Real-Time Time-Frequency Two-Dimensional Imaging of Ultrafast Transient Signals in Solid-State Organic Materials

Abstract
:1. Introduction
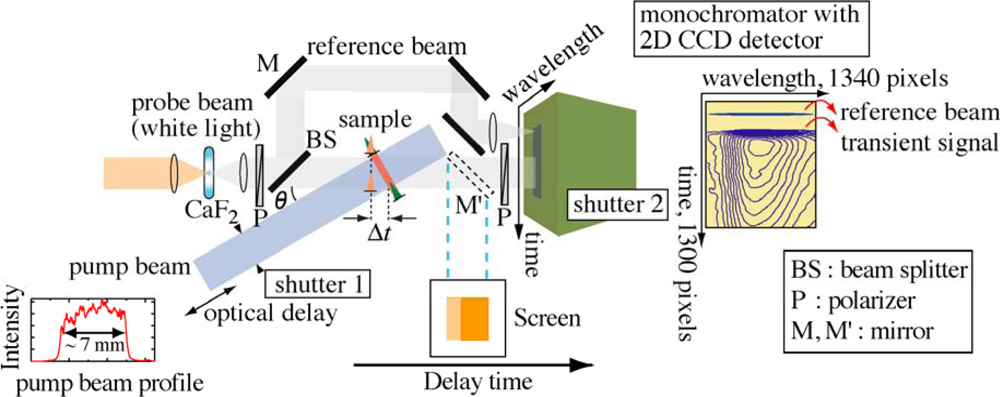
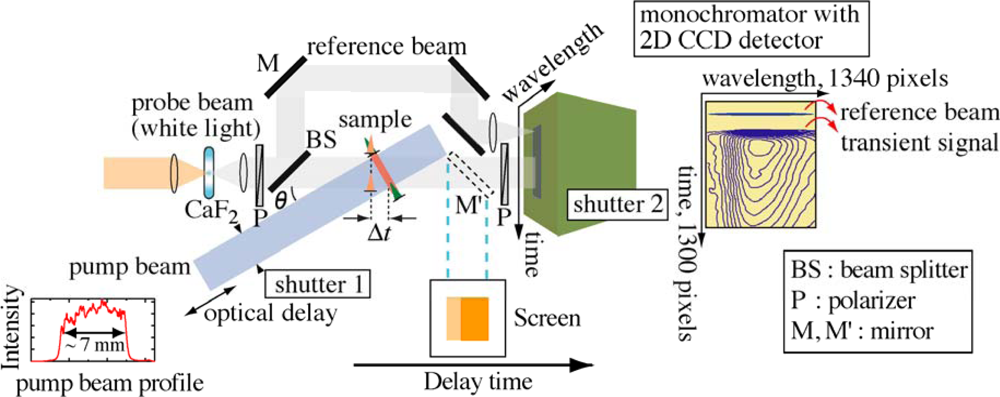
2. Real-Time Time-Frequency 2D Pump-Probe Imaging Spectroscopy
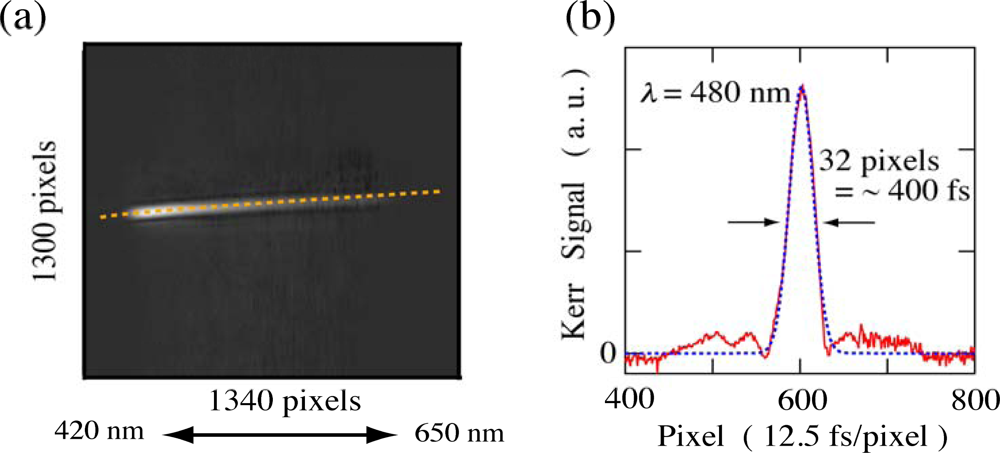
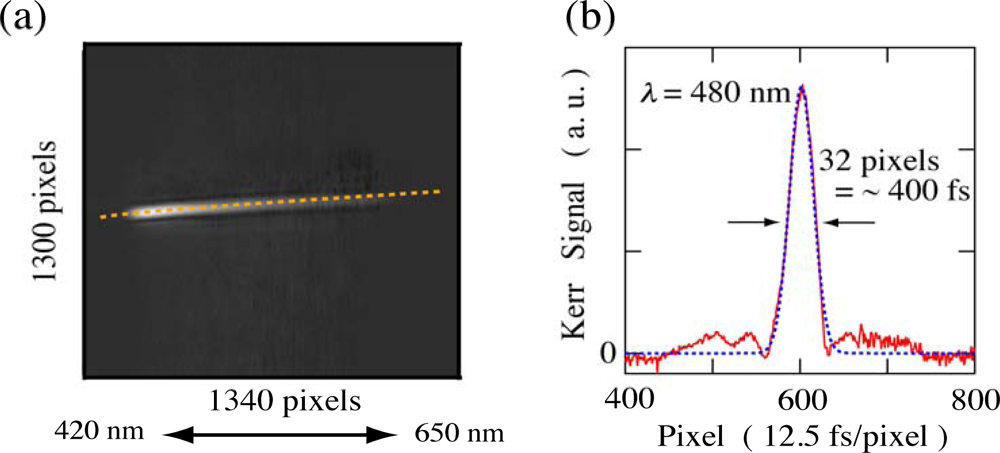
2.1. Principle of Femtosecond Real-Time Time-Frequency 2D Imaging Spectroscopy
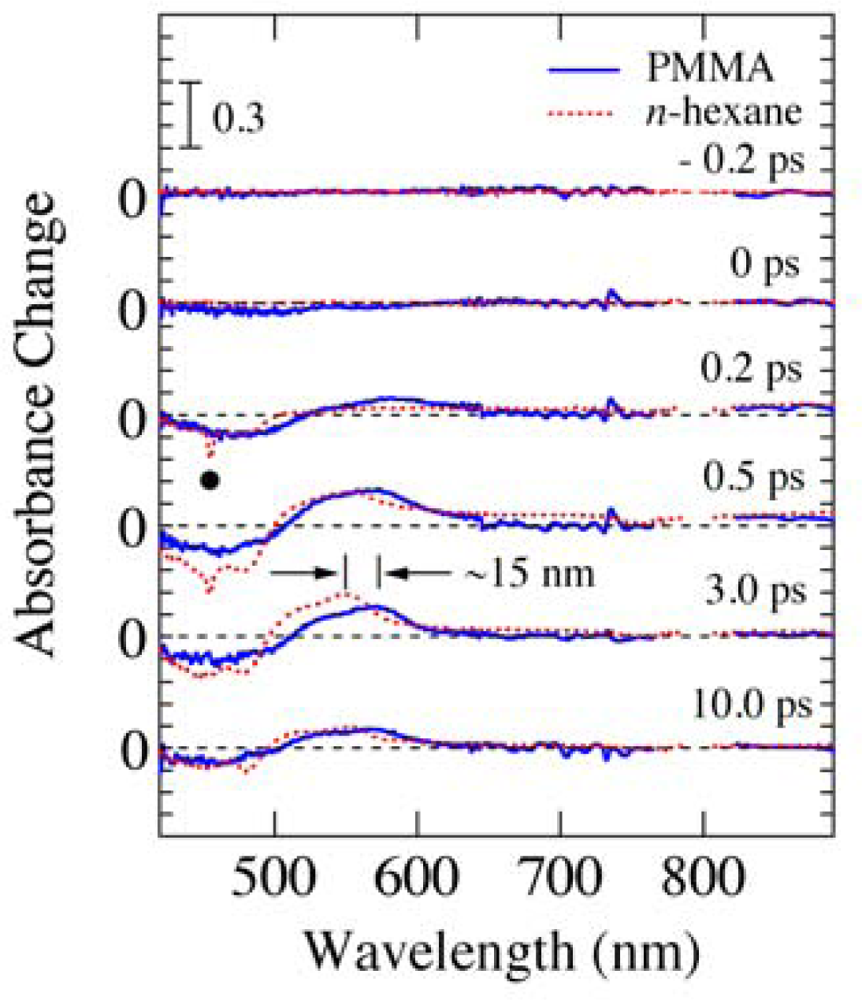
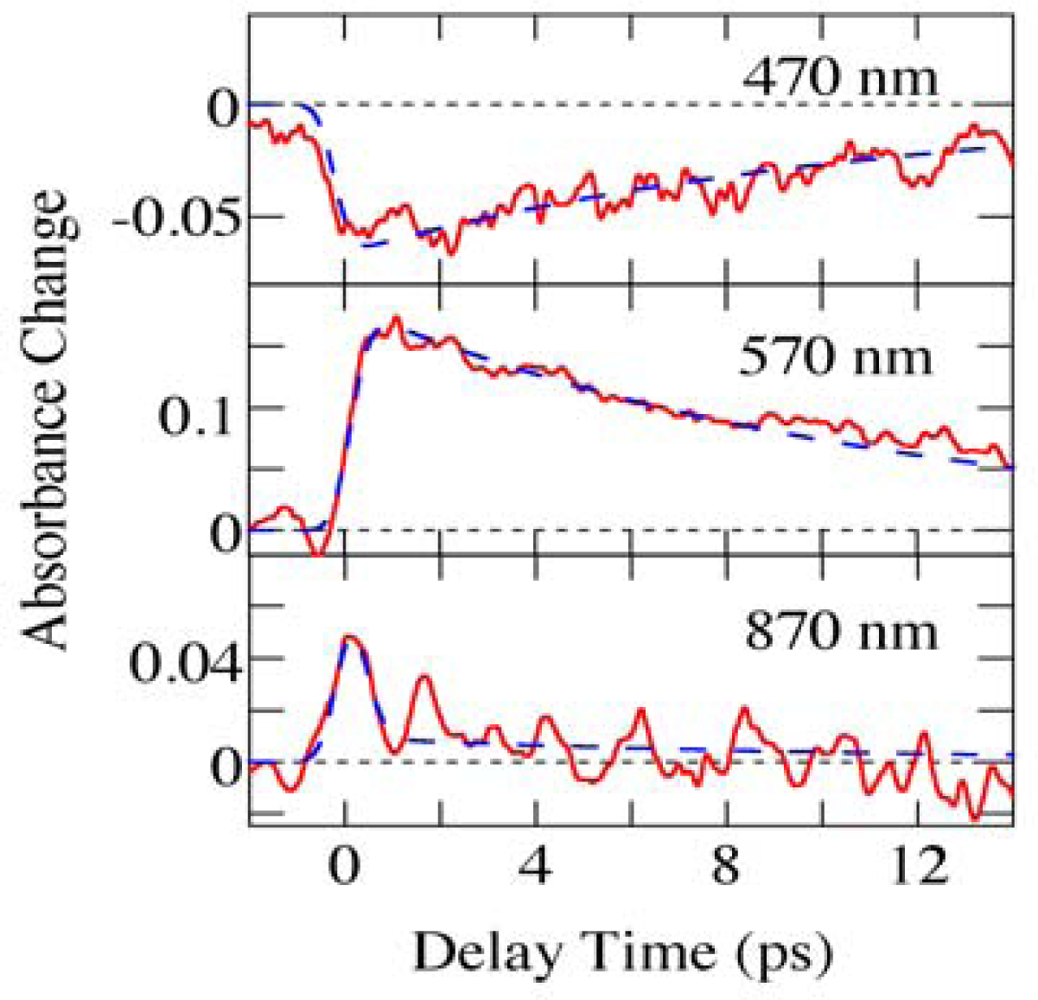
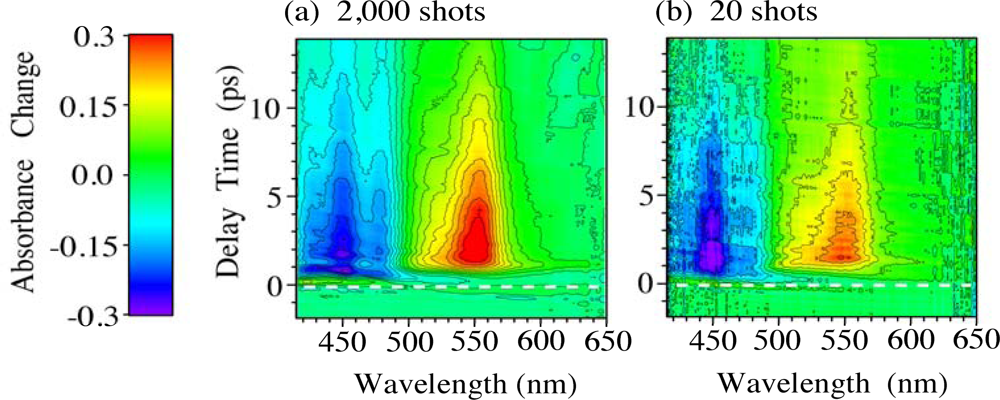
2.2. Time-Frequency 2D Imaging of Ultrafast Transient Absorption of β-Carotene in Solution
3. Photodegradation Processes of β-Carotene in Solid Films
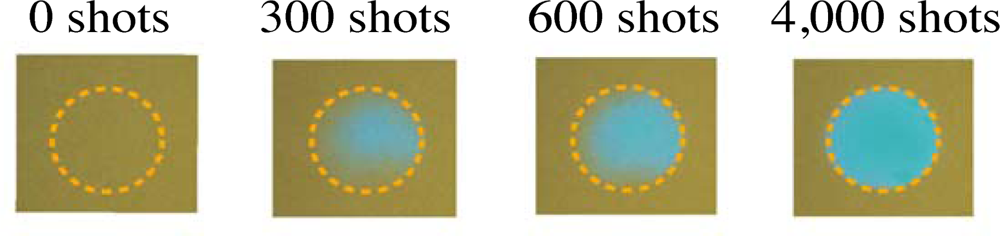
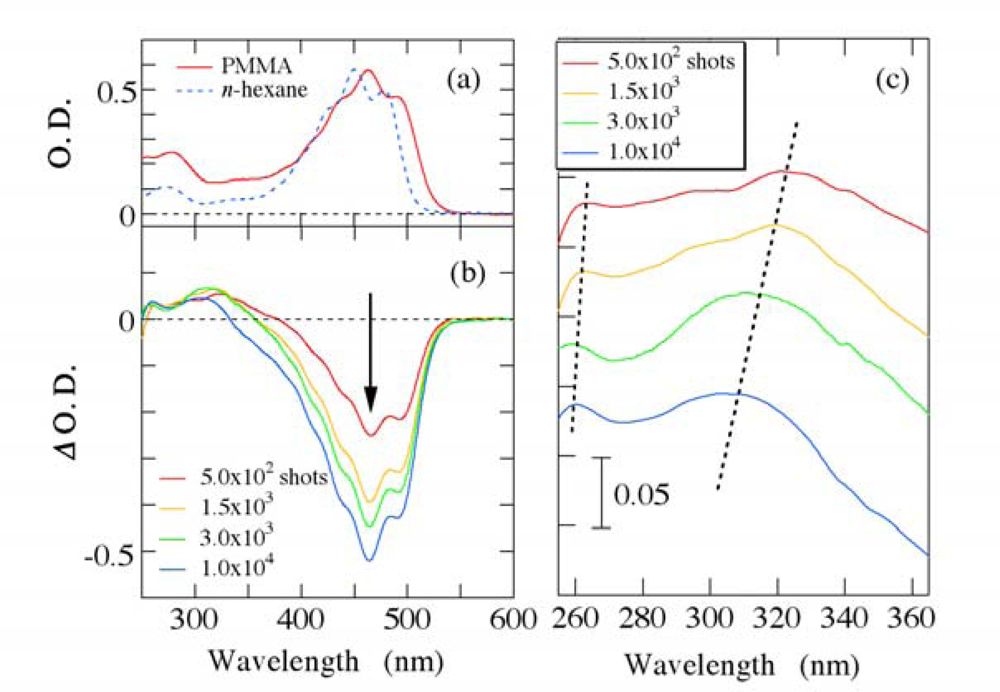
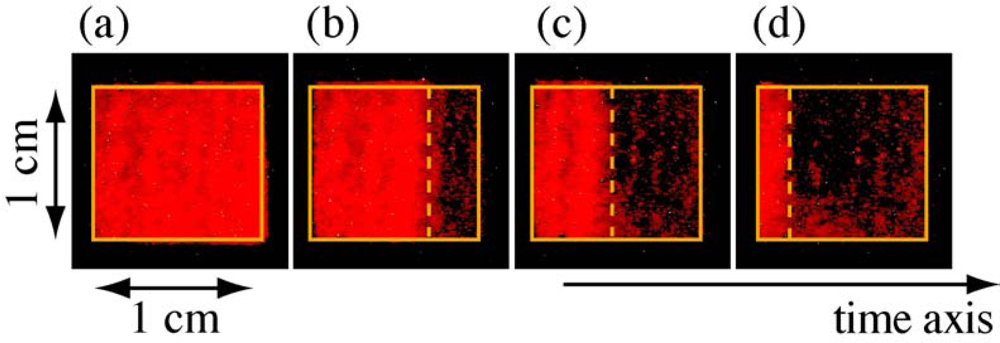
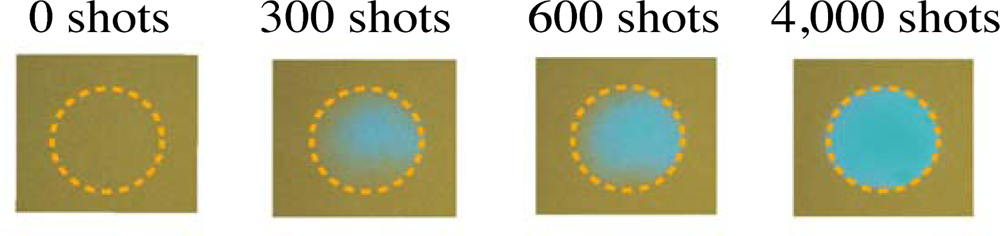
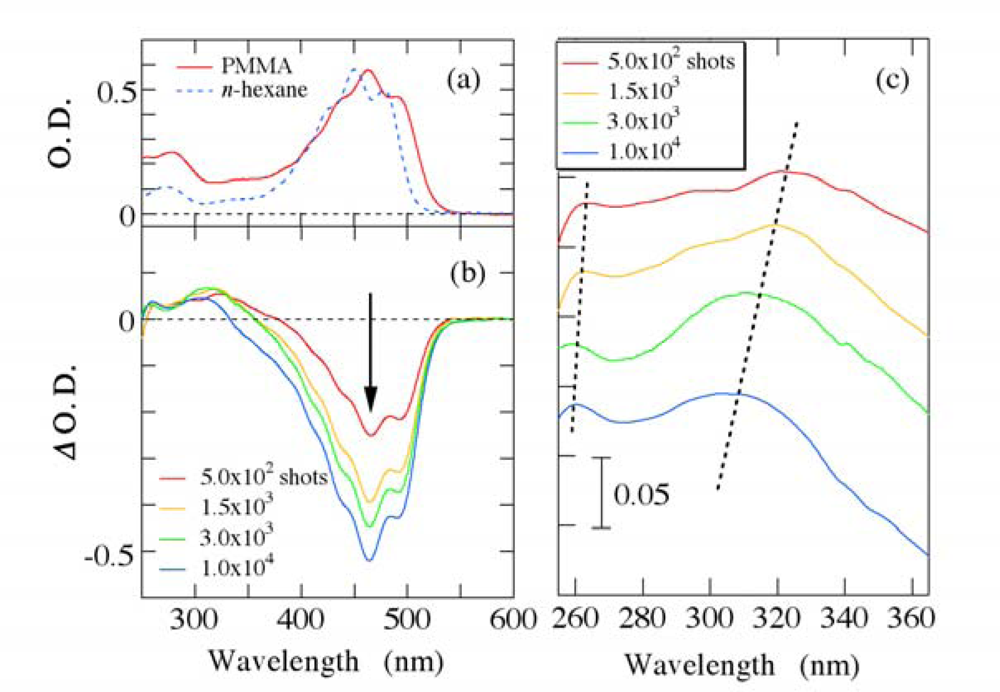
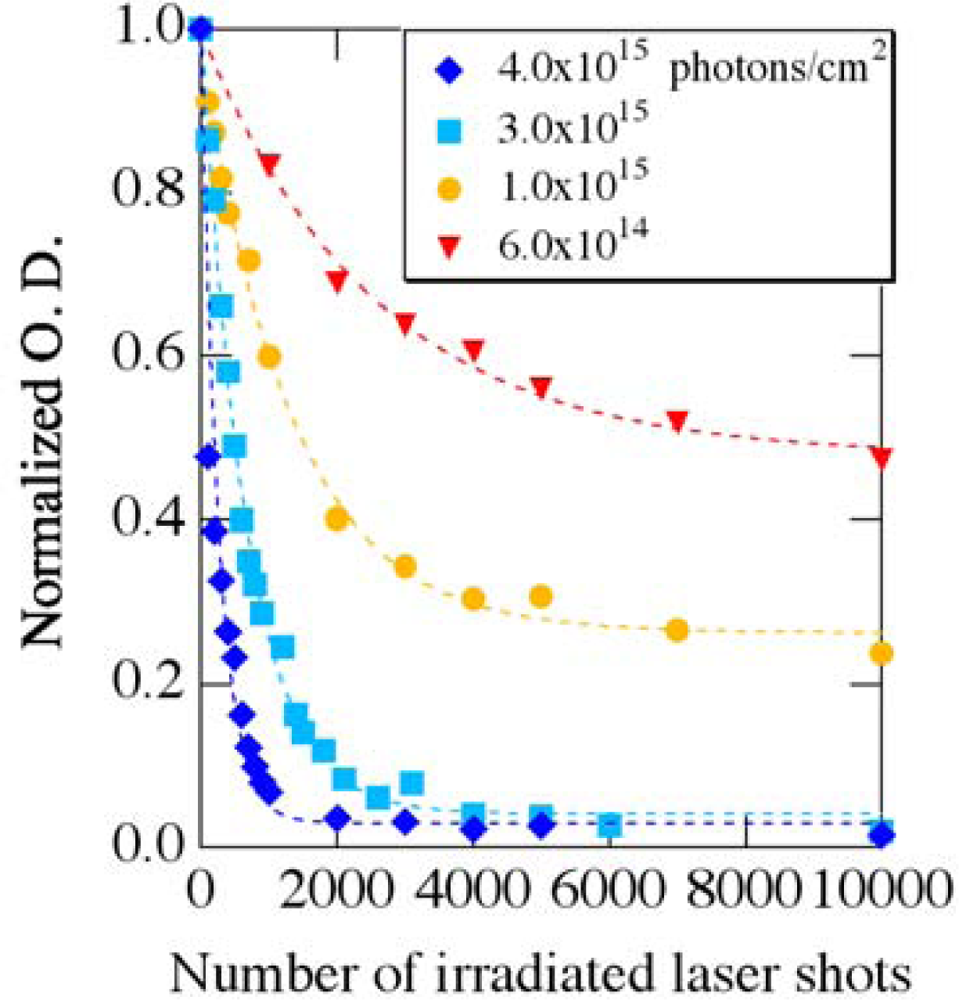
3.1. Photobleaching for Steady-State Absorption Spectra
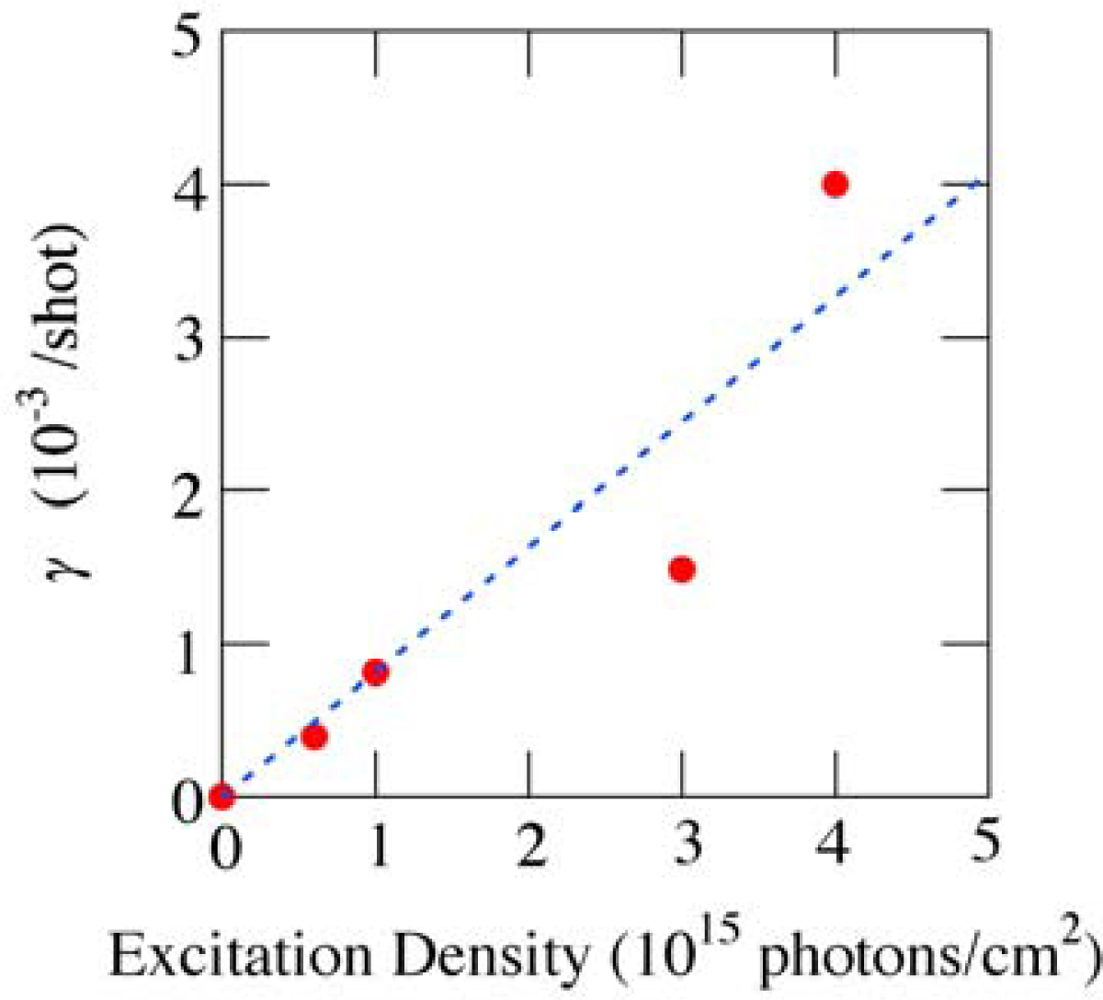
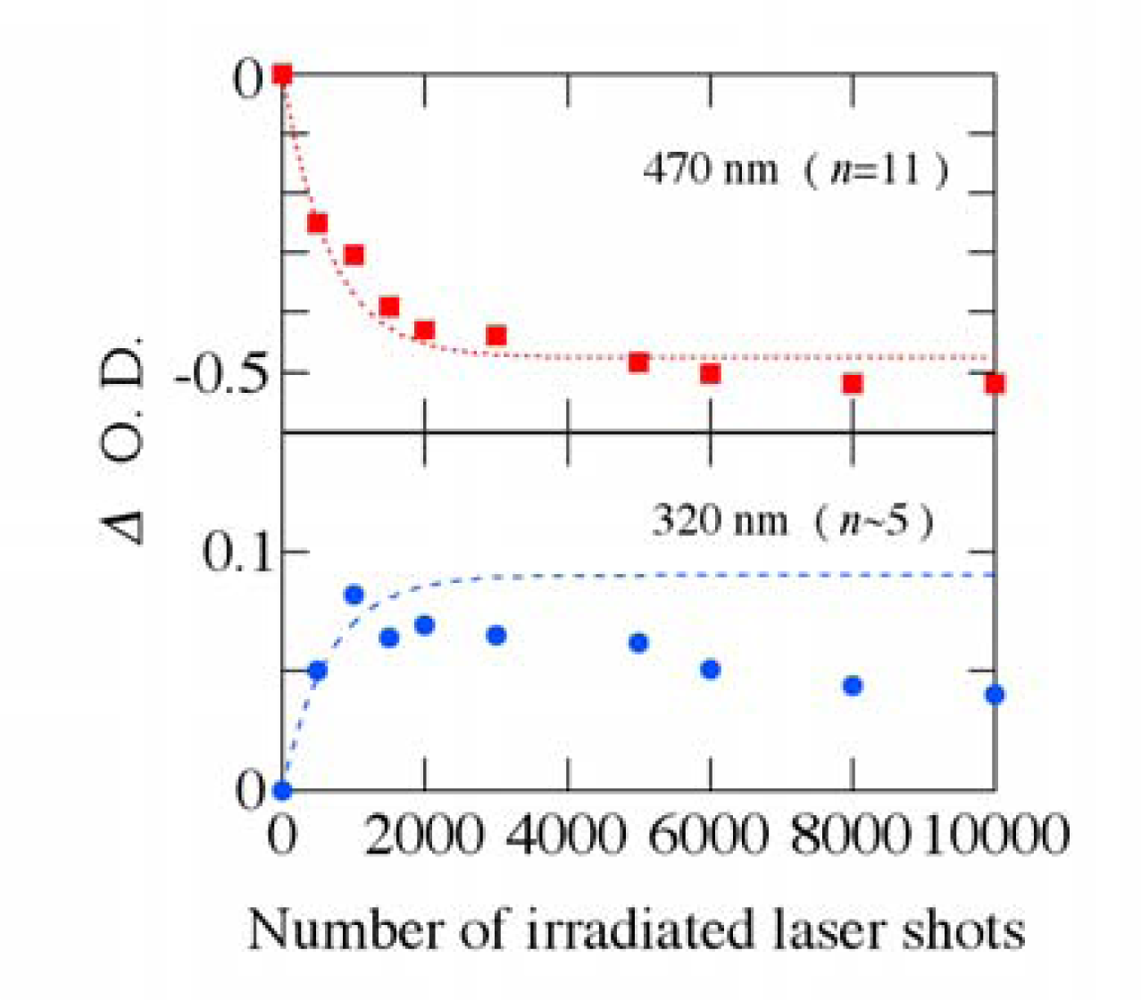
3.2. Possible Mechanism for the Photodegradation of β-Carotene in Solid Films
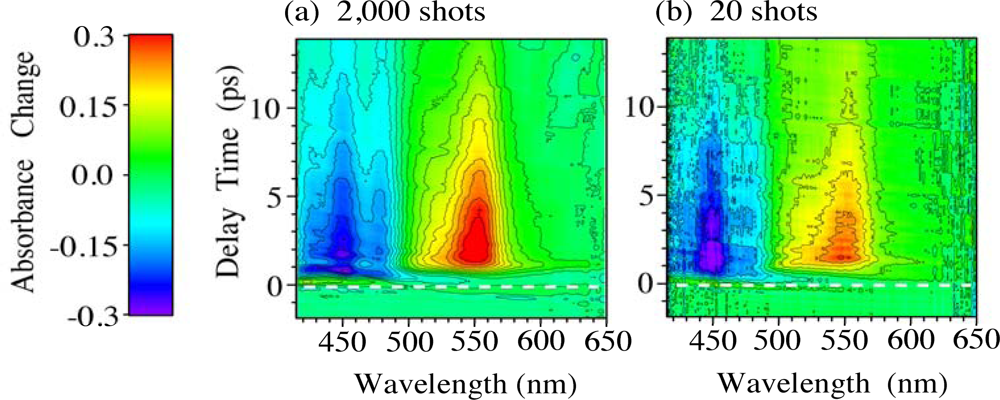
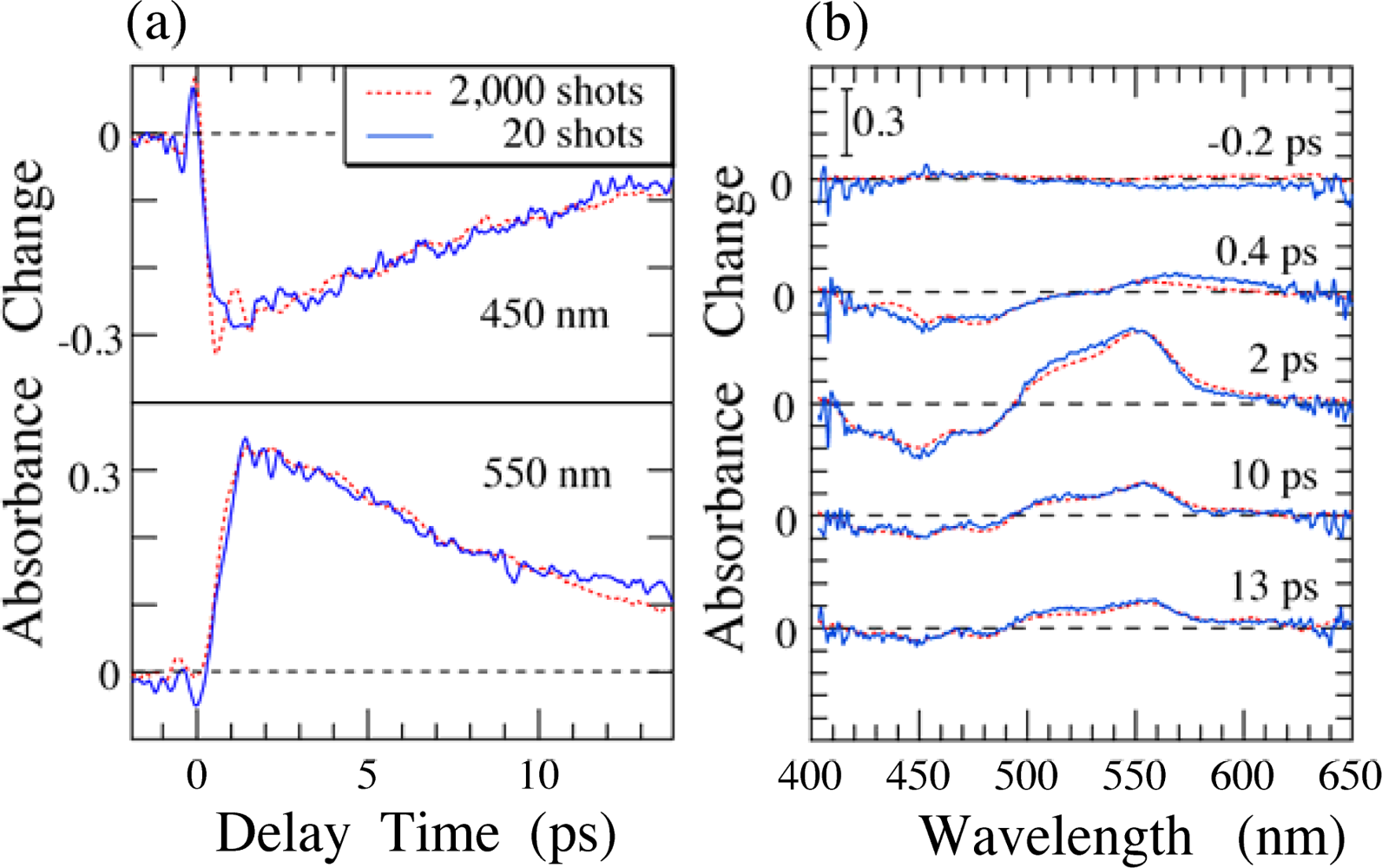
