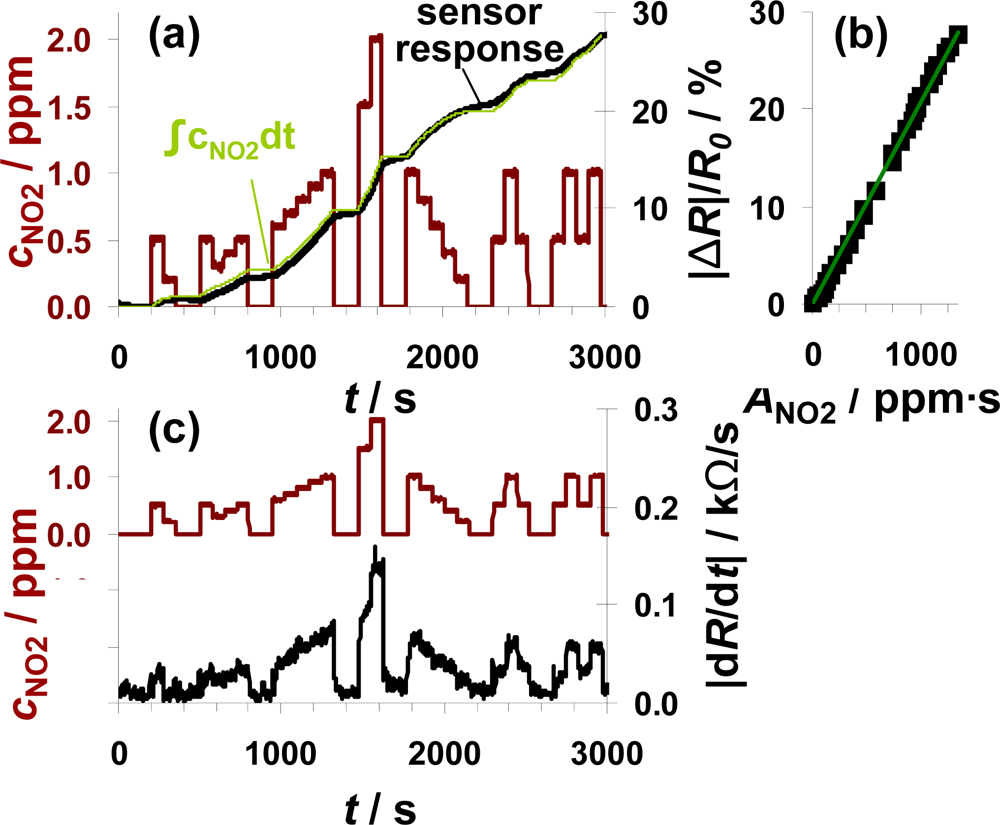
To check, whether the sensor can be used for air quality monitoring of NO2, the sensor test bench was modified to establish NO2 concentrations down to 0.2 ppm. A sensor sample (100 μm Pt-IDE) with an average sensitivity was selected for this test. The total amount of about 1,340 ppm·s NO2 was added to the gas stream in several steps of 75 s each with NO2 concentrations ranging from 200 ppb to 2 ppm. The red line in Figure 7(a) reflects the curve of the dosed NO2 concentrations with the focus on the sub-ppm range. The ppm-values are calculated from the mass flow controller outputs. As in the previous figures, |ΔR|/R0 increases in the presence of NO2 demonstrating that the sensor responds to very small levels of NO2 in the sub-ppm range (e.g., 200 ppb). The curve of the signal derivative |dR/dt| in Figure 7(b) was calculated according to Equation (7) and smoothed by a moving average of three values. Even in this sub-ppm range, the slope |dR/dt| correlates with the actual NO2 concentration demonstrating that the integrating properties are only negligibly affected by the actual analyte concentration and that the lowest levels tested can be detected. In periods with 200 ppb NO2, the signal changes by about 24 Ω/s. To clarify the amount-detecting properties, the curve of the sensor response |ΔR|/R0 (in black) is directly compared to the expected sensor response in light green in Figure 7(a). This expected curve has been obtained by multiplying the actual fraction of the dosed amount of NO2 (integration of the concentration curve) with the end value of the sensor response at 1,340 ppm·s. The measured and the calculated curves coincide very well. Up to about 1,500 s, the curve of the measured sensor response is slightly below the expected curve but exceeds it in the second part. These slight deviations might be due to a slightly increased sensitivity at higher concentrations or by small inaccuracies in the gas dosing system. Interestingly, the sensor response increases in the absence of NO2, which cannot be explained by desorption of formerly stored NOx. More likely, impreciseness of the modified gas mixing system (non-flushed dead volume in the NOx dosing unit) is responsible for this signal increase. Unfortunately, no additional information could be obtained from the chemiluminescence detector downstream of the sensor, since its resolution is 0.1 ppm. The characteristic line in Figure 7(b) confirms the integrating properties even in the area of small amounts since there is a linear correlation between |ΔR|/R0 and ANO2 in the measured range up to 1,340 ppm·s with a sensitivity of 0.021%/ppm·s.
