
2-(tert-Butyl)-4-phenyloxetane



Abstract
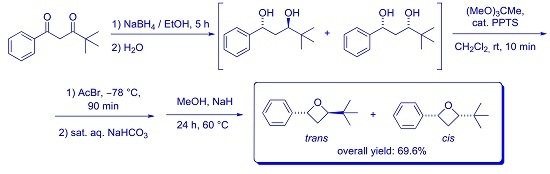
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
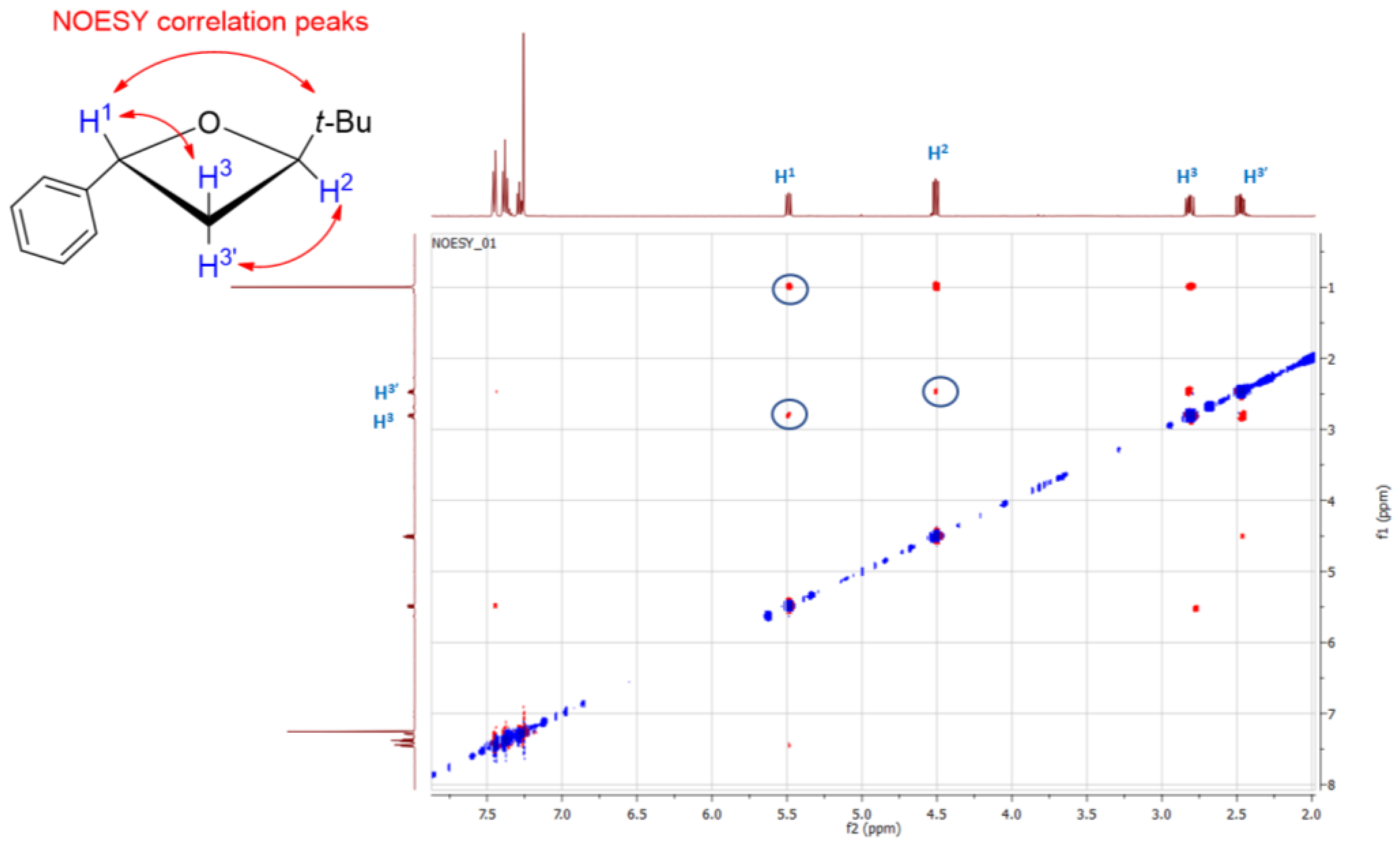
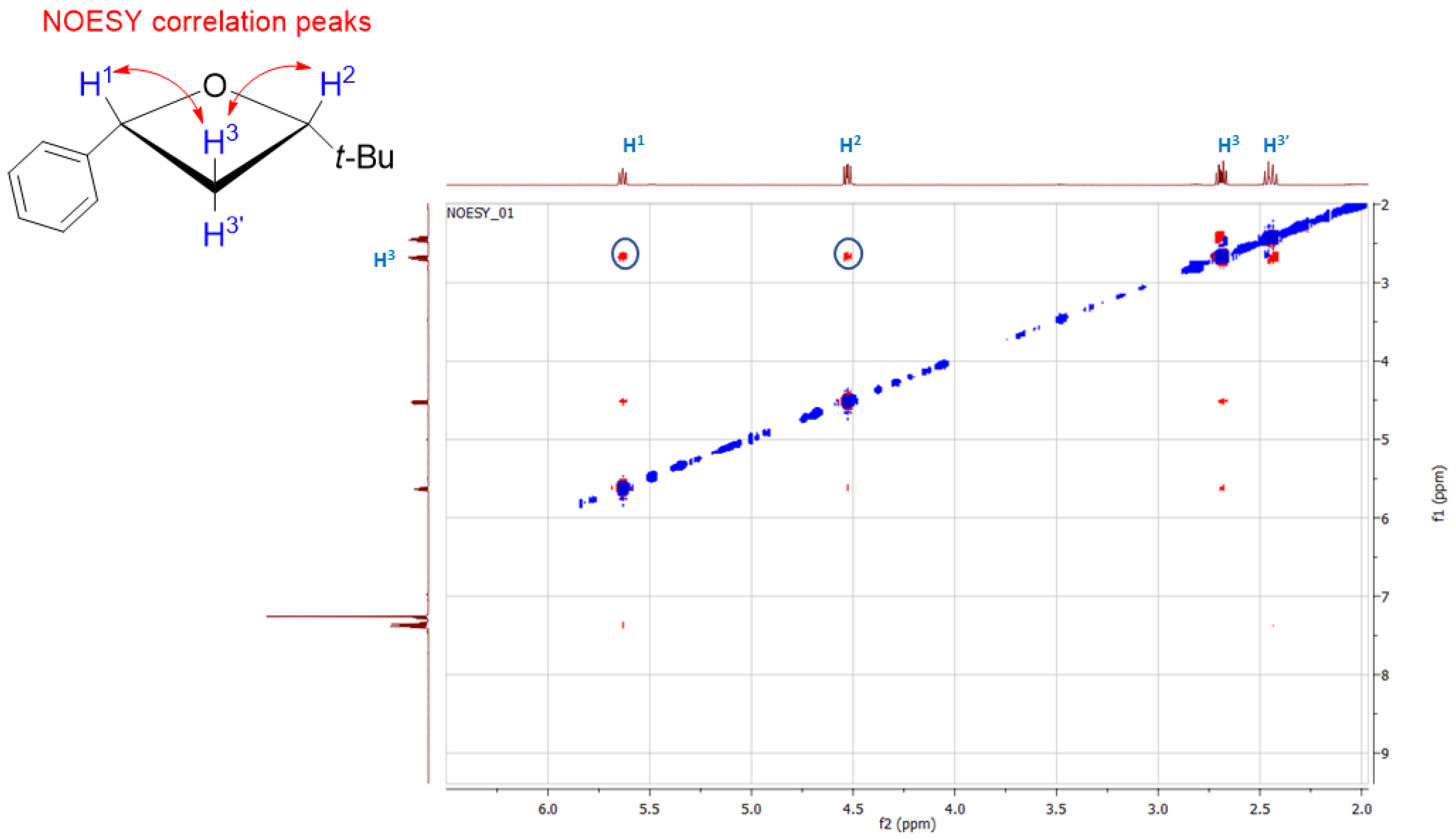
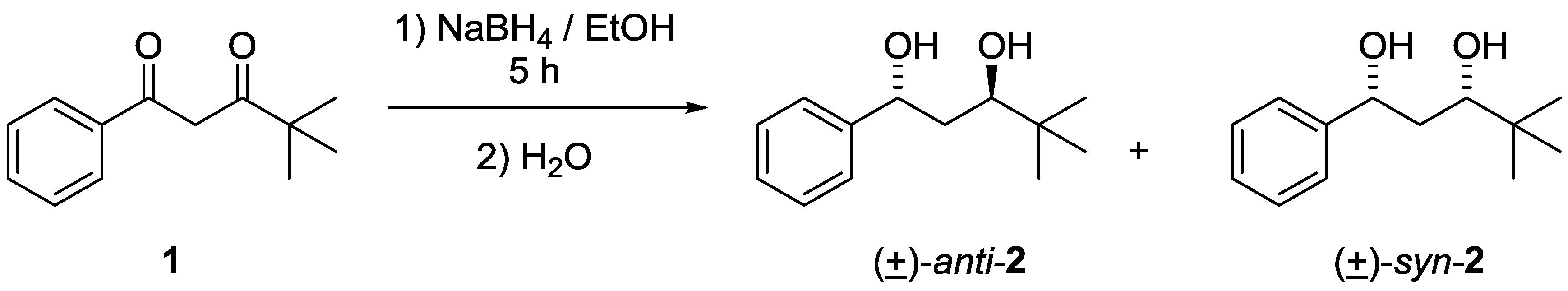
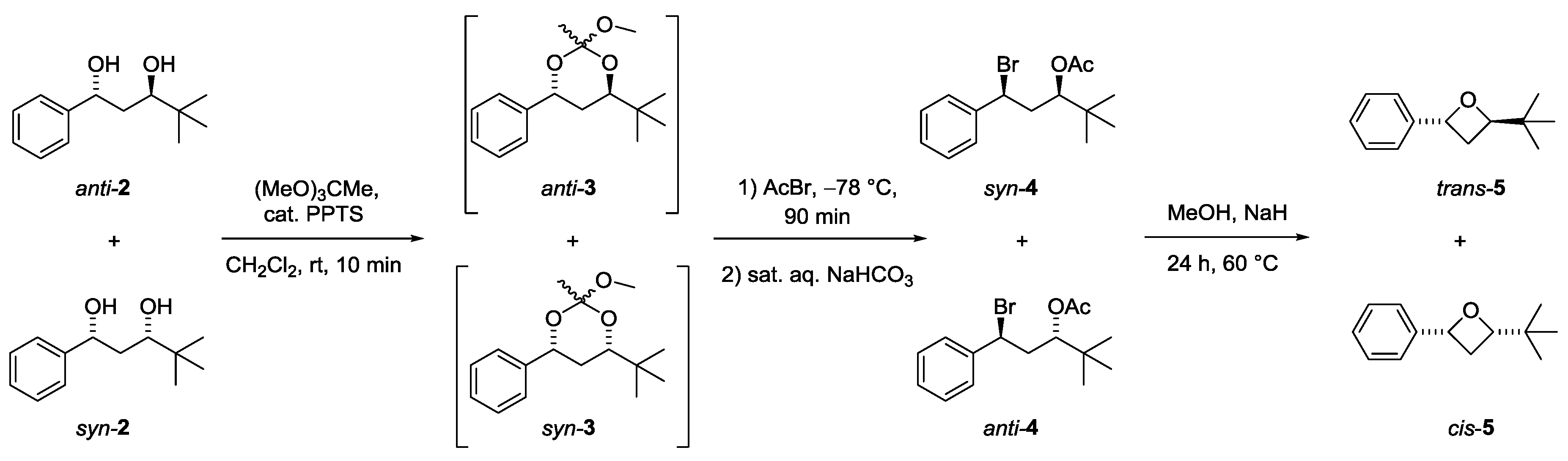
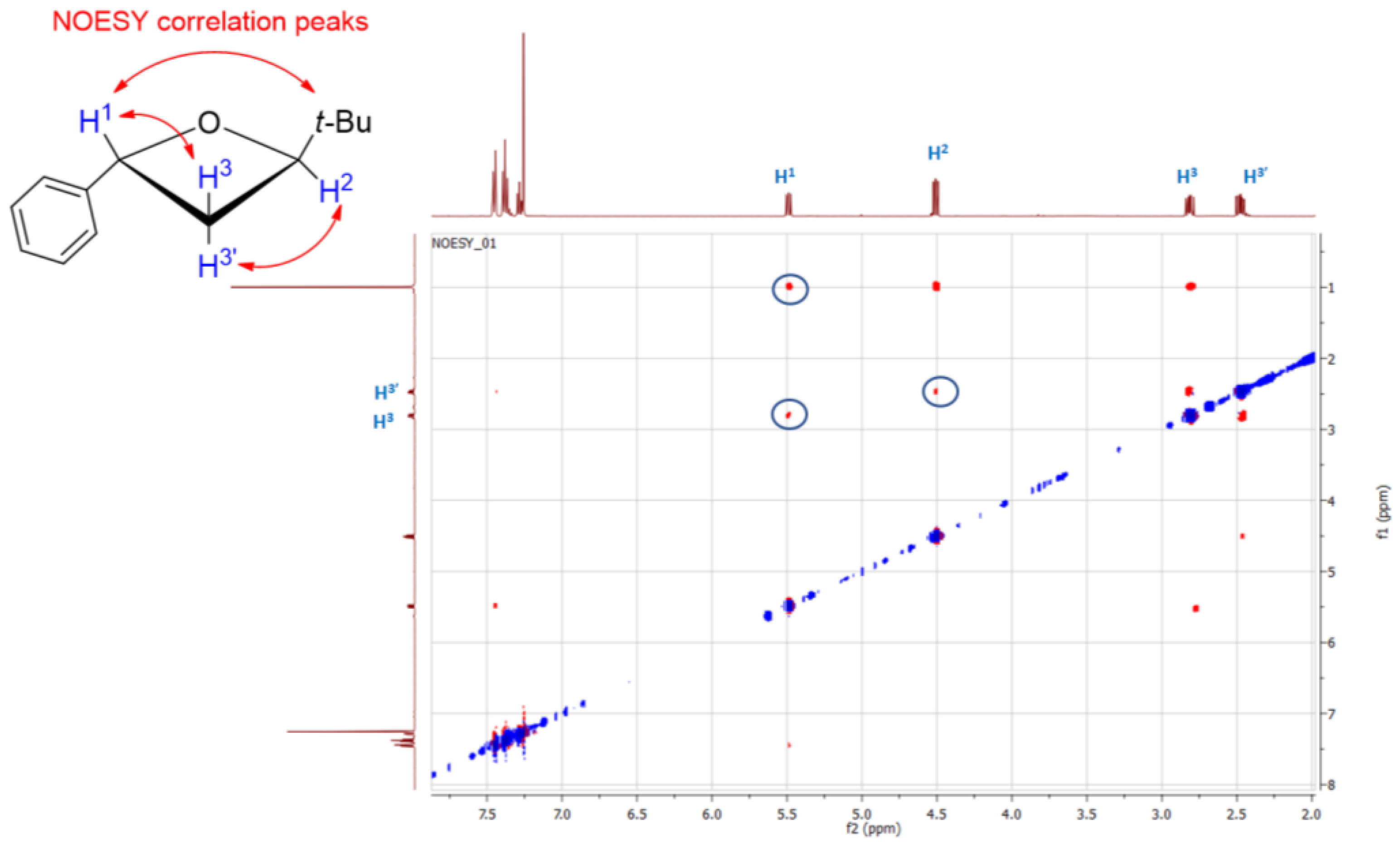
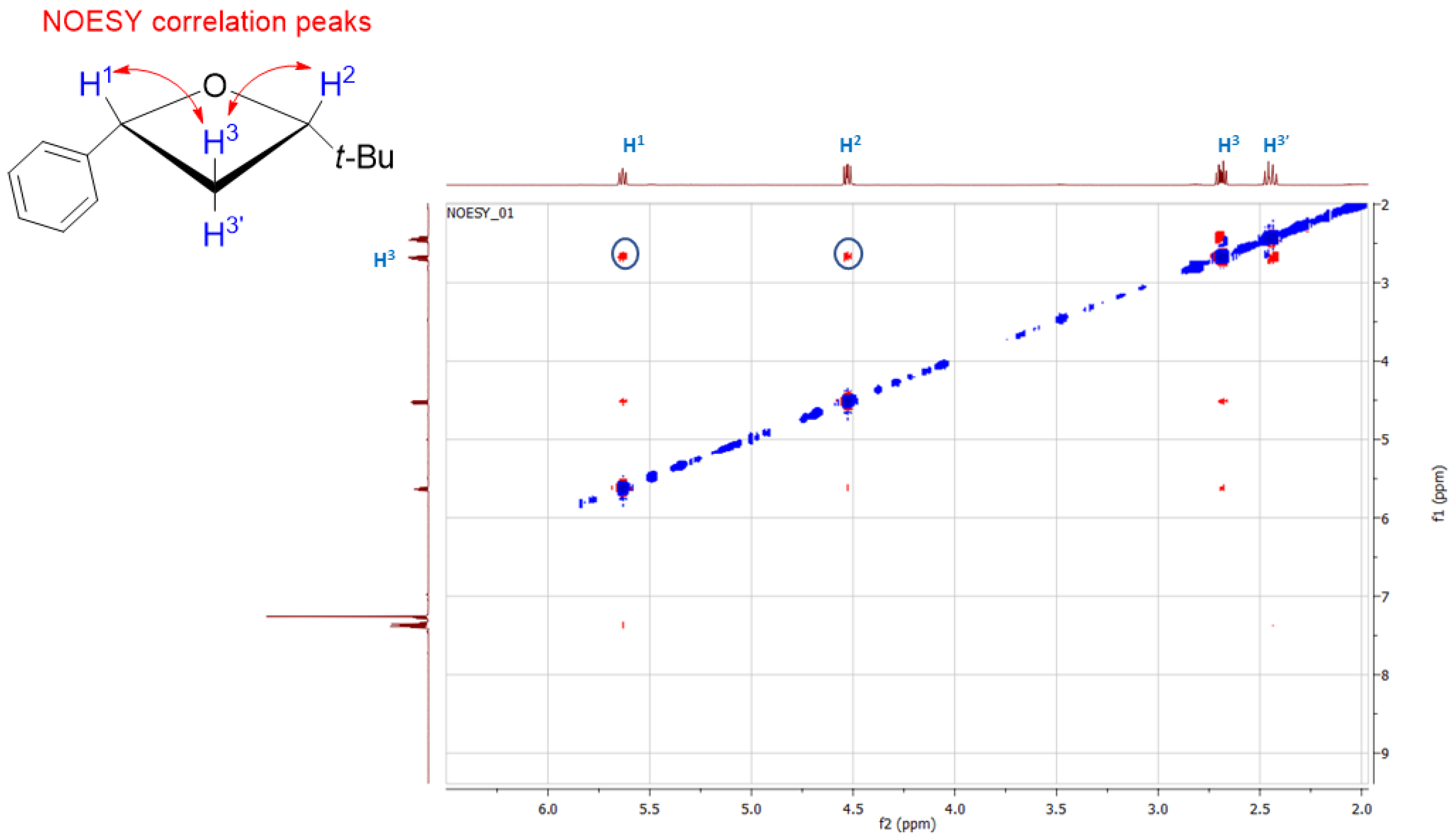
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of anti- and syn-4,4-dimethyl-1-phenylpentane-1,3-diols 2
3.2. Synthesis of trans- and cis-2-(tert-Butyl)-4-phenyloxetanes 5
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burkhard, J.A.; Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Carreira, M. Oxetanes as versatile elements in drug discovery and synthesis. Angew. Chem. Int. Ed. 2010, 49, 9052–9067. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Hasegawa, S.; Harada, T.; Tomisawa, T.; Fujii, A.; Takita, T. Oxetanocin, a novel nucleoside from bacteria. J. Antibiot. 1986, 39, 1623–1625. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Caggon, P.; McPhall, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lee, D.; Graf, T.N.; Phifer, S.S.; Nakanishi, Y.; Burgess, J.P.; Riswan, S.; Setyowati, F.M.; Saribi, A.M.; Soejarto, D.D.; et al. A Hexacyclic ent-Trachylobane Diterpenoid Possessing an Oxetane Ring from Mitrephora glabra. Org. Lett. 2005, 7, 5709–5712. [Google Scholar] [CrossRef] [PubMed]
- Wuitschik, G.; Rogers-Evans, M.; Buckl, A.; Bernasconi, M.; Märki, M.; Godel, T.; Fischer, H.; Wagner, B.; Parrilla, I.; Schuler, F.; et al. Spirocyclic Oxetanes: Synthesis and Properties. Angew. Chem. Int. Ed. 2008, 47, 4512–4515. [Google Scholar] [CrossRef] [PubMed]
- Carreira, E.M.; Fessard, T.C. Four-Membered Ring-Containing Spirocycles: Synthetic Strategies and Opportunities. Chem. Rev. 2014, 114, 8257–8322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Z.; Sun, J. Catalytic asymmetric nucleophilic openings of 3-substituted oxetanes. Org. Biomol. Chem. 2014, 12, 6028–6032. [Google Scholar] [CrossRef] [PubMed]