4. Time-Frequency 2D Imaging of Transient Absorption of β-Carotene in Solid Films
5. Conclusions
Acknowledgments
References and Notes
- Wakeham, G.P.; Nelson, K.A. Dual-echelon single-shot femtosecond spectroscopy. Opt. Lett 2000, 25, 505–507. [Google Scholar]
- Dhar, L.; Fourkas, J.T.; Nelson, K.A. Pulse-length-limited ultrafast pump-probe spectroscopy in a single shot. Opt. Lett 1994, 19, 643–645. [Google Scholar]
- Poulin, P.R.; Nelson, K.A. Irreversible organic crystalline chemistry monitored in real time. Science 2006, 291, 1756–1760. [Google Scholar]
- Furukawa, N.; Mair, C.E.; Kleiman, V.D.; Takeda, J. Femtosecond real-time pump-probe imaging spectroscopy. Appl. Phys. Lett 2004, 85, 4645–4647. [Google Scholar]
- Makishima, Y.; Furukawa, N.; Ishida, A.; Takeda, J. Femtosecond real-time pump-probe imaging spectroscopy implemented on a single shot basis. Jpn. J. Appl. Phys 2006, 45, 5986–5989. [Google Scholar]
- Furukawa, N.; Mair, C.E.; Kleiman, V.D.; Takeda, J. Direct visualization of transient absorption by real-time pump-probe imaging spectroscopy. In Ultrafast Phenomena XIV (Springer Series in Chemical Physics 79); Kobayashi, T., Okada, T., Kobayashi, T., Nelson, K.A., Silvestri, S.D., Eds.; Springer Berlin Heidelberg: New York, NY, USA, 2005; pp. 133–135. [Google Scholar]