The measurement range of 1,340 ppm·s and the NO2 concentration of 0.2 ppm are in a good agreement with the European and German air quality standards. As mentioned above, the permitted 1-hour value of NO2 is 200 μg/m3. This results in an average NO2 concentration of about 0.1 ppm. The results in Figure 7 demonstrate that the accumulating type NOx sensor at this stage of development is capable to detect 0.2 ppm of NO2 over at least 50 min without regeneration step in between. The permitted level of about 0.1 ppm for 1 h results in a total amount of 360 ppm·s NO2. According to the data in Figure 7, one remains in the linear range for almost four hours (3.7 h) if 200 μg/m3 (air quality threshold, see introduction) are in the sensor ambient. The permitted annual mean value of 40 μg/m3 could be monitored for about 18.5 hours.
4.5. Temperature Dependence of the Sensor Performance
Another interesting point is how the temperature dependence of the storage behavior of LNTs (kinetic limitations at low temperatures, decreased nitrate stability at high temperatures) affects the performance of the integrating NOx sensor based on an LNT layer.
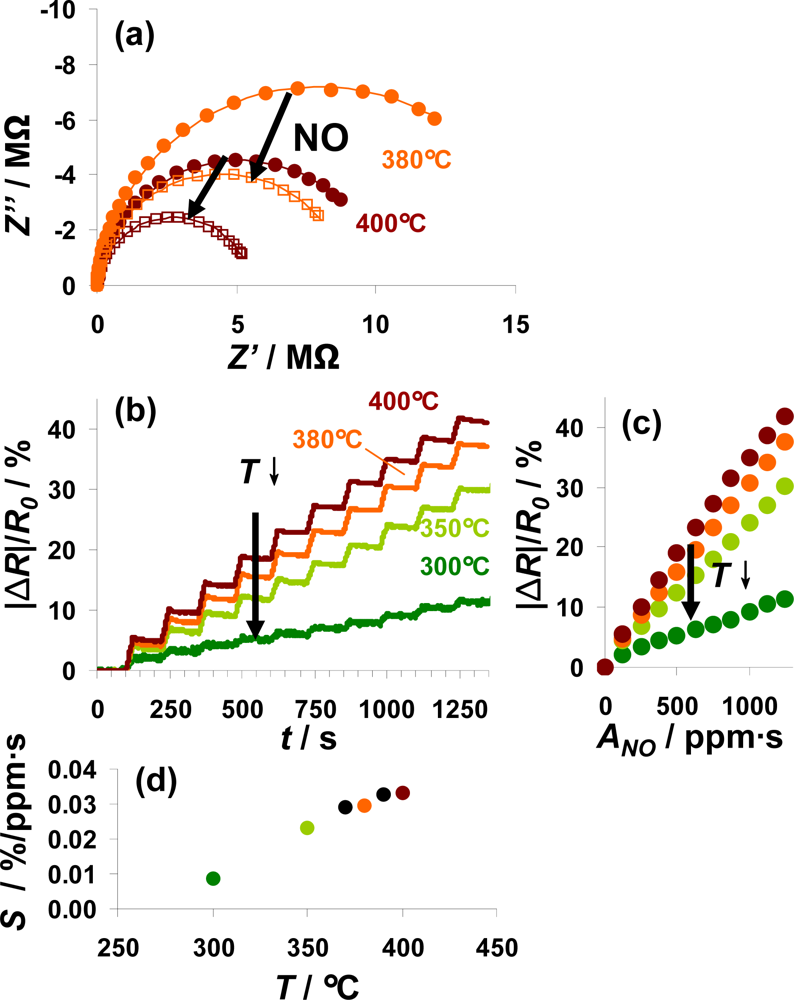
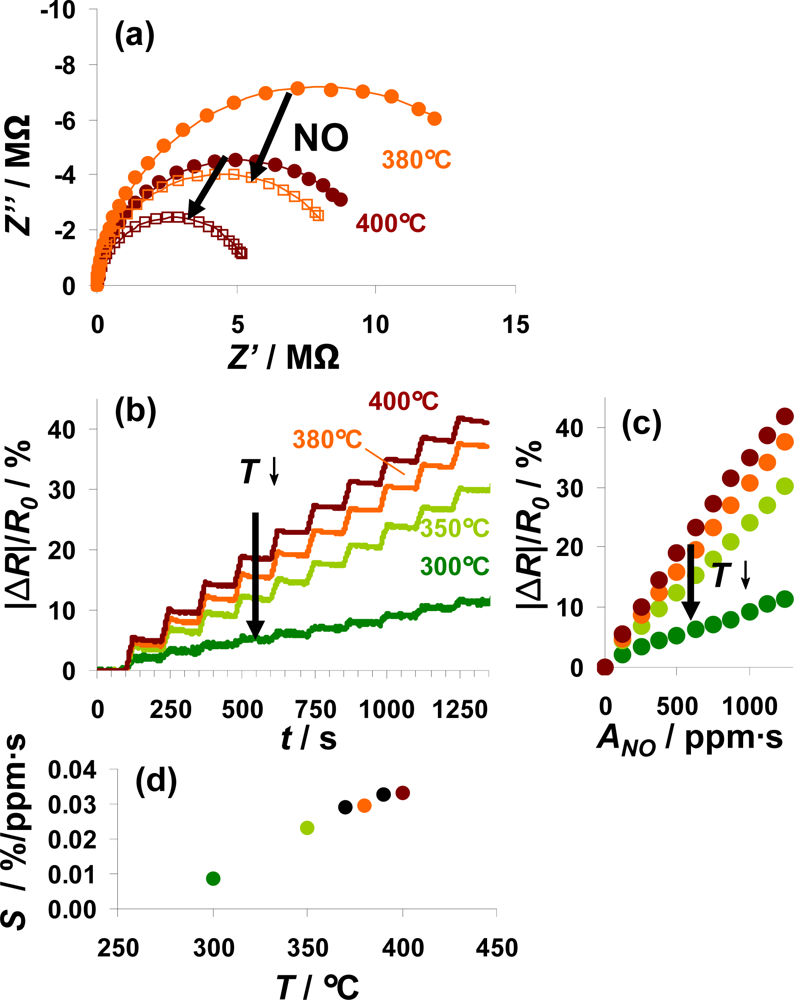
In Figure 8, the sensor responses of a 100 μm Pt-IDE sensor on NO steps at various temperatures from 300 to 400 °C are compared. For all investigated temperatures, the electrical behavior in the unloaded and partly NOx-loaded state can be described by an R||C element. The diameter of the semicircle in the Nyquist-plot, reflecting the resistance, decreased during NO loading as shown in Figure 8(a) for 380 °C (orange) and 400 °C (red line). Data are shown for both prior (solid points) and after (open symbols) exposure to 1,250 ppm·s NO. The