- Malapit, C.A.; Howell, A.R. Recent Applications of Oxetanes in the Synthesis of Heterocyclic Compounds. J. Org. Chem. 2015, 80, 8489–8495. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.A.; Croft, R.A.; Davis, O.A.; Doran, R.; Morgan, K.F. Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev. 2016, 116, 12150–12233. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, M.; Racioppi, R. Oxetane Synthesis through the Paternò-Büchi Reaction. Molecules 2013, 18, 11384–11428. [Google Scholar]
- Jenkinson, S.F.; Fleet, G.W.J. Oxetanes from the Ring Contraction of α-Triflates of γ-Lactones: Oxetane Nucleosides and Oxetane Amino Acids. Chimia 2011, 65, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Butova, E.D.; Barabash, A.V.; Petrova, A.A.; Kleiner, C.M.; Schreiner, P.R.; Fokin, A.A. Stereospecific Consecutive Epoxide Ring Expansion with Dimethylsulfoxonium Methylide. J. Org. Chem. 2010, 75, 6229–6235. [Google Scholar] [CrossRef] [PubMed]
- Coppi, D.I.; Salomone, A.; Perna, F.M.; Capriati, V. 2-Lithiated-2-phenyloxetane: a new attractive synthon for the preparation of oxetane derivatives. Chem. Commun. 2011, 47, 9918–9920. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.F.; Hollingsworth, I.A.; Bull, J.A. 2-(Aryl-sulfonyl)oxetanes as designer 3-dimensional fragments for fragment screening: synthesis and strategies for functionalisation. Chem. Commun. 2014, 50, 5203–5205. [Google Scholar] [CrossRef] [PubMed]
- Geden, J.V.; Beasley, B.O.; Clarkson, G.J.; Shipman, M. Asymmetric Synthesis of 2-Substituted Oxetan-3-ones via Metalated SAMP/RAMP Hydrazones. J. Org. Chem. 2013, 78, 12243–12250. [Google Scholar] [CrossRef] [PubMed]
- Coppi, D.I.; Salomone, A.; Perna, F.M.; Capriati, V. Exploiting the lithiation-directing ability of oxetane for the regioselective preparation of functionalized 2-aryloxetane scaffolds under mild conditions. Angew. Chem. Int. Ed. 2012, 51, 7532–7536. [Google Scholar] [CrossRef] [PubMed]
