The New Unified Theory of ATP Synthesis/Hydrolysis and Muscle Contraction, Its Manifold Fundamental Consequences and Mechanistic Implications and Its Applications in Health and Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Unified Theory of the ATP Cycle in ATP Synthesis/Hydrolysis and Muscle Contraction
2.1. Specific Details of Energy Distribution among the Elementary Steps of Binding, Bond Cleavage, and Product Release in the ATP Synthesis/Hydrolysis Catalytic Cycle
2.2. Contradictory Assumptions and Gross Inconsistencies among Previous Models as Seen from the Viewpoint of the Unified Theory
2.3. Further Fundamental Differences between the Torsional Mechanism of ATP Synthesis and the Binding Change Mechanism
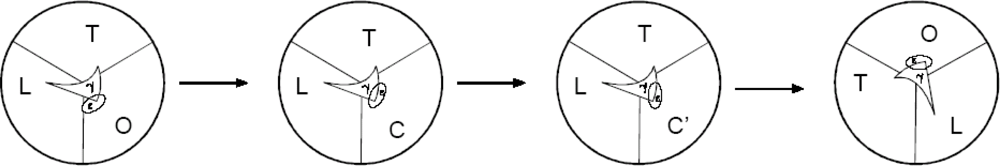
2.4. Further Development of the Torsional Mechanism of ATP Synthesis/Hydrolysis, the Rotation-Uncoiling-Tilt (RUT) Energy Storage Mechanism of Muscle Contraction and the Unified Theory
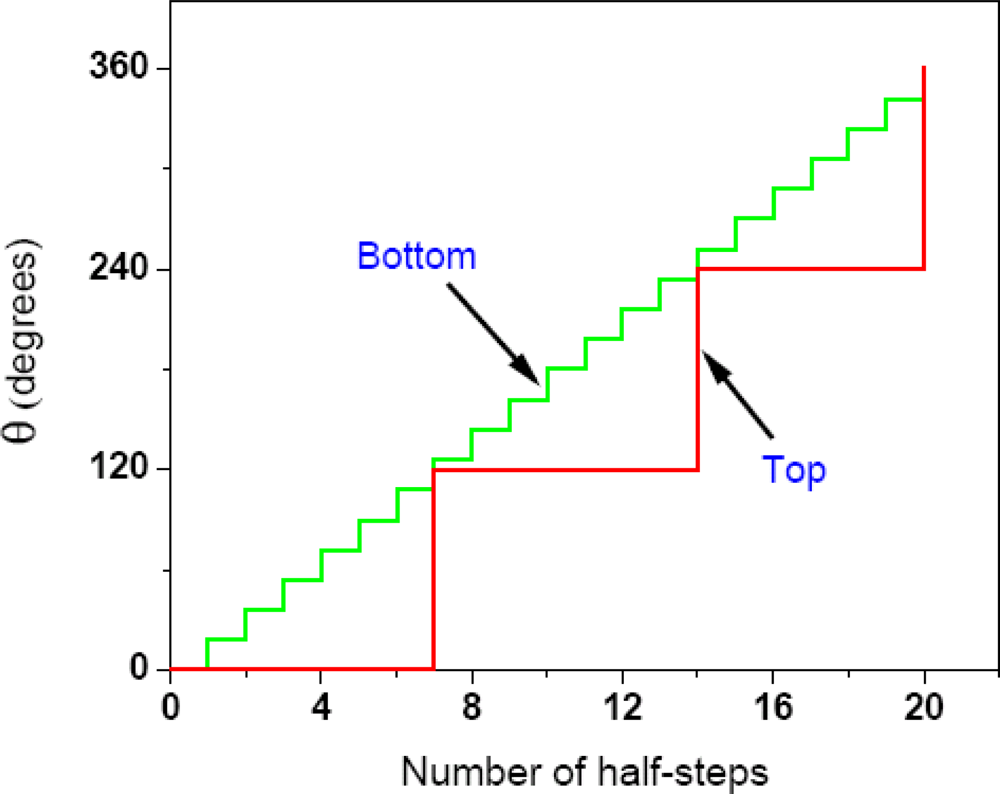
2.4.1. Complete Details of Quantized Release and Utilization of Energy in the Synthesis Mode and its Mechanistic Implications
2.4.2. Quantized Release and Utilization of Energy in the Hydrolysis Mode and Its Mechanistic Implications for Muscle Contraction
2.4.3. Complete Details of Quantized Release and Utilization of Energy in the Hydrolysis Mode and its Mechanistic Implications for F1-ATPase
2.5. Removal of the Various Inconsistencies in Previous Models by the Unified Theory
2.5.1. Models of Muscle Contraction and Motility
2.5.2. Models in Bioenergetics
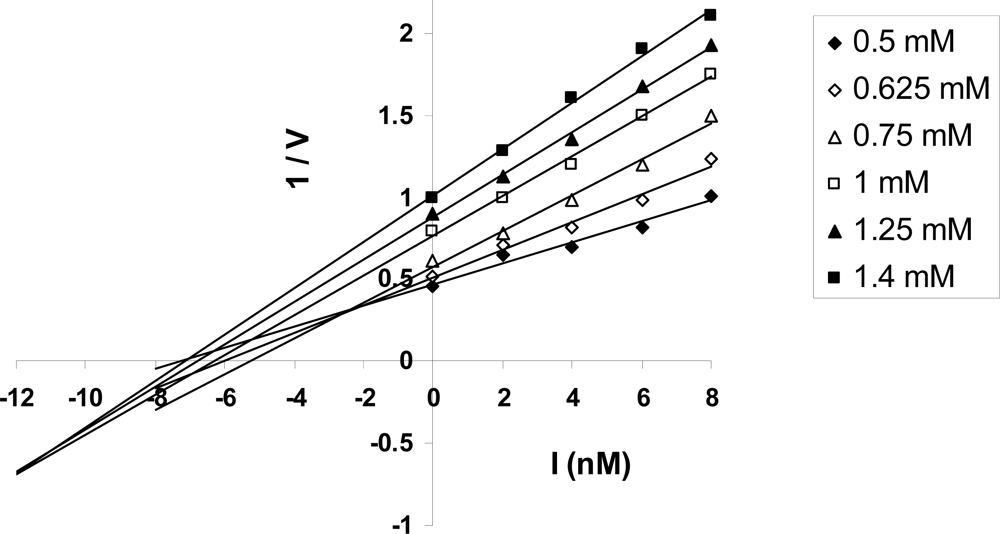
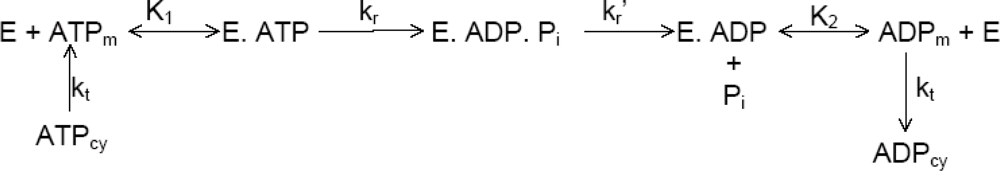
2.6. Kinetic Analysis of ATP Hydrolysis by F1-ATPase
3. Consistency of Current Mechanisms of ATP Synthesis and Muscle Contraction with Key Experimental Data
3.1. Bioenergetics
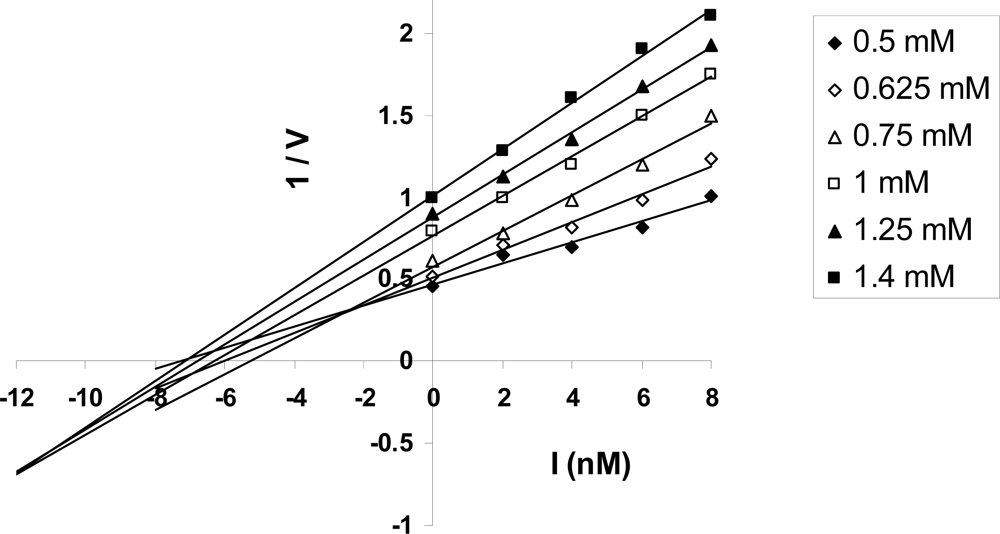
3.1.1. Nucleotide Binding Affinity (Kd) Measurements at the Catalytic Sites of F1-ATPase
3.1.2. Structure of Mitochondrial F1-ATPase with Nucleotide Bound to All Three Catalytic Sites