conductivity of the sensitive layer in the unloaded state depends on temperature (a detailed analysis gives an activation energy of 0.9 eV). Correspondingly, the resistance of the loaded state also decreases with temperature. The sensor response, |ΔR|/R0, as a function of time is depicted in Figure 8(b) for ten steps of 5 ppm NO for 25 s alternating with 0 ppm for 100 s. This plot shows that the sensor exhibits integrating properties from 300 °C to 400 °C with a reduced sensor response at lower temperatures. Extracting the characteristic lines from these data (Figure 8(c)) demonstrates the linearity of the sensor response up to 400 °C as well as the enhanced sensitivity at higher temperatures. Plotting the sensitivity S calculated from the characteristic lines according to Equation (6) as a function of the temperature (Figure 8(d)) points out that the sensitivity to NO correlates nearly linearly with the temperature from 300 to 370 °C. Further rise in the temperature up to 400 °C results in a slightly reduced increase in the sensitivity. As reported in [16], the decreased stability of the formed nitrates at higher temperatures deteriorates the holding abilities of the storage sites and therefore the accumulating behavior of the sensitive layer. At lower temperatures, the kinetics limits the storage rate being reflected by the sensitivity, whereas the linear measurement range is restricted by the stability of the nitrates. Therefore, the sensitivity in the linear detection range increases with temperature whereas this range gets smaller at elevated temperatures. To adapt the measurement range of the integrating NOx sensor to the requirements of the application, the measurement temperature is an effective tool as it is an easy parameter to adjust.
