Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Taxonomic Revision of the Clostridiales Is a Work in Progress
1.2. Lachnospiraceae and Ruminococcaceae are Active Members of the Gut Environment
1.3. The Complexity of Plant Material Poses Challenges for Bacterial Decomposition
1.4. Genomic Clues to Fibrolytic Function in Gut Environments
2. Experimental Section
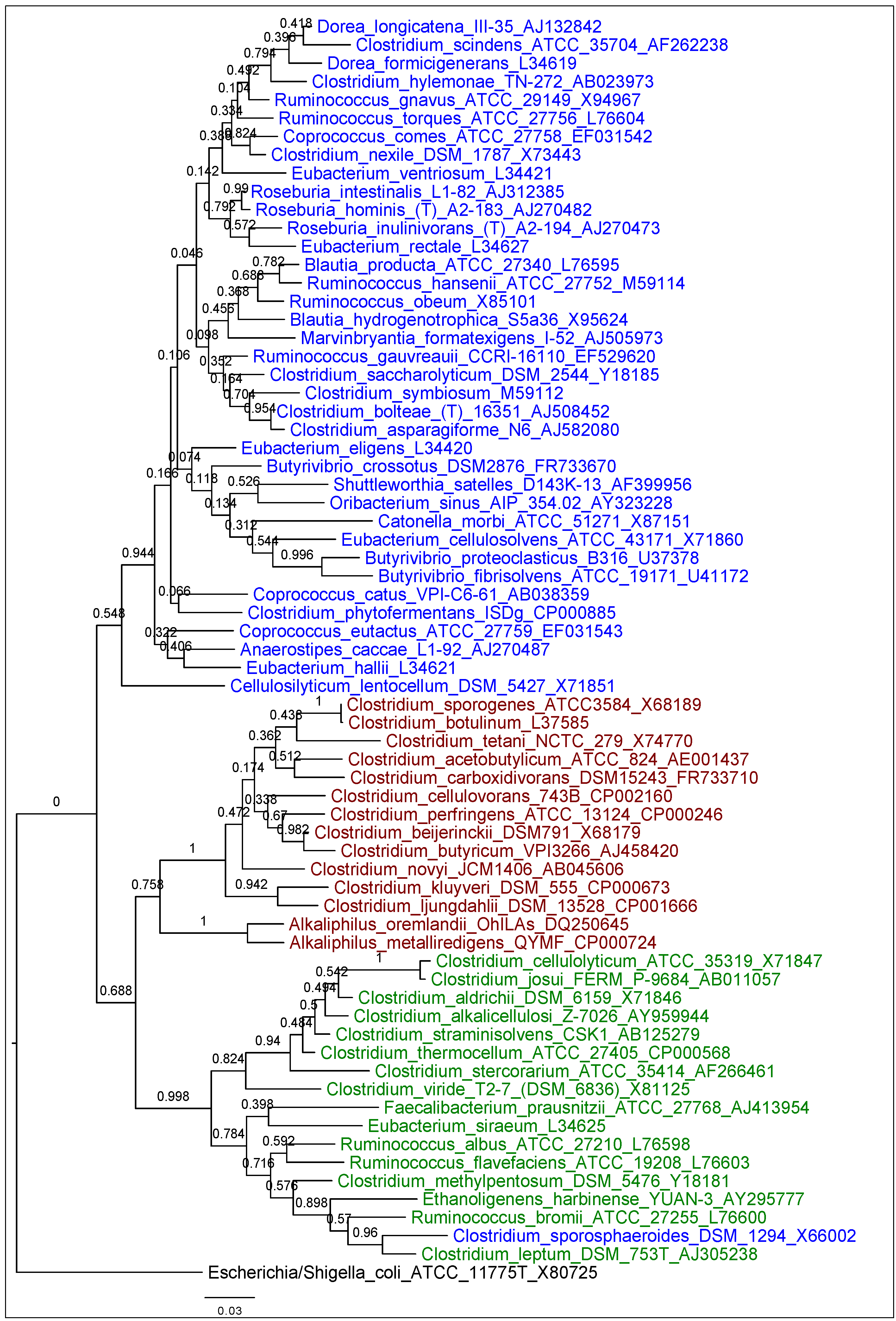
2.1. Phylogenetic Arrangement of Lachnospiraceae, Clostridiaceae, and Ruminococcaceae
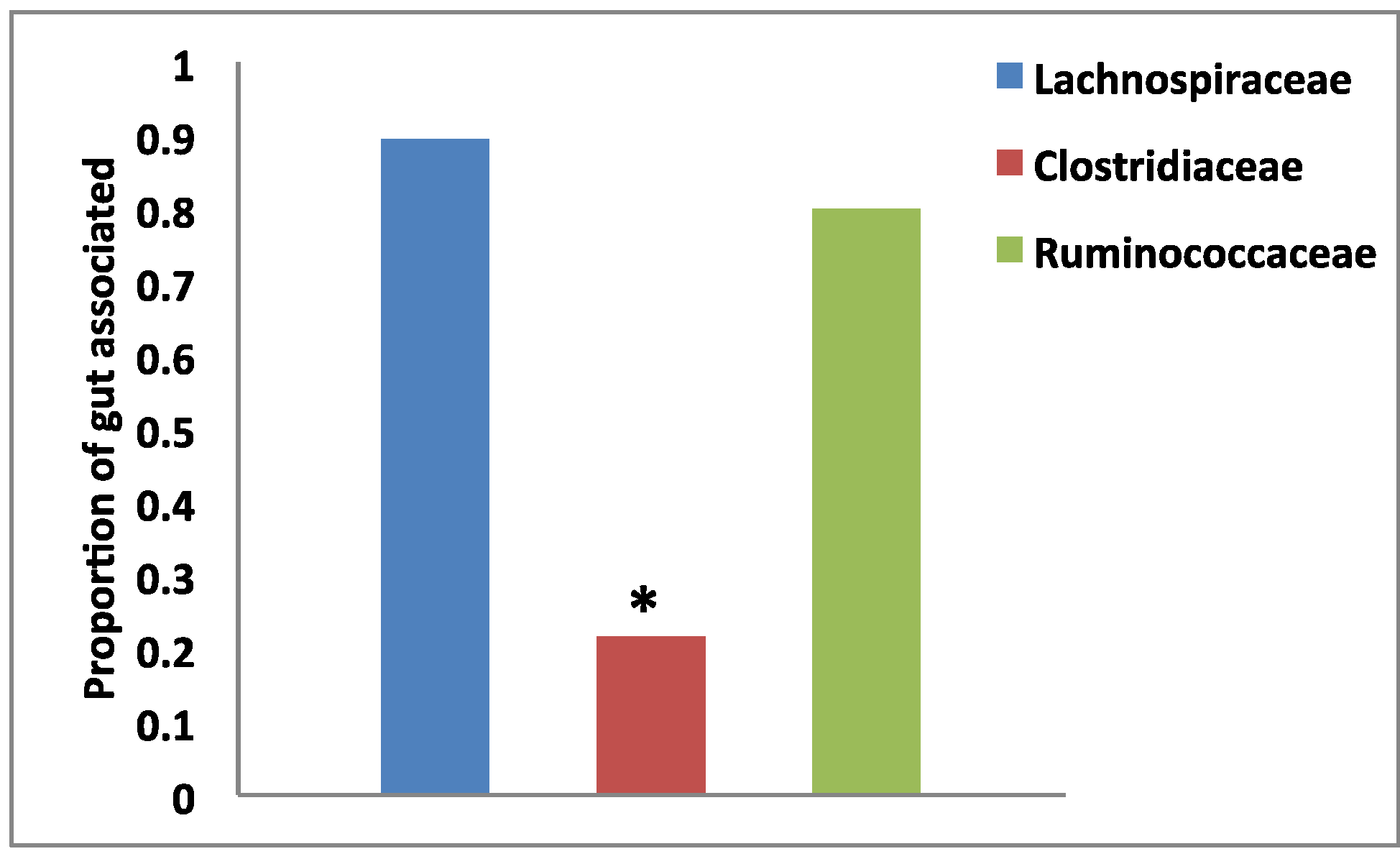
2.2. Habitat Association by Group
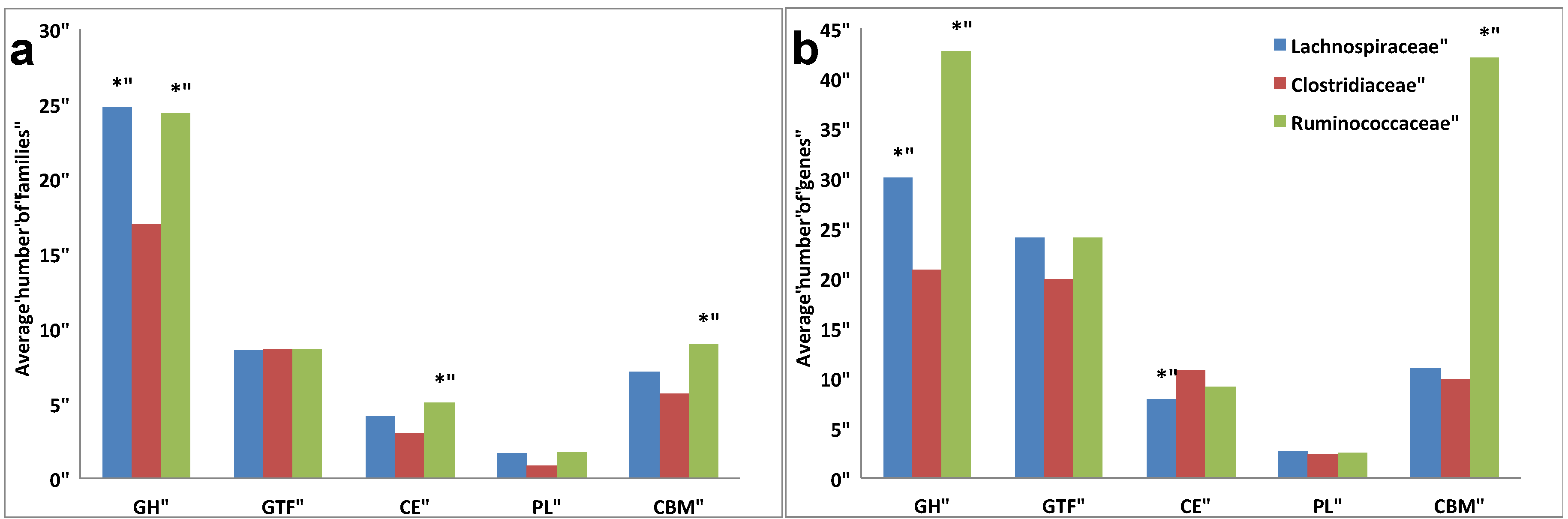
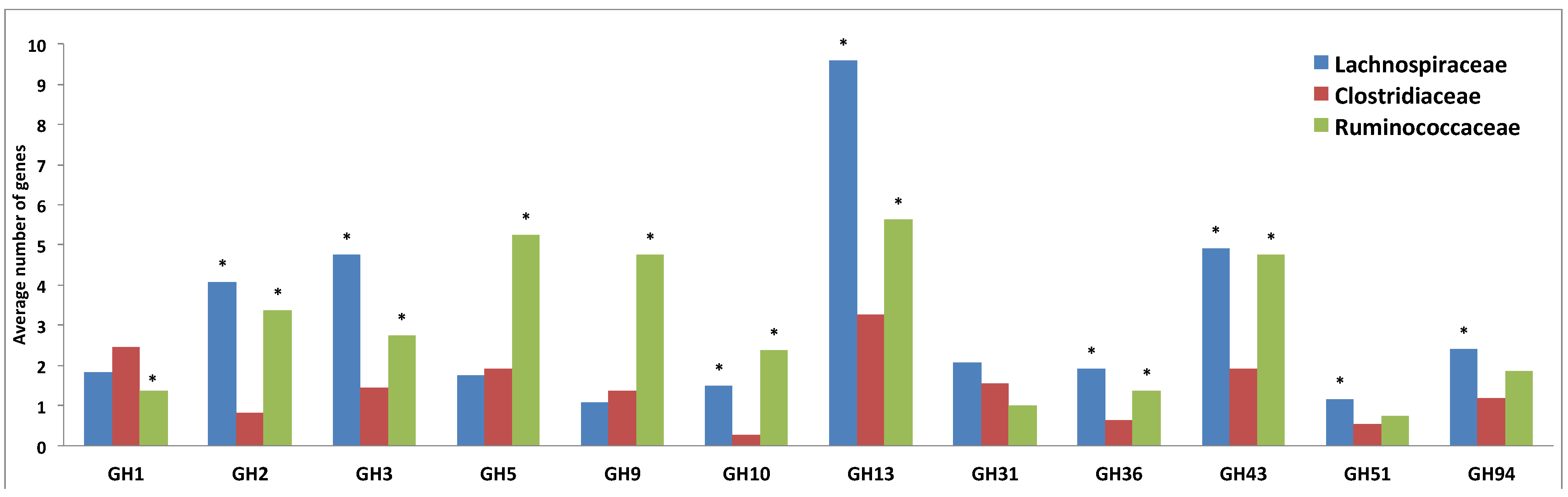
2.3. Comparative Analysis of Carbohydrate-Active Enzymes
2.4. Comparative Analysis of Sugar Transport Genes
2.5. Comparative Analysis of Metabolic Pathways
3. Results and Discussion
3.1. Phylogenetic Arrangement of Lachnospiraceae, Clostridiaceae, and Ruminococcaceae

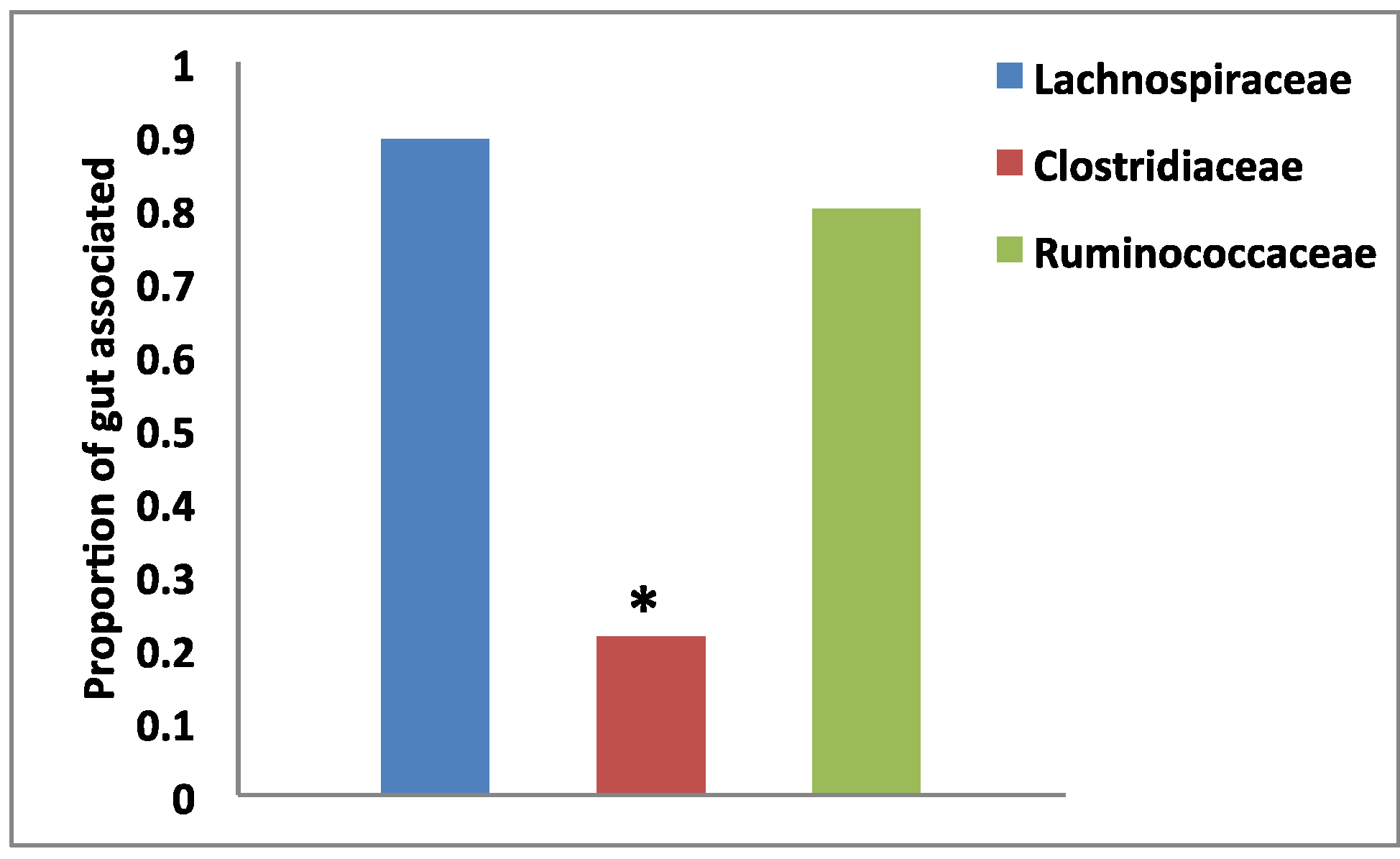
3.2. Habitat Association by Group
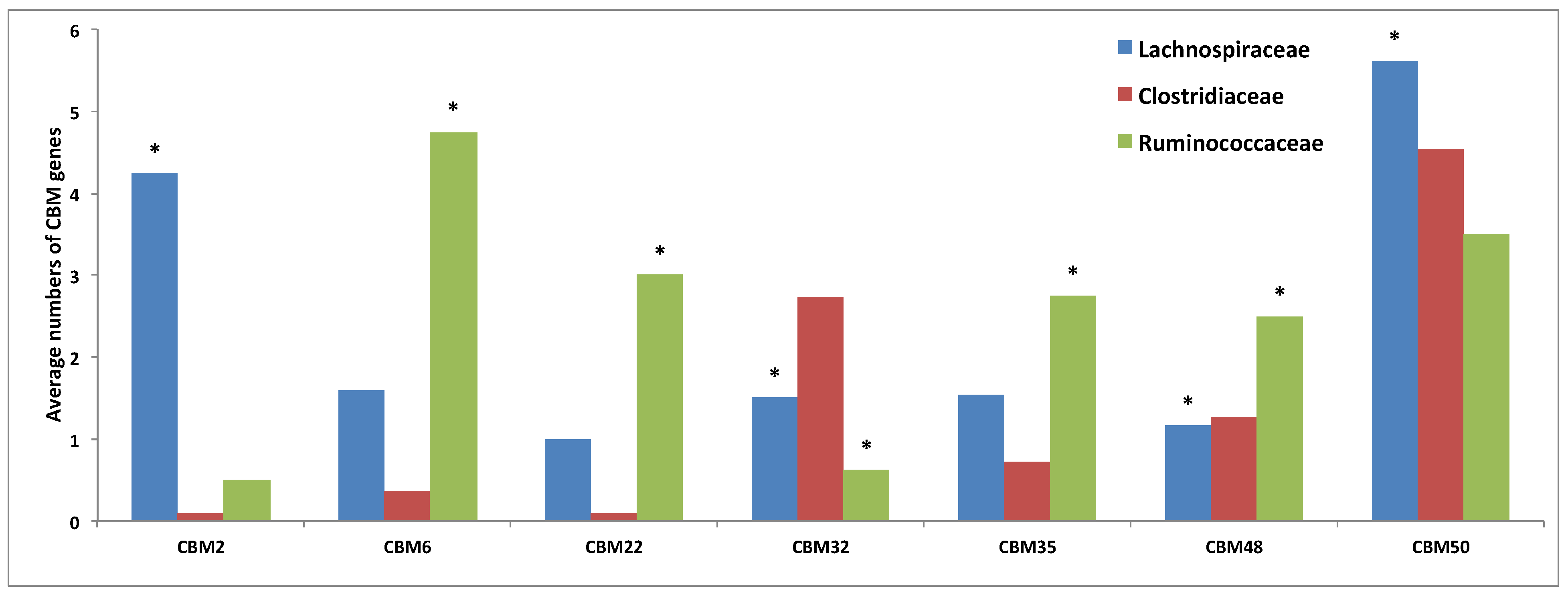
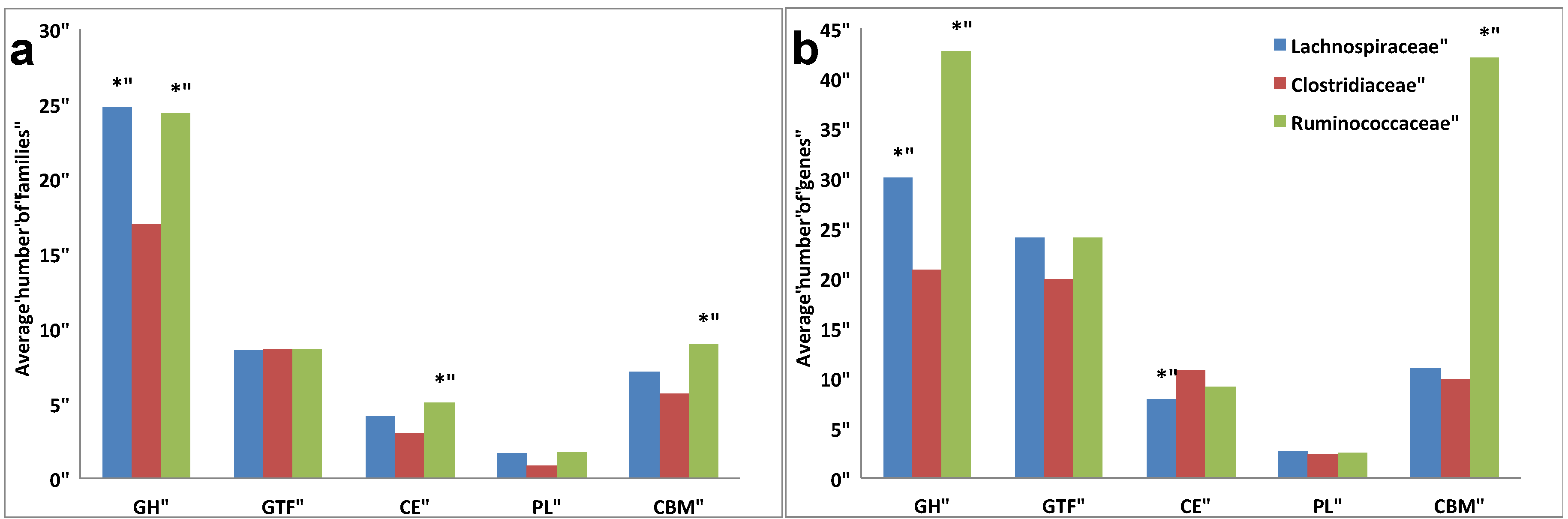