3.2. Muscle Contraction
3.2.1. Contraction Characteristics and ATPase Activity of Muscle Fibers in the Presence of Antibody to Myosin S-2
3.2.2. Single Molecule Imaging
4. Fundamental Consequences of the Torsional Mechanism, the Rotation-Uncoiling-Tilt Energy Storage Mechanism, and the Unified Theory for Molecular Mechanisms of ATP Synthesis/Hydrolysis and Muscle Contraction
4.1. Violations of the First Law of Thermodynamics by Previous Mechanisms of ATP Synthesis and Muscle Contraction
4.2. Violations of the Second Law of Thermodynamics by Previous Mechanisms of ATP Synthesis and Muscle Contraction
4.3. Mechanistic H+/O, H+/ATP and P/O Ratios, Efficiency of Oxidative Phosphorylation, and the Overall Energy Balance of Cellular Bioenergetics
5. Beyond the Chemiosmotic Theory: Details of the Molecular Mechanism of Energy Transduction in the Membrane-bound FO Portion of F1FO-ATP Synthase
5.1. Several Lines of Biochemical Evidence that Cannot be Accounted for by the Chemiosmotic Theory but Are Logically Explained by the Torsional Mechanism of Energy Transduction and ATP Synthesis
5.2. Establishment of the Type of Inhibition Found with Potent, Specific Anion Channel Blockers Such as DIDS and its Logical Explanation
5.3. Explanation of Rate Enhancement of ATP Synthesis at Very High Inhibitor Concentrations
5.4. Precise Explanation of Uncoupling Action by Weak Acid Anion Uncouplers of Oxidative Phosphorylation
5.5. The Way Forward
6. Applications of the Unified Theory to Other (Processive) Molecular Motors Crucial to Cell Life
7. Possible Applications of the Torsional Mechanism of ATP Synthesis and the Unified Theory in Apoptosis, Cell Death and Disease
8. Experimental Section
8.1. Isolation of Thylakoid Membranes
8.2. Estimation of Chlorophyll Content
8.3. Measurement of ATP Synthesis
8.4. Estimation and Calculation of Rates of ATP Synthesis
9. Prospects for Future Research
10. Conclusions
Acknowledgments
References