- Takeuchi, S.; Tahara, T. Time-wavelength two-dimensional femtosecond fluorescence imaging. Opt. Lett 2004, 29, 313–315. [Google Scholar]
- Frank, H.A.; Cogdell, R.J. Carotenoid in photosynthesis. Photochem. Photobiol 1996, 63, 257–264. [Google Scholar]
- Polívka, T.; Sundström, V. Ultrafast dynamics of carotenoid excited states-from solution to natural and artificial systems. Chem. Rev 2004, 104, 2021–2072. [Google Scholar]
- Zhang, J.P.; Fujii, R.; Koyama, Y.; Rondonuwu, F.S.; Watanabe, Y.; Mortensen, A.; Skibsted, L.H. The 1Bu-type singlet state of β-carotene as a precursor of the radical cation found in chloroform solution by sub-picosecond time-resolved absorption spectroscopy. Chem. Phys. Lett 2001, 348, 235–241. [Google Scholar]
- Nishimura, K.; Rondonuwu, F.S.; Fujii, R.; Akahane, J.; Koyama, Y.; Kobayashi, T. Sequential singlet internal conversion of 1Bu+→3Ag−→1Bu−→2Ag−→(1Ag−ground) in all-trans-spirilloxanthin revealed by two-dimensional sub-5-fs spectroscopy. Chem. Phys. Lett 2004, 392, 68–73. [Google Scholar]
- Cerullo, G.; Polli, D.; Lanzani, G.; Silvestri, S.D.; Hashimoto, H.; Cogdell, R.J. Photosynthetic light harvesting by carotenoids: detection of an intermediate excited state. Science 2002, 298, 2935–2938. [Google Scholar]