- Rouquet, G.; Blakemore, D.C.; Ley, S.V. Highly regioselective lithiation of pyridines bearing an oxetane unit by n-butyllithium. Chem. Commun. 2014, 50, 8908–8911. [Google Scholar] [CrossRef] [PubMed]
- Aftab, T.; Carter, C.; Hart, J.; Nelson, A. A Method for the Stereospecific Conversion of 1,3-Diols into Oxetanes. Tetrahedron Lett. 1999, 40, 8679–8683. [Google Scholar] [CrossRef]
- Aftab, T.; Carter, C.; Christlieb, M.; Hart, J.; Nelson, A. Stereospecific conversion of (1R*,3S*)- and (1R*,3R*)-3-cyclohexyl-1-phenylpropane-1,3-diol into the corresponding 2,4-disubstituted oxetanes. J. Chem. Soc. Perkin Trans. 1 2000, 711–722. [Google Scholar] [CrossRef]
- Vitale, P.; Perna, F.M.; Agrimi, G.; Scilimati, A.; Salomone, A.; Cardellicchio, C.; Capriati, V. Asymmetric chemoenzymatic synthesis of 1,3-diols and 2,4-disubstituted aryloxetanes by using whole cell biocatalysts. Org. Biomol. Chem. 2016, 14, 11438–11445. [Google Scholar] [CrossRef] [PubMed]