According to the results shown above, a typical measurement procedure would be as follows: The sensor is thermally regenerated at a temperature above 500 °C by decomposing the formed nitrates, and as soon as the sensor is cooled down to its operation temperature, the new value of R0 is defined. This periodically re-defining of R0 avoids long-term zero value drifts of the sensor signal, which is a common drawback of state-of-the-art, resistive gas sensors.
The disadvantage is that during regeneration, the sensors remain blind. In other words, the time the sensor needs to cool down from T > 500 °C to operation temperature should be as short as possible. With ceramic sensors as shown in Figure 3, typically a thermal time constant in the 10 s ranges is observed [41]. However, reducing this “sensor blind time” to about 1 s is possible using ceramic hot plates as originally designed in [42] and demonstrated in [43]. Applying the sensors on top of silicon micro hotplates as described for instance in [41,44–46], the sensor blind time can be reduced to below 100 ms. After regeneration, the next measurement period follows. Data are delivered until the saturation effects become noticeable (i.e., at about 30% of the maximum resistance change). At that point, the next regeneration is initiated.
It is a specific feature of this setup that the measurement range can be self-adapted with this concept. If the saturation level is reached very soon after the last regeneration, this is a strong indication for a high mean NOx concentration requiring an extended measurement range. Then, after the following regeneration period, the sensor may be cooled to a lower operation temperature which reduces the sensor’s sensitivity as illustrated in Figure 8(d), and R0 is set to the new value. In other words, the measurement range can be self-adapted—without losing linearity—taking advantage of the temperature dependency of the sensitivity.
It is noteworthy to mention that all classical metal oxide sensors have a temperature dependence of the zero-NOx offset that will severely affect their accuracy, especially when low NOx concentration shall be measured. The integrating sensor does too, but in the instantaneous mode (via the derivative) the NOx offset is zero per definition. The sensitivity changes with temperature, but not with the instantaneous NOx offset. This is unique for a resistive or impedimetric sensor.
4.6. Selectivity and Cross-Sensitivities to Other Gas Components
The accuracy of a gas sensor is often restricted by its selectivity to the analyte. In the lean automotive exhaust as well as in the ambient air, the gas components will vary (in particular O2, CO2 and H2O concentrations), necessitating the study of their influences on the sensor signal.
The sensitivity of our integrating NOx sensor to NOx is based on the carbonate-nitrate conversion. These reactions [including NO oxidation and NO2 storage according to Equations (3) and (2)] are influenced by the concentrations of O2 and CO2. In addition to the influence of CO2, H2O was also found to affect the catalytic performance of a K-based LNT [47]. Therefore, it is expected that variations in all three gas components might lead to cross-sensitivities. Initial studies on the influence of CO2 and O2 on the response of the integrating NOx sensor were performed [15]:
The presence of oxygen is essential for the accumulation of NOx in the sensitive layer, but variation from 2.5% to 7.5% O2 was found to have no influence on the sensor signal.