- Nath, S. The Molecular Mechanism of ATP synthesis by F1F0-ATP Synthase: A Scrutiny of the Major Possibilities. Adv. Biochem. Eng. Biotechnol 2002, 74, 65–98. [Google Scholar]
- Nath, S. Molecular Mechanisms of Energy Transduction in Cells: Engineering Applications and Biological Implications. Adv. Biochem. Eng. Biotechnol 2003, 85, 125–180. [Google Scholar]
- Senior, AE; Nadanaciva, S; Weber, J. The Molecular Mechanism of ATP synthesis by F1F0-ATP Synthase. Biochim. Biophys. Acta 2002, 1553, 188–211. [Google Scholar]
- Huxley, AF. Cross-bridge Action: Present Views, Prospects, and Unknowns. J. Biomech 2000, 33, 1189–1195. [Google Scholar]
- Geeves, MA; Holmes, KC. The Molecular Mechanism of Muscle Contraction. Adv. Protein Chem 2005, 71, 161–193. [Google Scholar]
- Mitchell, P. Chemiosmotic Coupling in Oxidative and Photosynthetic Phosphorylation. Biol. Rev 1966, 41, 445–502. [Google Scholar]
- Boyer, PD. The Binding Change Mechanism for ATP Synthase – Some Probabilities and Possibilities. Biochim. Biophys. Acta 1993, 1140, 215–250. [Google Scholar]
- Huxley, HE. The Mechanism of Muscular Contraction. Science 1969, 164, 1356–1366. [Google Scholar]
- Huxley, AF. Muscular Contraction. J. Physiol 1974, 243, 1–43. [Google Scholar]
- Nath, S. A Novel Systems Biology/Engineering Approach Solves Fundamental Molecular Mechanistic Problems in Bioenergetics and Motility. Process Biochem 2006, 41, 2218–2235. [Google Scholar]
- Nath, S. The Torsional Mechanism of Energy Transduction and ATP Synthesis as a Breakthrough in our Understanding of the Mechanistic, Kinetic and Thermodynamic Details. Thermochim. Acta 2004, 422, 5–17. [Google Scholar]
- Nath, S; Jain, S. The Detailed Molecular Mechanism of ATP Synthesis in the F0 Portion of ATP Synthase Reveals a Non-Chemiosmotic Mode of Energy Coupling. Thermochim. Acta 2002, 394, 89–98. [Google Scholar]
- Jain, S; Nath, S. Catalysis by ATP Synthase: Mechanistic, Kinetic and Thermodynamic Characteristics. Thermochim. Acta 2001, 378, 35–44. [Google Scholar]
