Introduction
Bio-orthogonal chemistry provides an absolutely remarkable method for biomolecular studies of proteins, glycans, and lipids in living systems without interfering with natural biochemical processes [
1]. From the chemical point of view, the development of bioorthogonal reactions imposes special and restrictive conditions, such as selectivity, biological inertness, strong covalent bonds, high reaction rates, and biocompatibility of the chemical reactions [
2].
The classical methodologies to perform a reliable chemical ligation are the well-documented Staudinger ligation [
3], the Huisgen 1,3-dipolar cycloaddition of azides (Click Reactions) [
4], as well as the Diels-Alder (DA) reactions of suitably design diene systems [
5].
The use of reduced molecular weight organic molecules as chemical probes represents an outbreaking task in modern organic chemistry research [
6]. Typically, these molecules are analogs of the bio-interacting substrate containing a fluorescent tag for imaging analysis [
7]. In the search of novel synthetic approaches to be applied at Activity-Based Protein Profiling (ABPP) [
8], we describe here the rapid and selective synthesis of an anthracene-isoxazolyl derivative containing a protected aminoacidic residue.
Results and Discussion
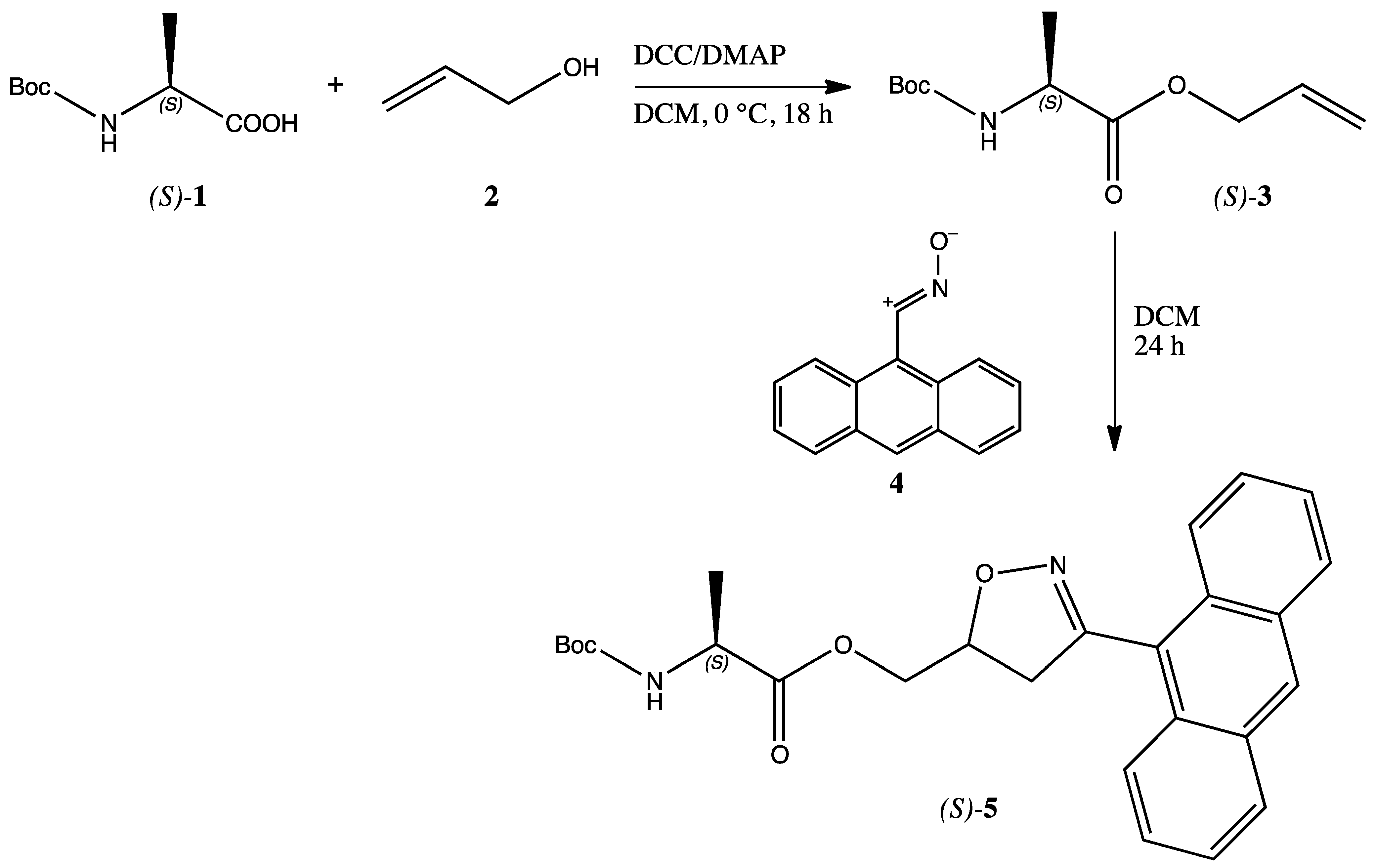
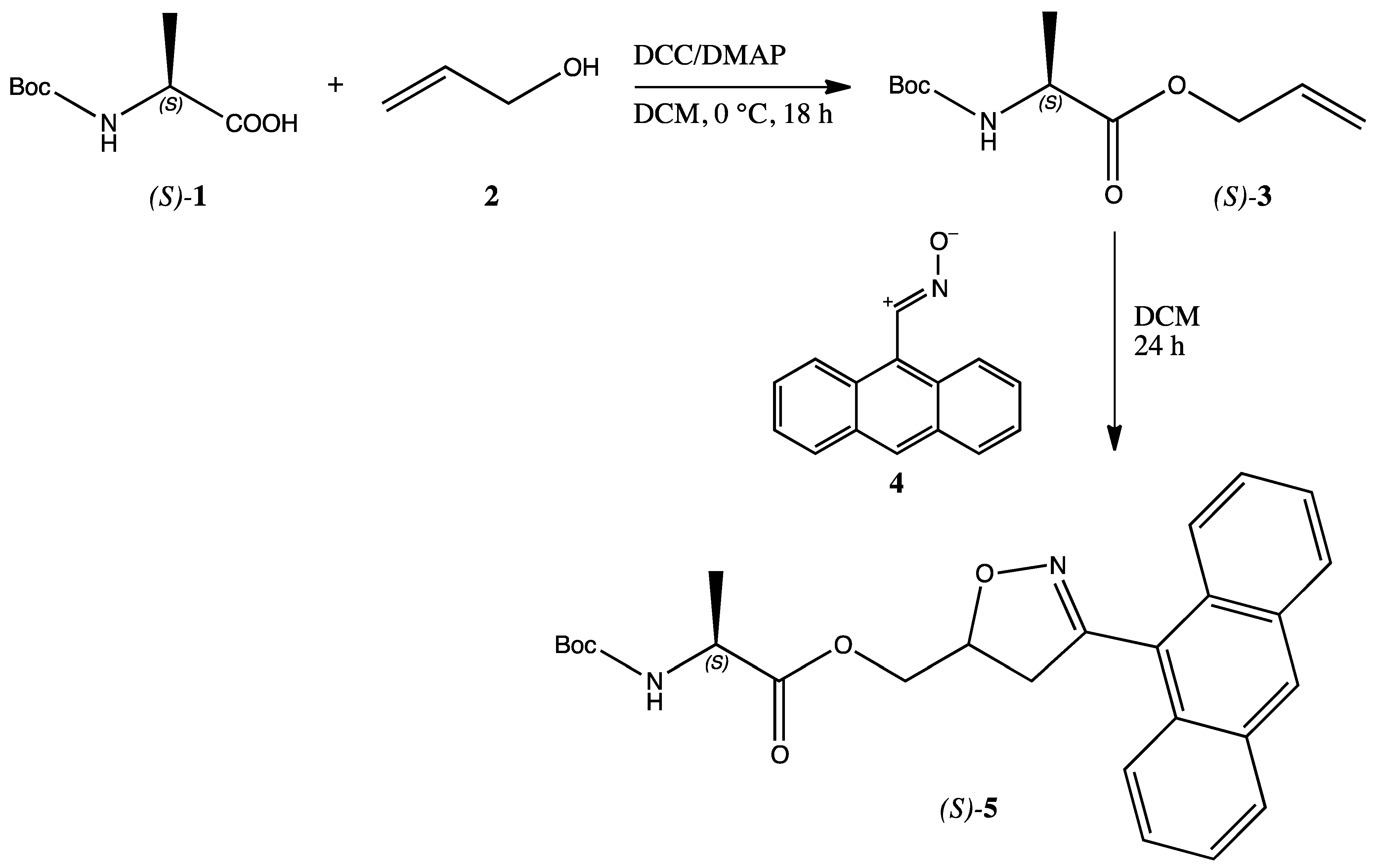
Commercially available
N-Boc protected (
S)-alanine
1 was converted into the corresponding allyl ester
3 through the typical DCC/DMAP coupling procedure with allyl alcohol
2 [
9]. The white solid ester
3 was isolated through column chromatography and obtained in 75% yield and submitted to the 1,3-dipolar cycloaddition with a slight excess (1.2 equiv.) of freshly prepared [
10] anthracenenitrile oxide
4 in dichloromethane (DCM) solution. The reaction was conducted at room temperature for 24 h. Chromatographic separation of the worked-up reaction mixture afforded the expected single regioisomer
5 as a dark yellow solid (m.p. 68–70 °C from diisopropyl ether/petrol ether) in 60% yield (
Scheme 1).