- Polli, D.; Cerullo, G.; Lanzani, G.; Silvestri, S.D.; Yanagi, K.; Hashimoto, H.; Cogdell, R.J. Conjugation length dependence of internal conversion in carotenoids: role of the intermediate state. Phys. Rev. Lett 2004, 93, 163002. [Google Scholar]
- Larsen, D.S.; Papagiannakis, E.; van Stokkum, I.H.M.; Vengris, M.; Kennis, J.T.M.; van Grondelle, R. Excited state dynamics of β-carotene explored with dispersed multi-pulse transient absorption. Chem. Phys. Lett 2003, 381, 733–742. [Google Scholar]
- Pendon, Z.D.; Gibson, G.N.; van der Hoef, I.; Lugtenburg, J.; Frank, H.A. Effect of isomer geometry on the steady-state absorption spectra and femtosecond time-resolved dynamics of carotenoids. J. Phys. Chem. B 2005, 109, 21172–21179. [Google Scholar]
- Wohlleben, W.; Buckup, T.; Hashimoto, H.; Cogdell, R.J.; Herek, J.L.; Motzkus, M. Pump-deplete-probe spectroscopy and the puzzle of carotenoid dark states. J. Phys. Chem. B 2004, 108, 3320–3325. [Google Scholar]
- Kosumi, D.; Komukai, M.; Hashimoto, H.; Yoshizawa, M. Ultrafast dynamics of all-trans-β-carotene explored by resonant and nonresonant photoexcitations. Phys. Rev. Lett 2005, 95, 213601. [Google Scholar]