- Vitale, P.; Perna, F.M.; Perrone, M.G.; Scilimati, A. Screening on the use of Kluyveromyces marxianus CBS 6556 growing cells as enantioselective biocatalysts for ketone reductions. Tetrahedron: Asymmetry 2011, 22, 1985–1993. [Google Scholar] [CrossRef]
- Vitale, P.; D’Introno, C.; Perna, F.M.; Perrone, M.G.; Scilimati, A. Kluyveromyces marxianus CBS 6556 growing cells as a new biocatalyst in the asymmetric reduction of substituted acetophenones. Tetrahedron: Asymmetry 2013, 24, 389–394. [Google Scholar] [CrossRef]
- Perna, F.M.; Ricci, M.A.; Scilimati, A.; Mena, M.C.; Pisano, I.; Palmieri, L.; Agrimi, G.; Vitale, P. Cheap and environmentally sustainable stereoselective arylketones reduction by Lactobacillus reuteri whole cells. J. Mol. Catal. B: Enzym. 2016, 124, 29–37. [Google Scholar] [CrossRef]
- Lavandera, I.; Kern, A.; Ferreira-Silva, B.; Glieder, A.; de Wildeman, S.; Kroutil, W. Stereoselective Bioreduction of Bulky-Bulky Ketones by a Novel ADH from Ralstonia sp. J. Org. Chem. 2008, 73, 6003–6005. [Google Scholar] [CrossRef] [PubMed]