CO2 variations were also found to be negligible in the concentration range from 1.5% to 10%.
The effect of H2O on the sensing characteristics has not been investigated yet. However, it is clear that the automotive exhaust components O2, H2O, and CO2 do not vary independently. They depend on the air-to-fuel ratio, in other words an increasing O2 concentration comes along with a decreasing CO2 and H2O concentration. Therefore, the sensor signal dependency on the air-to-fuel ratio will be subject of further studies.
LNTs are known to suffer from sulfur poisoning [34]: SO2, which origins from the sulfur content in diesel fuel, competes with NO2 for the storage sites, forming highly stable sulfates and leading to the deactivation of the storage capacity. Additionally, sulfur oxides occupy the catalytically active precious metal sites, lowering the oxidizing properties of the catalyst. Therefore, BaCO3 can be applied successfully as a sulfur adsorber layer to prevent thick film exhaust gas sensors from sulfation [48]. Toops et al. [49] report that the temperature dependent SO2 deactivation of a K-based LNT is mainly attributed to a loss in active storage sites. This loss in storage capacity after exposure to SO2 is expected to lower the sensitivity and measurement range of the integrating NOx sensor. The effect of SO2 on the sensing performance is currently under investigation.
5. Conclusions
The integrating sensing principle is suited for the reliable detection of low concentrations of analyte. Besides the dose (or the amount), the actual analyte concentration can be determined by calculating the signal derivative, offering a second functionality for the sensor. We present an accumulative-type impedimetric NOx sensor based on a NOx storage catalyst material as sensitive layer. The sensor can be operated at temperatures from 300 to 400 °C which is suited for different applications requiring mean value detection like air quality monitoring or OBD in the automotive exhaust. The sensitivity is neither dependent on the actual NOx concentration nor on the NO/NO2 ratio. Another benefit of this sensor compared to many concentration-detecting semiconducting devices is that the sensor responds and recovers fast. The determined response times or recovery times of 5 to 7 s are in the range of the gas exchange time of the test setup. Due to the accumulating properties, even small levels like 1,340 ppm·s or 200 ppb NO can be monitored time-continuously with a sufficient accuracy. The periodic regeneration of the storage sites ensures linearity, avoids a long-term base-line drift of the sensor signal via adjusting the zero value and enables a self-adaption of the linear measurement range taking advantage of the temperature dependency of the sensitivity.
Acknowledgments
The authors gratefully acknowledge the material preparation by Johnson Matthey (Shadab Mulla, Todd H. Ballinger, Hai-Ying Chen). Ralf Moos thanks the German Research Foundation (DFG) for supporting this work under grant numbers MO 1060/15-1.
References
- Sberveglieri, G. Recent developments in semiconducting thin-film gas sensors. Sens. Actuat. B 1995, 23, 103–109. [Google Scholar]
- Moos, R.; Sahner, K.; Fleischer, M.; Guth, U.; Barsan, N.; Weimar, U. Solid state gas sensor research in Germany—A status report. Sensors 2009, 9, 4323–4365. [Google Scholar]
- Korotcenkov, G. Metal oxides for solid-state gas sensors: What determines our choice? Mater. Sci. Eng. B 2007, 139, 1–23. [Google Scholar]
- Barsan, N.; Weimar, U. Understanding the fundamental principles of metal oxide based gas sensors; The example of CO sensing with SnO2 sensing in the presence of humidity. J. Phys. Condens. Matter 2003, 15, R813–R839. [Google Scholar]
- Gouma, P.I.; Kalyanasundaram, K. A selective nanosensing probe for nitric oxide. Appl. Phys. Lett 2008, 93. [Google Scholar] [CrossRef]
- Menil, F.; Coillard, V.; Lucat, C. Critical review of nitrogen monoxide sensors for exhaust gases of lean burn engines. Sens. Actuat. B 2000, 67, 1–23. [Google Scholar]
- Kannan, S.; Steinebach, H.; Rieth, L.; Solzbacher, F. Selectivity, stability and repeatability of In2O3 thin films towards NOx at high temperatures (≥500 °C). Sens. Actuat. B 2010, 148, 126–134. [Google Scholar]
- Sasaki, H.; Scholl, D.; Parsons, M.; Hiroshi, I.; Shiotani, K.; Visser, J.; Zawacki, G.; Kawai, T.; Teramoto, S.; Kubinski, D. Development of an Al2O3/ZrO2-composite high-accuracy NOx sensor. SAE Tech. Paper 2010. [Google Scholar] [CrossRef]
- Directive 2008/50/EC of the European Parliament and of the Council of 21 May 2008 on Ambient Air Quality and Cleaner Air for Europe. Off. J. EU 2008. L152/1..