The structure of compound 5 was assigned through a diligent examination of all the collected analytical and spectroscopic data. From the IR spectrum the presence of the NH of the Boc-protected aminoacidic residue is shown by a broad band centred at 3381 cm−1; the C=O groups are shown by the strong and intense bands at 1755 and 1694 cm−1. The 1H-NMR spectrum (d6-DMSO) shows the presence of the anthracene ring in the aromatic region with signals ranging from 7.62 ppm to 8.77 ppm. The presence of the newly formed isoxazoline ring is confirmed by the presence of a broad signal at 5.27 ppm relative to the CH-O of the heterocyclic ring, coupled with the exocyclic methylene group of which signals are found at 3.39 and 3.71 ppm (AB system). Another AB system constituted by two double triplets at 4.36 and 4.50 ppm corresponds to the isoxazoline methylene. The alanine moiety gave the expected signals of the methyl group at 1.33 ppm as a doublet coupled with the CH found at 4.20 ppm as typical quartet. The NH of the aminoacid was also found at 7.39 ppm as a triplet and exchanges with D2O. In order to corroborate the above reported attribution, COSY and NOESY experiments were performed. The 13C-NMR spectrum indicates that the cycloadduct 5 exists as an inseparable mixture of two diastereoisomers (1:1 ratio) due to the undefined chirality of the C5 isoxazoline carbon atom.
The stability of the anthracenenitrile oxide
4 is due to the known forbidden dimerization because of the high steric demanding structure of the anthracene moiety [
11]. This behavior allowed also to perform the cycloaddition reaction in water solution, the privileged solvent in biological systems, even with better yields (75% of product
5). The same steric effects are at work in determining the high regioselectivity of the reaction. Upon investigation on the composition of the crude reaction mixture by NMR techniques, no other compounds were identified in addition to the isolated cycloadduct
5, confirming the peculiar regiochemical outcome.
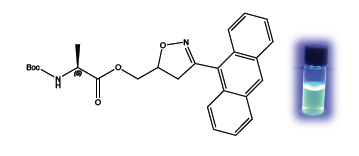
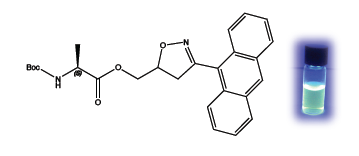
The fluorescence properties given by the anthryl residue to the cycloadduct
5 were determined by recording a fluorescence spectrum in methanol as solvent. The emission maximum is centered at 481 nm and is displayed in the cycloadduct, only.
Figure 1 shows the switching on of the cycloadduct
5 during the reaction, which is nearly completed in 12 h. Reference materials, the anthracenenitrile oxide
4, the isolated cycloadduct
5 and the alanine dipolarophile
3 are reported for comparison.
The present protocol represents a valuable and innovative methodology for the synthesis of fluorescence chemical probes for ligation chemistry.
Experimental
General - All melting points (m.p.) are uncorrected. Elemental analyses were done on an elemental analyzer available at the Department. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on a Bruker AVANCE 400 spectrometer (solvents specified). Chemical shifts are expressed in ppm from internal tetramethylsilane (δ) and coupling constants (J) are in Hertz (Hz). IR spectra (nujol mulls) were recorded on a spectrophotometer Perkin Elmer RX-1 available at the Department and absorbtions (ν) are in cm−1. Fluerescence spectrum was recorded on a Perkin Elmer LS-55.
Synthesis of cycloadduct 5 - To a solution of 204 mg (0.93 mmol) of anthracenenitrile oxide 4 in 50 mL of DCM, 100 mg (0.77 mmol) of N-Boc protected alanine allyl ester 3 were added portionwise under stirring at room temperature and the reaction was conducted overnight. After evaporation of the solvent at reduced pressure, the residue was submitted to column chromatography (Kieselgel 60, 70–230 mesh, Merck; eluent cyclohexane/ethyl acetate 9:1 to 1:1) to isolate the final product 5 as a dark yellow solid (m.p. 68–70 °C from diisopropyl ether/petrol ether) in 60% yield.
FT-IR: ѵN-H 3381cm−1; ѵC=O 1755, 1694 cm−1.
1H-NMR (400 MHz, δ DMSO): 1.33 (d, 3H, J = 7 Hz, CH3), 3.39, 3.71 (m, 1H + 1H, CH2-O), 4.20 (q, J = 7 Hz, 1H, CH-CH3), 4.36, 4.51 (dt, 1H + 1H, J = 13, 9 Hz, CH-CH2), 5.27 (bs, 1H, CH-O), 7.39 (t, J = 6 Hz, 1H, NH), 7.61 (m, 4H, Anth), 8.00 (m, 2H, Anth), 8.18 (d, J = 8 Hz, 2H, Anth), 8.77 (s, 1H, Anth).
13C-NMR (100 MHz, δ DMSO): 17.0 [16.9], 28.1, 42.0 [42.1], 49.1, 65.4 [65.2], 77.9 [78.0], 123.3, 124.7, 125.7, 126.9, 127.0, 128.6, 128.7, 129.25, 129.3, 130.7, 155.3, 155.8, [155.9], 173.3 [173.2]. In brackets are the signal of the minor diastereoisomer.
Elemental analysis: Calculated for C26H28N2O5 (MW = 448.20): C, 69.63; H, 6.29; N, 6.25. Found: C, 69.60; H, 6.30; N, 6.22.
The reaction was similarly performed in water as a solvent. Upon extraction of the organic compounds from the water phase with DCM, a residue was obtained upon evaporation of the organic solvent. From the reaction mixture, the expected product 5 was obtained in 75% yield, identical to the previously reported specimen.

{kind=link}
{kind=link}
{kind=link}