- Kosumi, D.; Yanagi, K.; Nishino, T.; Hashimoto, H.; Yoshizawa, M. Excitation energy dependence of excited states dynamics in all-trans-carotenes determined by femtosecond absorption and fluorescence spectroscopy. Chem. Phys. Lett 2005, 408, 89–95. [Google Scholar]
- Brodeur, A.; Chin, S.L. Band-gap dependence of the ultrafast white-light continuum. Phys. Rev. Lett 1998, 80, 4406–4409. [Google Scholar]
- Nagura, A.; Suda, A.; Kawano, H.; Obara, M.; Midorikawa, K. Generation and characterization of ultrafast white-light continuum in condensed media. Appl. Opt 2002, 41, 3735–3742. [Google Scholar]
- Fourkas, J.T.; Dhar, L.; Nelson, K.A.; Trebino, R. Spatially encoded, single-shot ultrafast spectroscopies. J. Opt. Soc. Am. B 1995, 12, 155–165. [Google Scholar]
- Takeda, J.; Nakajima, K.; Kurita, S.; Tomimoto, S.; Saito, S.; Suemoto, T. Time-resolved luminescence spectroscopy by the optical Kerr-gate method applicable to ultrafast relaxation processes. Phys. Rev. B 2000, 62, 10083–10087. [Google Scholar]
- Takeda, J.; Nakajima, K.; Kurita, S.; Tomimoto, S.; Saito, S.; Suemoto, T. Femtosecond optical Kerr gate fluorescence spectroscopy for ultrafast relaxation processes. J. Lumin 2000, (87–89), 927–929. [Google Scholar]