- BImSchV. Neununddreißigste Verordnung zur Durchführung des Bundes-Immissionsschutz-gesetzes (Verordnung über Luftqualitätsstandards und Emissionshöchstmengen—39. BImSchV). Bundesgesetzblatt Jahrgang 2010 ausgegeben zu Bonn am 5. August. 2010, Nr.. 40, 1065.
- Haugen, J.-E.; Tomic, O.; Kvaal, K. A calibration method for handling the temporal drift of solid state gas-sensors. Anal. Chim. Acta 2000, 407, 23–39. [Google Scholar]
- Capone, S.; Epifani, M.; Francioso, L.; Kaciulis, S.; Mezzi, A.; Siciliano, P.; Taurino, A.M. Influence of electrodes ageing on the properties of the gas sensors based on SnO2. Sens. Actuat. B 2006, 115, 396–402. [Google Scholar]
- Cantalini, C.; Wlodarski, W.; Sun, H.T.; Atashbar, M.Z.; Passacantando, M.; Santucci, S. NO2 response of In2O3 thin film gas sensors prepared by sol–gel and vacuum thermal evaporation techniques. Sens. Actuat. B 2000, 65, 101–104. [Google Scholar]
- Yuan, L.; Hyodo, T.; Shimizu, Y.; Egashira, M. Preparation of mesoporous and/or macroporous SnO2-based powders and their gas-sensing properties as thick film sensors. Sensors 2011, 11, 1261–1276. [Google Scholar]
- Geupel, A.; Schönauer, D.; Röder-Roith, U.; Kubinski, D.J.; Mulla, S.; Ballinger, T.H.; Chen, H.-Y.; Visser, J.H.; Moos, R. Integrating nitrogen oxide sensor: A novel concept for measuring low concentrations in the exhaust gas. Sens. Actuat. B 2010, 145, 756–761. [Google Scholar]
- Geupel, A.; Kubinski, D.J.; Mulla, S.; Ballinger, T.H.; Chen, H.-Y.; Visser, J.H.; Moos, R. Integrating NOx sensor for automotive exhausts—A novel concept. Sens. Lett 2011, 9, 311–315. [Google Scholar]
- Beulertz, G.; Groß, A.; Moos, R.; Kubinski, D.J.; Visser, J.H. Determining the total amount of NOx in a gas stream—Advances in the accumulating gas sensor principle. Sens. Actuat. B 2012. accepted,. [Google Scholar] [CrossRef]
- Moos, R.; Wedemann, M.; Spörl, M.; Reiß, S.; Fischerauer, G. Direct catalyst monitoring by electrical means: An overview on promising novel principles. Top. Catal 2009, 52, 2035–2040. [Google Scholar]
- Shu, J.H.; Wikle, H.C.; Chin, B.A. Passive chemiresistor sensor based on iron (II) phthalocyanine thin films for monitoring of nitrogen dioxide. Sens. Actuat. B 2010, 148, 498–503. [Google Scholar]
- Li, J.; Lu, Y.; Ye, Q.; Cinke, M.; Han, J.; Meyyappan, M. Carbon nanotube sensors for gas and organic vapor detection. Nano Lett 2003, 3, 929–933. [Google Scholar]
- Moos, R. Catalysts as sensors—A promising novel approach in automotive exhaust gas aftertreatment. Sensors 2010, 10, 6773–6787. [Google Scholar]
- Rodriguez-Gonzalez, L.; Rodriguez-Castellon, E.; Jimenez-Lopez, A.; Simon, U. Correlation of TPD and impedance measurements on the desorption of NH3 from zeolite H-ZSM-5. Solid State Ion 2008, 179, 1968–1973. [Google Scholar]
- Rodriguez-Gonzalez, L.; Simon, U. NH3-TPD measurements using a zeolite-based sensor. Meas. Sci. Technol 2010, 21. [Google Scholar] [CrossRef]