- Capriati, V.; Perna, F.M.; Salomone, A. “The Great Beauty” of organolithium chemistry: A land still worth exploring. Dalton Trans. 2014, 43, 14204–14210. [Google Scholar] [CrossRef] [PubMed]
- Mansueto, R.; Perna, F.M.; Salomone, A.; Florio, S.; Capriati, V. Dynamic resolution of lithiated ortho-trifluoromethyl styrene oxide and the effect of chiral diamines on the barrier to enantiomerisation. Chem. Commun. 2013, 49, 4911–4913. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perna, F.M.; Vitale, P.; Summa, S.; Capriati, V. 2-(tert-Butyl)-4-phenyloxetane. Molbank 2017, 2017, M930. https://doi.org/10.3390/M930
Perna FM, Vitale P, Summa S, Capriati V. 2-(tert-Butyl)-4-phenyloxetane. Molbank. 2017; 2017(1):M930. https://doi.org/10.3390/M930
Chicago/Turabian StylePerna, Filippo Maria, Paola Vitale, Simona Summa, and Vito Capriati. 2017. "2-(tert-Butyl)-4-phenyloxetane" Molbank 2017, no. 1: M930. https://doi.org/10.3390/M930
APA StylePerna, F. M., Vitale, P., Summa, S., & Capriati, V. (2017). 2-(tert-Butyl)-4-phenyloxetane. Molbank, 2017(1), M930. https://doi.org/10.3390/M930