- Kinoshita, S.; Ozawa, H.; Kanematsu, Y.; Tanaka, I.; Sugimoto, N.; Fujiwara, S. Efficient optical Kerr shutter for femtosecond time-resolved luminescence spectroscopy. Rev. Sci. Instrum 2000, 71, 3317–3322. [Google Scholar]
- Arzhantsev, S.; Maroncelli, M. Design and characterization of a femtosecond fluorescence spectrometer based on optical Kerr gating. Appl. Spectrsc 2005, 59, 206–220. [Google Scholar]
- Nagae, H.; Kuki, M.; Zhang, J.P.; Sashima, T.; Mukai, Y.; Koyama, Y. Vibronic coupling through the in-phase, C=C stretching mode plays a major role in the 2Ag− to 1Ag− internal conversion of all-trans-β-Carotene. J. Phys. Chem. A 2000, 104, 4155–4166. [Google Scholar]
- Akai, I.; Miyanari, K.; Shimamoto, T.; Fujii, A.; Nakao, H.; Okada, A.; Kanemoto, K.; Karasawa, T.; Hashimoto, H.; Ishida, A.; Yamada, A.; Katayama, I.; Takeda, J.; Kimura, M. Rapid energy transfer in a dendrimer having π-conjugated light-harvesting antennas. New J. Phys 2008, 10, 125024. [Google Scholar]
- Ishida, A.; Makishima, Y.; Okada, A.; Akai, I.; Kanemoto, K.; Karasawa, T.; Kimura, M.; Takeda, J. Time–frequency two-dimensional mapping of rapid energy transfer in light-harvesting star-shaped dendrimers. J. Lumin 2008, 128, 771–773. [Google Scholar]