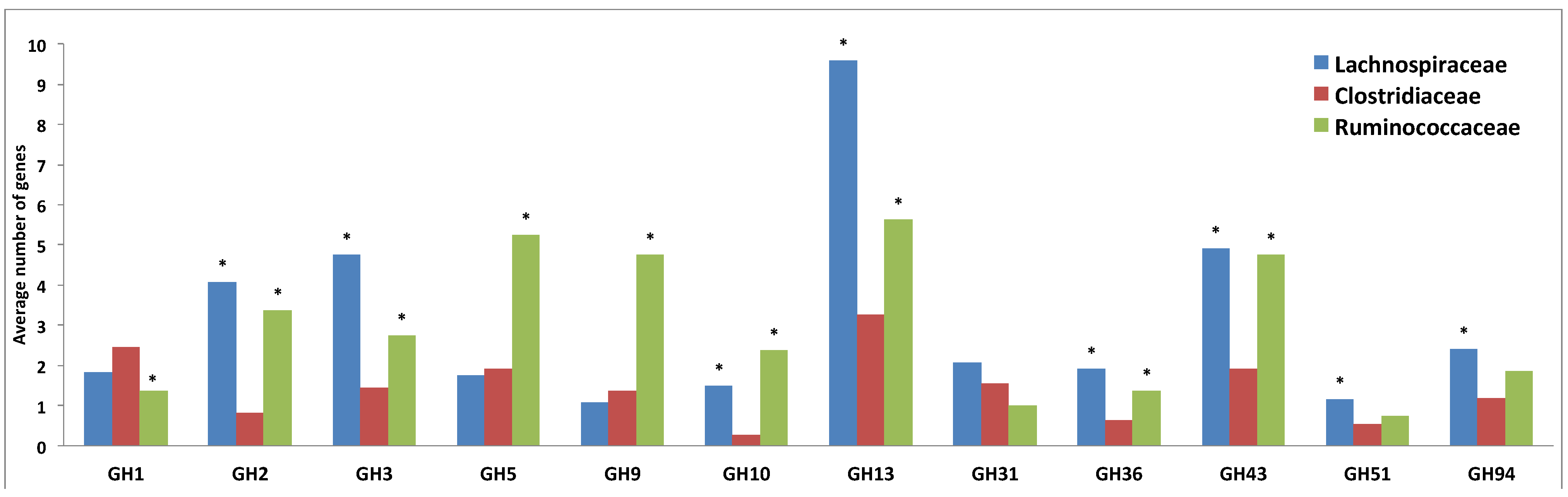
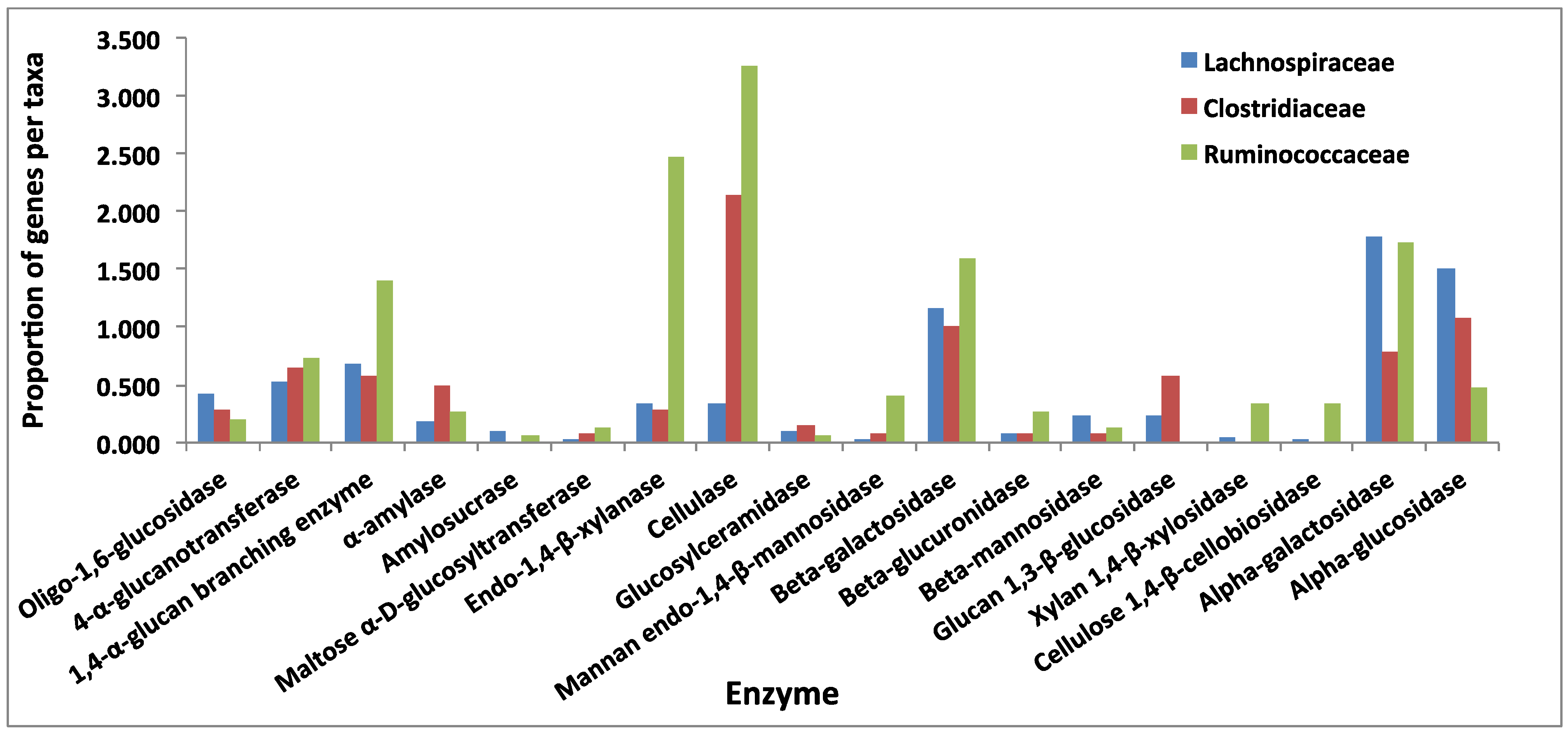
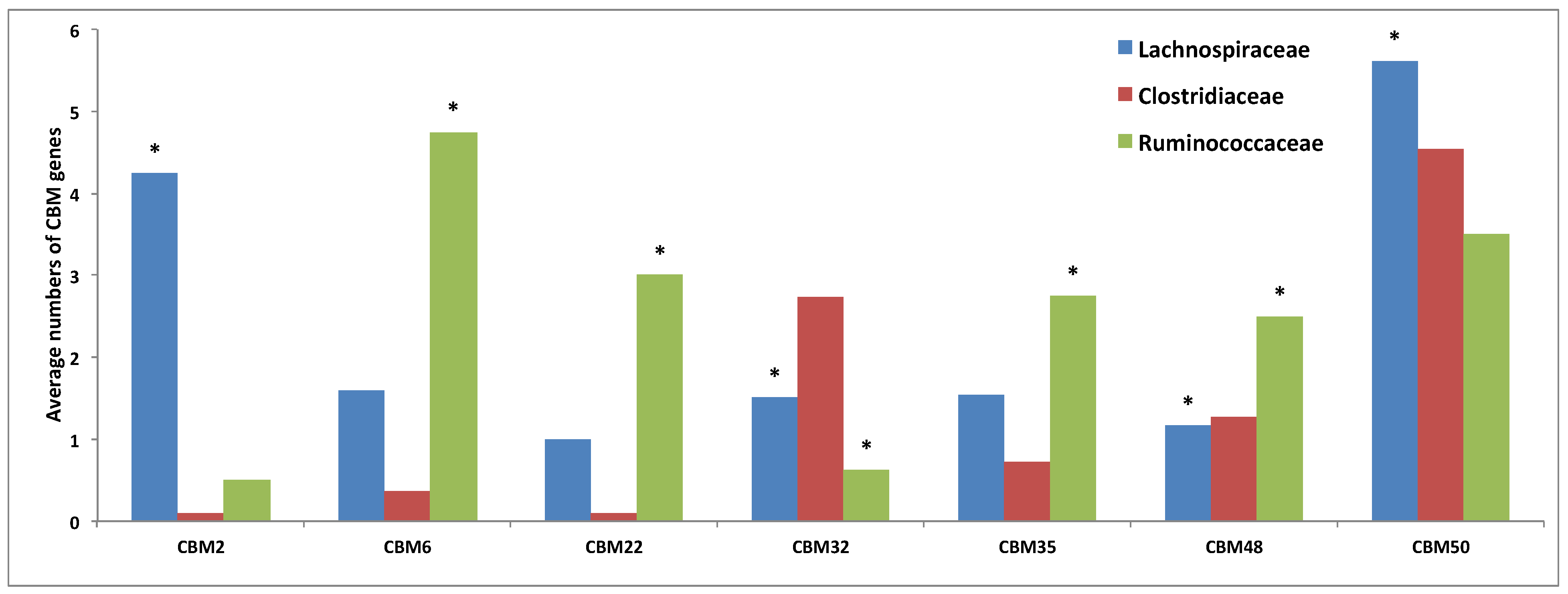
3.3. Comparative Analysis of Carbohydrate-Active Enzymes
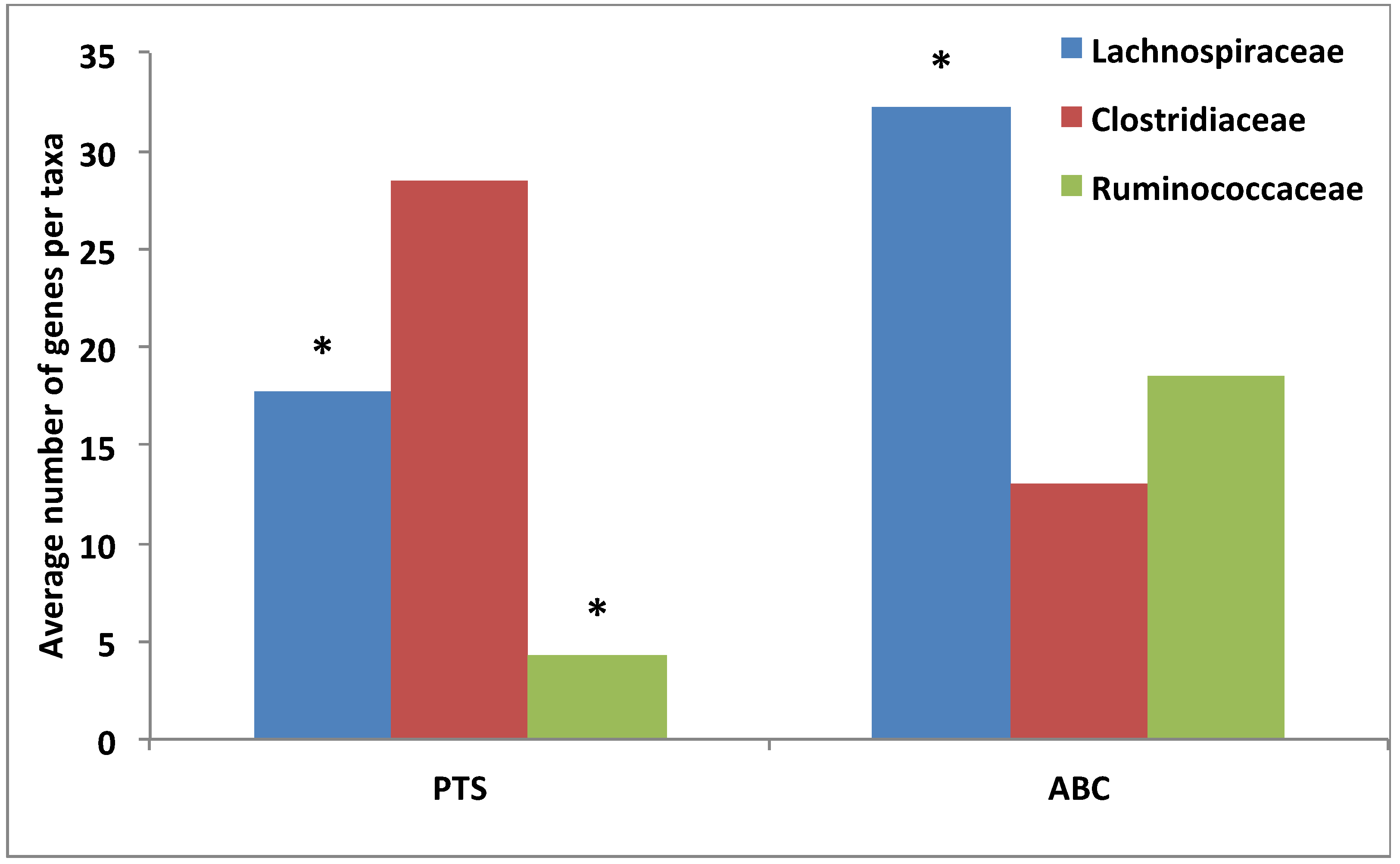
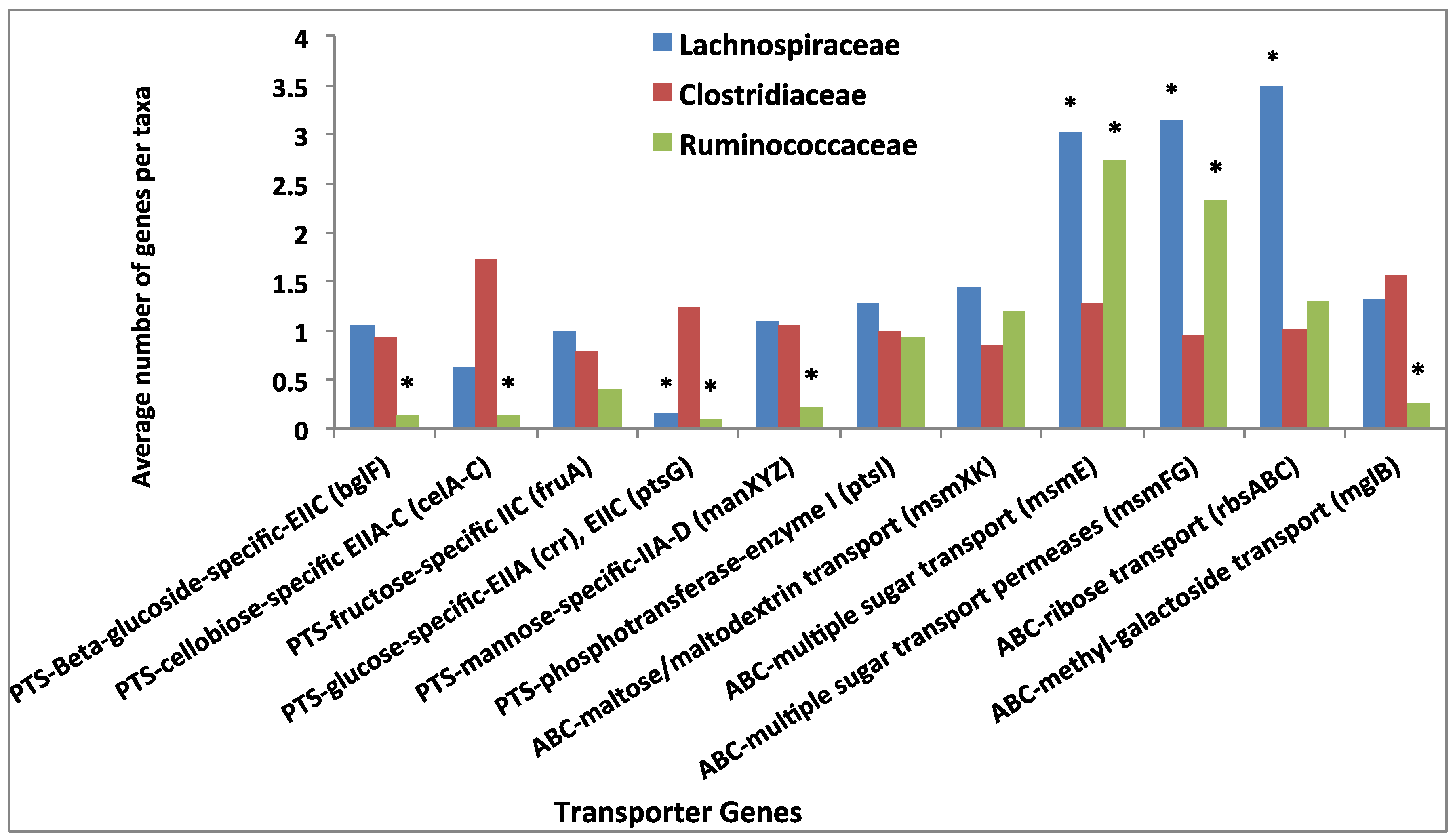
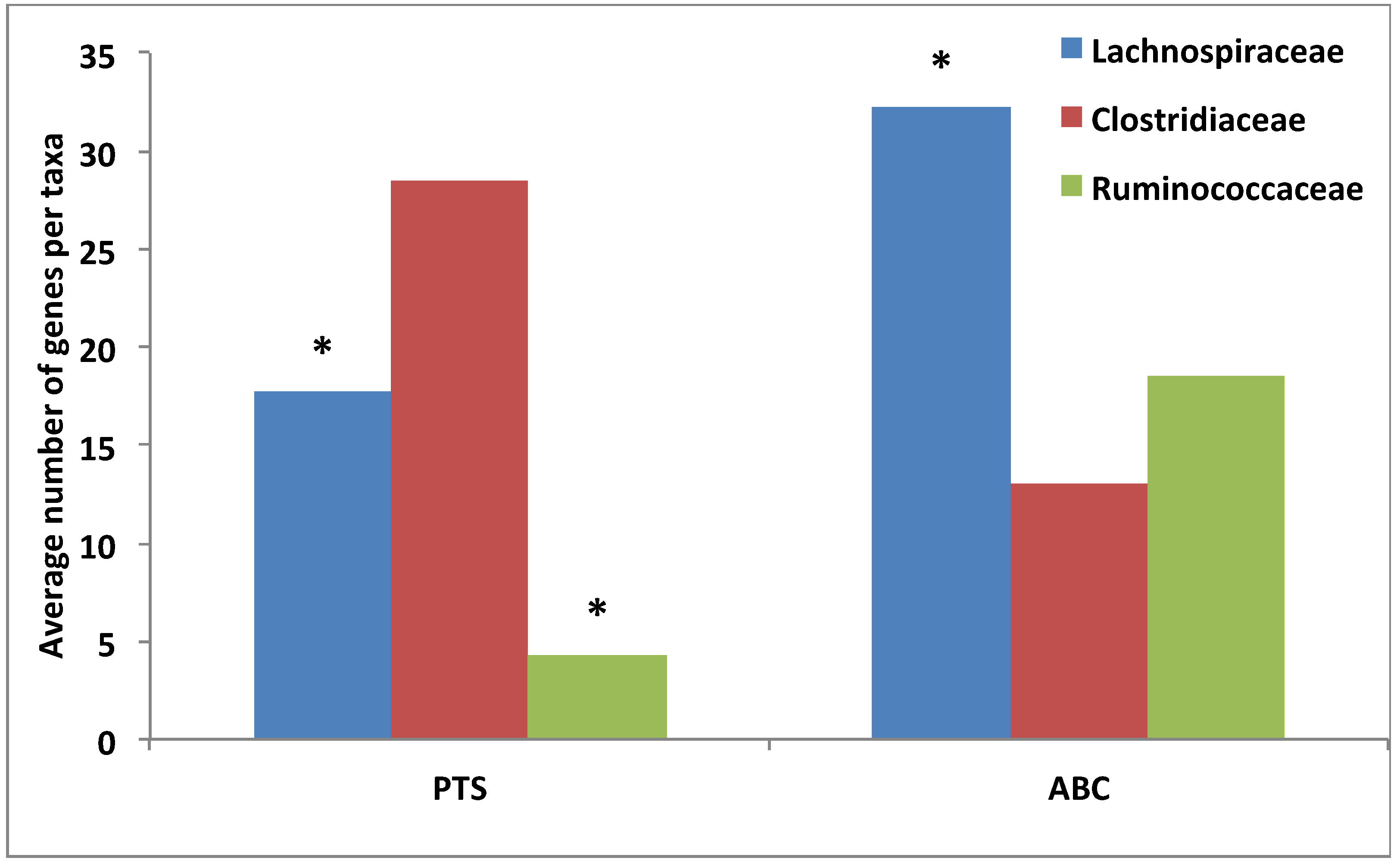
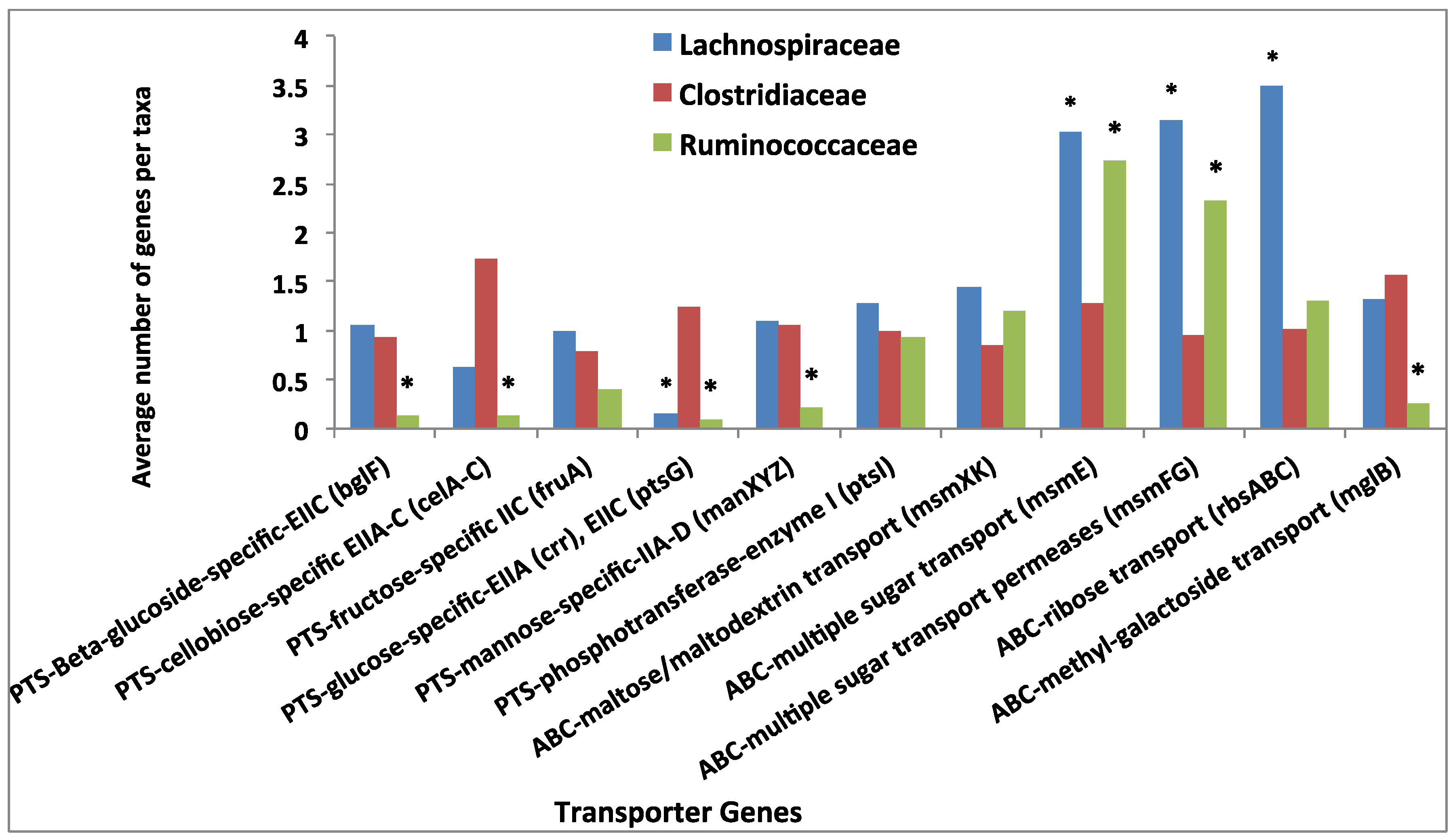
3.4. Comparative Analysis of Transporter Proteins
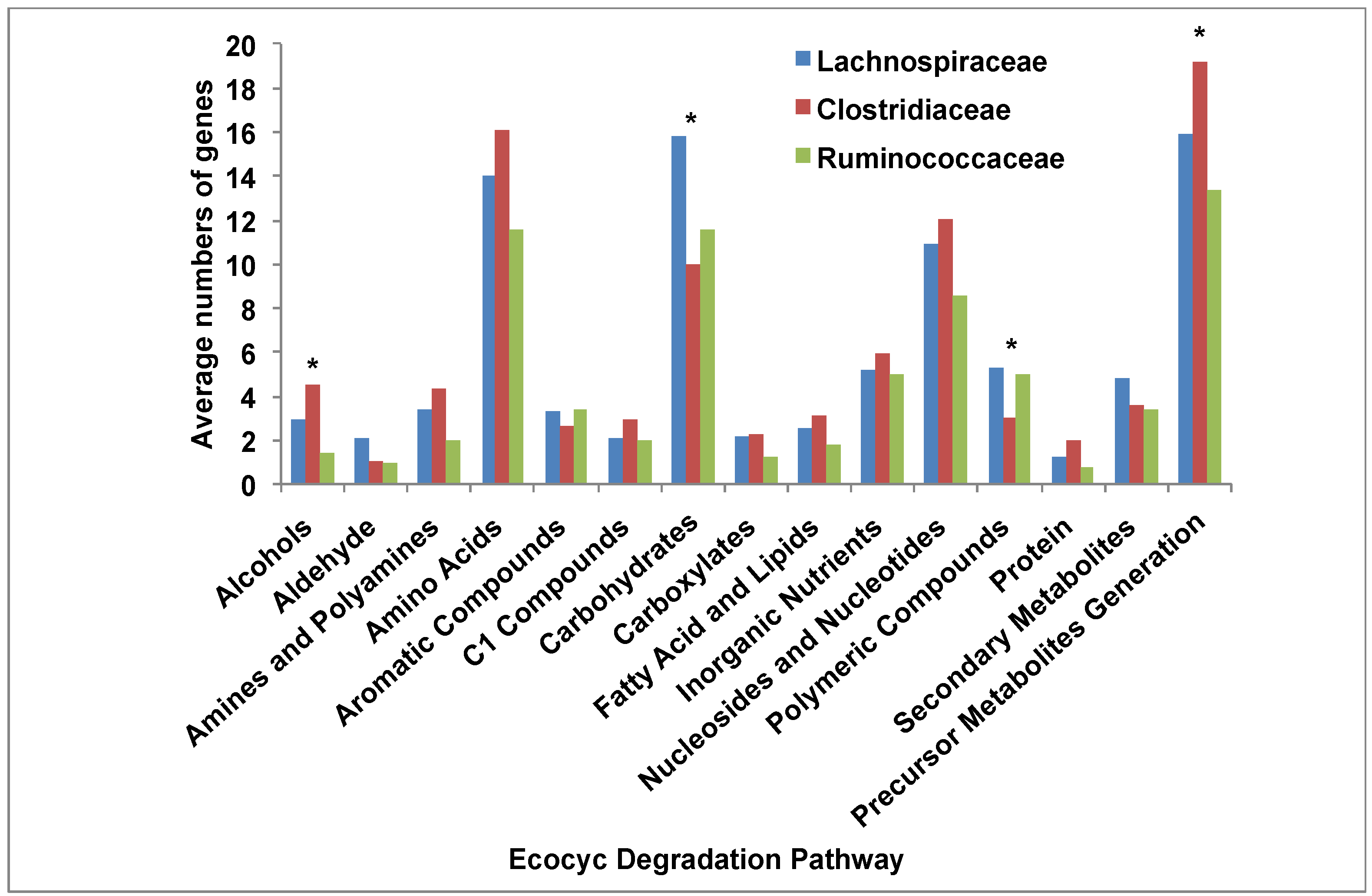
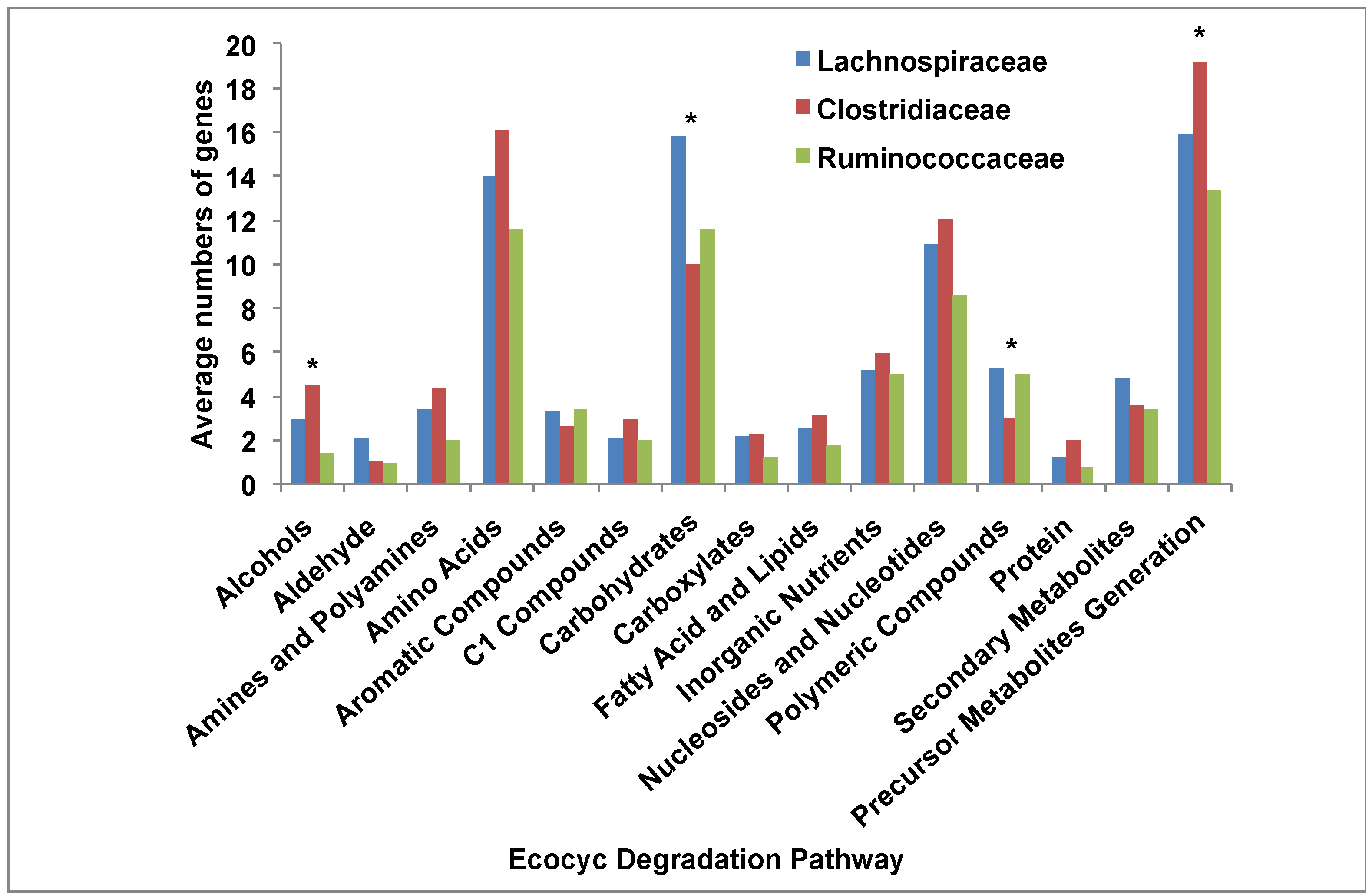
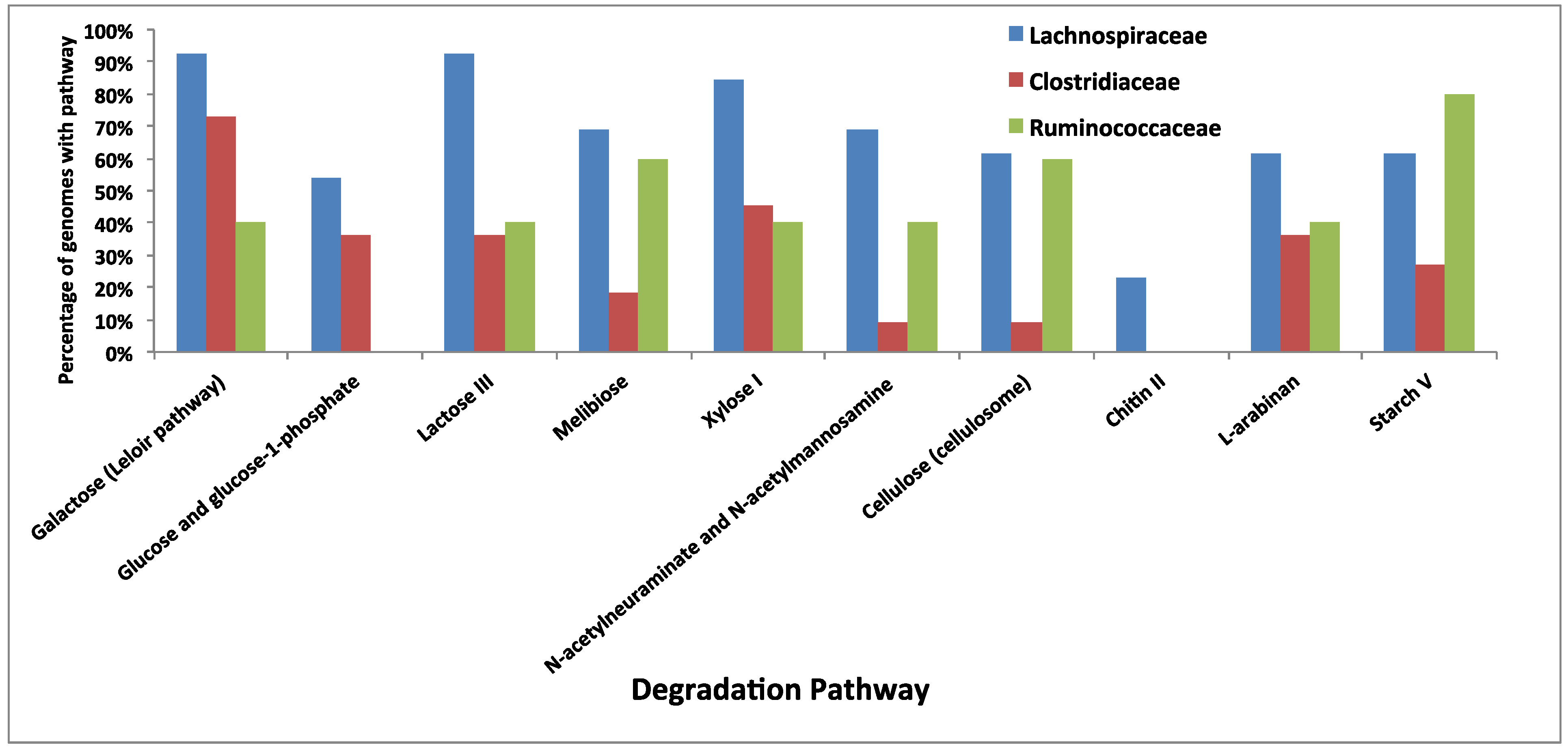
3.5. Comparative Analysis of Metabolic Pathways
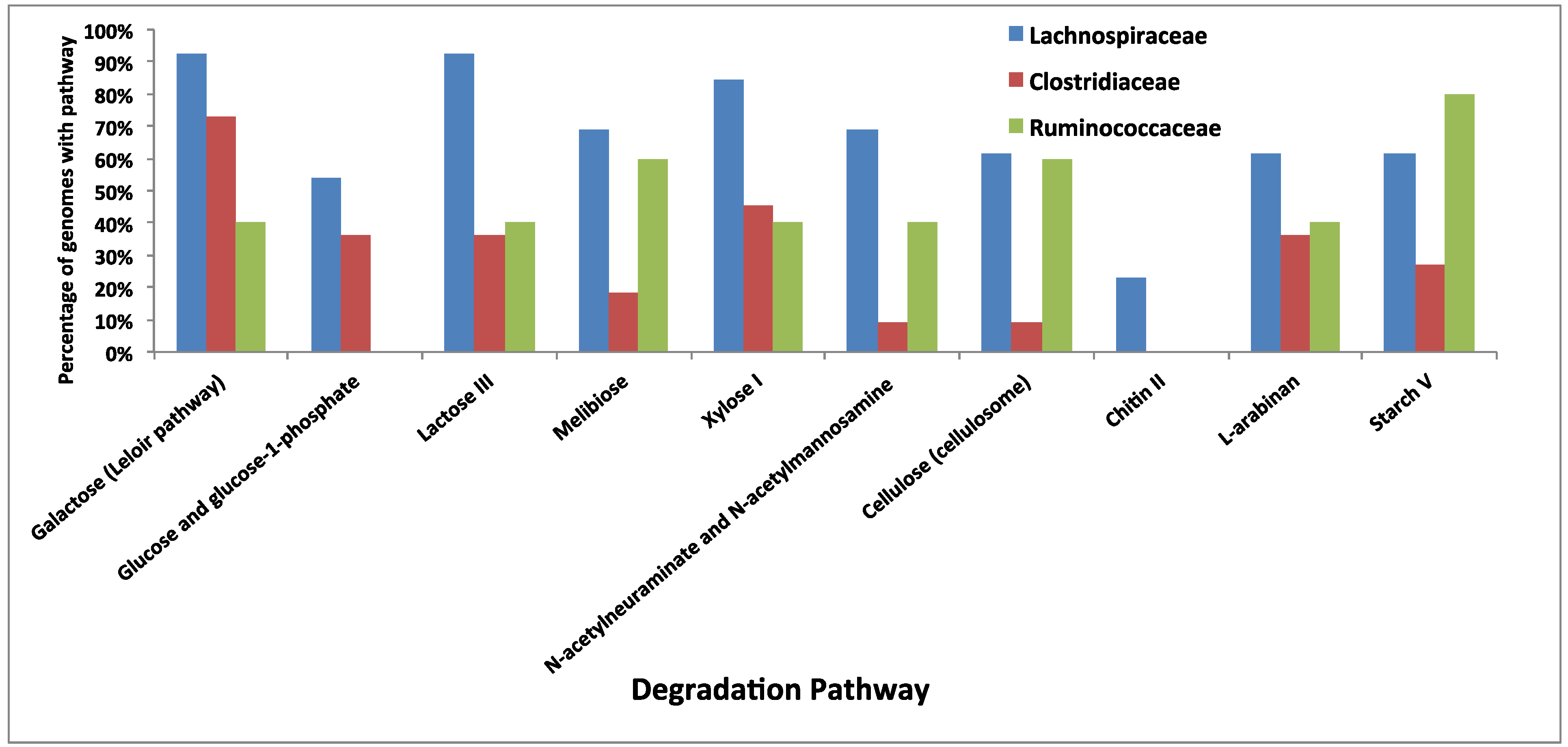
4. Conclusions
Acknowledgments
Conflict of Interest
Supplementary Materials
References and Notes
- Collins, M.D.; Lawson, P.A.; Willems, A.; Cordoba, J.J.; Fernandez-Garayzabal, J.; Garcia, P.; Cai, J.; Hippe, H.; Farrow, J.A.E. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol. 1994, 44, 812–826. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Rainey, F.A. Phylogenic relationships. In The Clostridia: Molecular Biology and Pathogenesis; Rood, J.I., McClane, B.A., Songer, J.G., Titball, R.W., Eds.; Academic Press: New York, NY, USA, 1997. [Google Scholar]