- Matsuguchi, M.; Kadowaki, Y.; Tanaka, M. A QCM-based NO2 gas detector using morpholine-functional cross-linked copolymer coatings. Sens. Actuat. B 2005, 108, 572–575. [Google Scholar]
- Jung, W.; Sahner, K.; Leung, A.; Tuller, H.L. Acoustic wave-based NO2 sensor: Ink-jet printed active layer. Sens. Actuat. B 2009, 141, 485–490. [Google Scholar]
- Maruo, Y.Y.; Kunioka, T.; Akaoka, K.; Nakamura, J. Development and evaluation of ozone detection paper. Sens. Actuat. B 2009, 135, 575–580. [Google Scholar]
- Small, W., IV; Maitland, D.J.; Wilson, T.S.; Bearinger, J.P.; Letts, S.A.; Trebes, J.E. Development of a prototype optical hydrogen gas sensor using a getter-doped polymer transducer for monitoring cumulative exposure: Preliminary results. Sens. Actuat. B 2009, 139, 375–379. [Google Scholar]
- Kubinski, D.J.; Visser, J.H. Sensor and method for determining the ammonia loading of a zeolite SCR catalyst. Sens. Actuat. B 2008, 130, 425–429. [Google Scholar]
- Varshney, C.K.; Singh, A.P. Passive samplers for NOx monitoring: A critical review. Environmentalist 2003, 23, 127–136. [Google Scholar]
- Helwig, A.; Müller, G.; Garrido, J.A.; Eickhoff, M. Gas sensing properties of hydrogen-terminated diamond. Sens. Actuat. B 2008, 133, 156–165. [Google Scholar]
- Fremerey, R.; Reiß, S.; Geupel, A.; Fischerauer, G.; Moos, R. Determining of the NOx loading of an automotive lean NOx trap by directly monitoring of the electrical properties of the catalyst material itself. Sensors 2011, 11, 8261–8280. [Google Scholar]
- Roy, S.; Baiker, A. NOx Storage catalysis: From mechanism und materials properties to storage-reduction performance. Chem. Rev 2009, 109, 4054–4091. [Google Scholar]
- Takeuchi, M.; Matsumoto, S. NOx storage-reduction catalysts for gasoline engines. Top. Catal 2004, 28, 151–156. [Google Scholar]
- Epling, W.S.; Campbell, L.E.; Yezerets, A.; Currier, N.W.; Parks, J.E., II. Overview of the fundamental reactions and degradation mechanism of NOx storage/reduction catalysts. Catal. Rev 2004, 46, 163–245. [Google Scholar]
- Lesage, T.; Saussey, J.; Malo, S.; Hervieu, M.; Hedouin, C.; Blanchard, G.; Daturi, M. Operando FTIR study of NOx storage over a Pt/K/Mn/Al2O3-CeO2 catalyst. Appl. Catal. B 2007, 72, 166–177. [Google Scholar]
- Dawody, J.; Skoglundh, M.; Wall, S.; Fridell, E. Role of Pt-precursor on the performance of Pt/BaCO3/Al2O3·NOx storage catalysts. J. Mol. Catal. A 2005, 225, 259–269. [Google Scholar]
- Nova, I.; Castoldi, L.; Lietti, L.; Tronconi, E.; Forzatti, P.; Prinetto, F.; Ghiotti, G. NOx adsorption study over Pt-Ba/alumina catalysts: FT-IR and pulse experiments. J. Catal 2004, 222, 377–388. [Google Scholar]
- Chen, H.-Y.; Mulla, S.; Ballinger, T.H. NOx storage Materials for Sensor Applications. U.S. Patent 20100146935. 2010. [Google Scholar]
- Schütze, A.; Pieper, N.; Zacheja, J. Quantitative ozone measurement using a phthalocyanine thin-film sensor and dynamic signal evaluation. Sens. Actuat. B 1995, 23, 215–217. [Google Scholar]