- Macpherson, A.N.; Gillbro, T. Solvent dependence of the ultrafast S2-S1 internal conversion rate of β-carotene. J. Phys. Chem. A 1998, 102, 5049–5058. [Google Scholar]
- Lewis, G.N.; Calvin, M. The color of organic substances. Chem. Rev 1939, 25, 273–328. [Google Scholar]
- Ferguson, L.N. Relationships between absorption spectra and spectra and chemical constitution of organic molecules. Chem. Rev 1948, 43, 385–446. [Google Scholar]
- Horie, K.; Hirao, K.; Kenmochi, N.; Mita, I. Photochromic reactions of spirobenzopyran, azobenzene, and fulgide at 4 K in polymer films. Makromol. Chem. Rapid Commun 1988, 9, 267–273. [Google Scholar]
- Takeda, J.; Nakayama, N.; Nagase, N.; Tayu, T.; Kainuma, K.; Kurita, S.; Yokoyama, Y.; Kurita, Y. Inter-molecular interaction of photochromic furylfulgide dispersed in polymer film. Chem. Phys. Lett 1992, 198, 609–614. [Google Scholar]
- Victor, J.G.; Torkelson, J.M. On measuring the distribution of local free volume in glassy polymers by photochromic and fluorescence techniques. Macromolecules 1987, 20, 241–2250. [Google Scholar]
- Furukawa, N.; Takeda, J. Ultrafast internal conversion of all-trans-β-carotene studied by femtosecond spectroscopy. Nonlinear Opt 2002, 29, 579–585. [Google Scholar]