- Garrity, G.M.; Bell, J.A.; Lilburn, T. The Proteobacteria, Part A, Introductory Essays: The Revised Road Map to the Manual. In Bergey’s Manual® of Systematic Bacteriology; Brenner, D.J., Krieg, N.R., Staley, J.T., Garrity, G.M., Eds.; Springer: New York, NY, USA, 2005. [Google Scholar]
- Ludwig, W.; Schleifer, K.-H.; Whitman, W.B. Revised road map to the phylum Firmicutes. In Bergey’s Manual® of Systematic Bacteriology; Vos, P., Garrity, G.M., Jones, D., Krieg, N.R., Ludwig, W., Rainey, F.A., Schleifer, K-H., Whitman, W.B., Eds.; Springer: New York, NY, USA, 2009. [Google Scholar]
- De Long, E.F. The microbial ocean from genomes to biomes. Nature 2009, 459, 200–206. [Google Scholar] [CrossRef]
- Jalanka-Tuovinen, J.; Salonen, A.; Nikkila, J.; Immonen, O.; Kekkonen, R.; Lahti, L.; Palva, A.; de Vos, W.M. Intestinal Microbiota in Healthy Adults: Temporal Analysis Reveals Individual and Common Core and Relation to Intestinal Symptoms. PLoS One 2011, 6, e23035. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P. A core gut microbiome in obese and lean twins. Nature 2008, 457, 480–484. [Google Scholar]
- Peris-Bondia, F.; Latorre, A.; Artacho, A.; Moya, A.; D’Auria, G. The Active Human Gut Microbiota Differs from the Total Microbiota. PLoS One 2011, 6, e22448. [Google Scholar]
- Tap, J.; Mondot, S.; Levenez, F.; Pelletier, E.; Caron, C.; Furet, J.; Ugarte, E.; Muñoz-Tamayo, R.; Paslier, D.L.E; Nalin, R. Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 2009, 11, 2574–2584. [Google Scholar] [CrossRef]
- Barcenilla, A.; Pryde, S.E.; Martin, J.C.; Duncan, S.H.; Stewart, C.S.; Henderson, C.; Flint, H.J. Phylogenetic Relationships of Butyrate-Producing Bacteria from the Human Gut. Appl. Environ. Microbiol. 2000, 66, 1654–1661. [Google Scholar] [CrossRef]
- Duncan, S.H.; Barcenilla, A.; Stewart, C.S.; Pryde, S.E.; Flint, H.J. Acetate utilization and butyryl coenzyme A (CoA): acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl. Environ. Microbiol. 2002, 68, 5186–5190. [Google Scholar] [CrossRef]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef]