- Forzatti, P.; Castoldi, L.; Nova, I.; Lietti, L.; Tronconi, E. NOx removal catalysis under lean conditions. Catal. Today 2006, 117, 316–320. [Google Scholar]
- Simon, I.; Barsan, N.; Bauer, M.; Weimar, U. Micromachined metal oxide gas sensors: Opportunities to improve sensor performance. Sens. Actuat. B 2001, 73, 1–26. [Google Scholar]
- Rettig, F.; Moos, R. Ceramic meso hot-plates for gas sensors. Sens. Actuat. B 2004, 103, 91–97. [Google Scholar]
- Kita, J.; Rettig, F.; Moos, R.; Drüe, K.-H.; Thust, H. Hot-plate gas sensors—Are ceramics better? Int. J. Appl. Ceram. Technol 2005, 2, 383–389. [Google Scholar]
- Faglia, G.; Comini, E.; Cristalli, A.; Sberveglieri, G.; Dori, L. Very low power consumption micromachined term CO sensors. Sens. Actuat. B 1999, 55, 140–146. [Google Scholar]
- Semancik, S.; Cavicchia, R.E.; Wheeler, M.C.; Tiffany, J.E.; Poirier, G.E.; Walton, R.M.; Suehle, J.S.; Panchapakesan, B.; DeVoe, D.L. Microhotplate platforms for chemical sensor research. Sens. Actuat. B 2001, 77, 579–591. [Google Scholar]
- Friedberger, A.; Kreisl, P.; Rose, E.; Müller, G.; Kühner, G.; Wöllenstein, J.; Böttner, H. Micromechanical fabrication of robust low-power metal oxide gas sensors. Sens. Actuat. B 2003, 93, 345–349. [Google Scholar]
- Toops, T.J.; Smith, D.B.; Epling, W.S.; Parks, J.E.; Partridge, W.P. Quantified NOx adsorption on Pt/K/gamma-Al2O3 and the effects of CO2 and H2O. Appl. Catal. B 2005, 58, 255–264. [Google Scholar]
- Rettig, F.; Moos, R.; Plog, C. Sulfur adsorber for thick-film exhaust gas sensors. Sens. Actuat. B 2003, 93, 36–42. [Google Scholar]
- Toops, T.J.; Smith Pihl, J.A. Sulfation of potassium-based lean NOx trap while cycling between lean and rich conditions: I. Microreactor study. Catal. Today 2008, 136, 164–172. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Groß, A.; Beulertz, G.; Marr, I.; Kubinski, D.J.; Visser, J.H.; Moos, R. Dual Mode NOx Sensor: Measuring Both the Accumulated Amount and Instantaneous Level at Low Concentrations. Sensors 2012, 12, 2831-2850. https://doi.org/10.3390/s120302831
Groß A, Beulertz G, Marr I, Kubinski DJ, Visser JH, Moos R. Dual Mode NOx Sensor: Measuring Both the Accumulated Amount and Instantaneous Level at Low Concentrations. Sensors. 2012; 12(3):2831-2850. https://doi.org/10.3390/s120302831
Chicago/Turabian StyleGroß, Andrea, Gregor Beulertz, Isabella Marr, David J. Kubinski, Jaco H. Visser, and Ralf Moos. 2012. "Dual Mode NOx Sensor: Measuring Both the Accumulated Amount and Instantaneous Level at Low Concentrations" Sensors 12, no. 3: 2831-2850. https://doi.org/10.3390/s120302831
APA StyleGroß, A., Beulertz, G., Marr, I., Kubinski, D. J., Visser, J. H., & Moos, R. (2012). Dual Mode NOx Sensor: Measuring Both the Accumulated Amount and Instantaneous Level at Low Concentrations. Sensors, 12(3), 2831-2850. https://doi.org/10.3390/s120302831