- Yoshizawa, M.; Aoki, H.; Hashimoto, H. Vibrational relaxation of the 2Ag− excited state in all-trans-β-carotene obtained by femtosecond time-resolved Raman spectroscopy. Phys. Rev. B 2001, 63, 180301(R). [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| solvents/matrix | Time constant (ps) | |
|---|---|---|
| internal conversion 11Bu+ → 21Ag− | recovery 21Ag− → 11Ag− | |
| PMMA*1) | 0.17 | 10.0 |
| n-hexane | 0.22*2), 0.28*3) | 9.4*2), 9.3*3) |
| benzene*2) | 0.25 | 9.1 |
| chloroform*3) | 0.20 | 9.8 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Takeda, J.; Ishida, A.; Makishima, Y.; Katayama, I. Real-Time Time-Frequency Two-Dimensional Imaging of Ultrafast Transient Signals in Solid-State Organic Materials. Sensors 2010, 10, 4253-4269. https://doi.org/10.3390/s100504253
Takeda J, Ishida A, Makishima Y, Katayama I. Real-Time Time-Frequency Two-Dimensional Imaging of Ultrafast Transient Signals in Solid-State Organic Materials. Sensors. 2010; 10(5):4253-4269. https://doi.org/10.3390/s100504253
Chicago/Turabian StyleTakeda, Jun, Akihiro Ishida, Yoshinori Makishima, and Ikufumi Katayama. 2010. "Real-Time Time-Frequency Two-Dimensional Imaging of Ultrafast Transient Signals in Solid-State Organic Materials" Sensors 10, no. 5: 4253-4269. https://doi.org/10.3390/s100504253