- Ding, S.-Y.; Rincon, M.T.; Lamed, R.; Martin, J.C.; McCrae, S.I.; Aurilia, V.; Shoham, Y.; Bayer, E.A.; Flint, H.J. Cellulosomal Scaffoldin-Like Proteins from Ruminococcus. flavefaciens. J. Bacteriol. 2001, 183, 1945–1953. [Google Scholar] [CrossRef]
- Fujimoto, T.; Imaeda, H.; Takahashi, K.; Kasumi, E.; Bamba, S.; Fujiyama, Y.; Andoh, A. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroen. Hepatol. 2013, 28, 613–619. [Google Scholar] [CrossRef]
- Leschine, S. Cellulose Degradation in Anaerobic Environments. Annu. Rev. Microbiol. 1995, 49, 399–426. [Google Scholar] [CrossRef]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial Cellulose Utilization: Fundamentals and Biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef]
- Brulc, J.M.; Antonopoulosb, D.A.; Millera, M.E.B.; Wilsona, M.K.; Yannarella, A.C.; Dinsdaled, E.A.; Edwardsd, R.E.; Frankh, E.D.; Emersoni, J.B.; Wacklini, P.; et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. USA 2009, 106, 1948–1953. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Composition and Metabolic Activities of Bacterial Biofilms Colonizing Food Residues in the Human Gut. Appl. Environ. Microbiol. 2006, 72, 6204–6211. [Google Scholar] [CrossRef]
- Cameron, E.A.; Maynard, M.A.; Smith, C.J.; Smith, T.J.; Koropatkin, N.M.; Martens, E.C. Multidomain Carbohydrate-binding Proteins Involved in Bacteroides thetaiotaomicron Starch Metabolism. J. Biol. Chem. 2012, 287, 34614–34625. [Google Scholar]
- Proctor, L.M. The Human Microbiome Project in 2011 and Beyond. Cell Host Microbe 2011, 10, 287–291. [Google Scholar] [CrossRef]
- Cole, J.R. The Ribosomal Database Project (RDP-II): Sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 2004, 33, D294–D296. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- FigTree. version 1.3.1. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 17 May 2013).
- Markowitz, V.M.; Chen, I.M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Gretchkin, Y. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2011, 40, D115–D122. [Google Scholar]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- R Core Team, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012; Version 2.15.1.
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S. Available online: http://www.stats.ox.ac.uk/pub/MASS4 (accessed on 17 May 2013).
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar]
- O’Hara, R.B.; Kotze, D.J. Do not log-transform count data. Methods Ecol. Evol. 2010, 1, 118–122. [Google Scholar] [CrossRef]
- The UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2011, 40, D71–D75. [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar]
- Keseler, I.M.; Mackie, A.; Peralta-Gil, M.; Santos-Zavaleta, A.; Gama-Castro, S.; Bonavides-Martinez, C.; Fulcher, C.; Huerta, A.M.; Kothari, A.; Krummenacker, M. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2012, 41, D605–D612. [Google Scholar]
- Karp, P.D. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005, 33, 6083–6089. [Google Scholar] [CrossRef]
- Saier, M.H. Families of transmembrane sugar transport proteins. Mol. Microbiol. 2000, 35, 699–710. [Google Scholar] [CrossRef]
- Brückner, R.; Titgemeyer, F. Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol. Lett. 2002, 209, 141–148. [Google Scholar] [CrossRef]
- Jojima, T.; Omumasaba, C.A.; Inui, M.; Yukawa, H. Sugar transporters in efficient utilization of mixed sugar substrates: current knowledge and outlook. Appl. Microbiol. Biotechnol. 2009, 85, 471–480. [Google Scholar]
- Stülke, J.; Hillen, W. Regulation of carbon catabolism in Bacillus species. Annu. Rev. Microbiol. 2000, 54, 849–880. [Google Scholar] [CrossRef]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities. Diversity 2013, 5, 627-640. https://doi.org/10.3390/d5030627
Biddle A, Stewart L, Blanchard J, Leschine S. Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities. Diversity. 2013; 5(3):627-640. https://doi.org/10.3390/d5030627
Chicago/Turabian StyleBiddle, Amy, Lucy Stewart, Jeffrey Blanchard, and Susan Leschine. 2013. "Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities" Diversity 5, no. 3: 627-640. https://doi.org/10.3390/d5030627