- Nath, S; Khurana, D. The Molecular Mechanism of the Contractile Cycle of Muscle. Curr. Sci 2001, 81, 78–82. [Google Scholar]
- Jain, S; Nath, S. Kinetic Model of ATP Synthase: pH Dependence of the Rate of ATP Synthesis. FEBS Lett 2000, 476, 113–117. [Google Scholar]
- Nath, S; Jain, S. Kinetic Modeling of ATP Synthesis by ATP Synthase and Its Mechanistic Implications. Biochem. Biophys. Res. Commun 2000, 272, 629–633. [Google Scholar]
- Nath, S; Rohatgi, H; Saha, A. The Catalytic Cycle of ATP Synthesis by Means of a Torsional Mechanism. Curr. Sci 2000, 78, 23–27. [Google Scholar]
- Nath, S; Rohatgi, H; Saha, A. The Torsional Mechanism of Energy Transfer in ATP Synthase. Curr. Sci 1999, 77, 167–169. [Google Scholar]
- Rohatgi, H; Saha, A; Nath, S. Mechanism of ATP Synthesis by Protonmotive Force. Curr. Sci 1998, 75, 716–718. [Google Scholar]
- Nath, S. A Thermodynamic Principle for the Coupled Bioenergetic Processes of ATP Synthesis. Pure Appl. Chem 1998, 70, 639–644. [Google Scholar]
- Nath, S. The Torsional Mechanism of Energy Transduction and ATP Synthesis: An Alternative to the Binding Change Mechanism. Biophys. J 2002, 82, 291a. [Google Scholar]
- Nath, S. A New Paradigm for Ion Translocation and Energy Transduction in Biological Systems. Biophys. J 2005, 88, 548a. [Google Scholar]
- Nath, S. Beyond the Chemiosmotic Theory: The Torsional Mechanism of Energy Transduction and ATP Synthesis as a New Paradigm in Bioenergetics. Biophys. J 2006, 90, 131a. [Google Scholar]
- Nath, S. A Thermodynamic Principle for the Coupled Nonequilibrium Processes of ATP Synthesis. Proceedings of the ISBC X: International Society for Biological Calorimetry, Monte Verita, Switzerland; 1997; p. i9. [Google Scholar]
- Nath, S. A Fundamental Thermodynamic Principle for Coupling in Oxidative Phosphorylation. Proceedings of the Sixteenth International Congress of Biochemistry and Molecular Biology, New Delhi; 1994; II, p. 390. [Google Scholar]
- Nath, S. A Novel Molecular Mechanism of Energy Transduction in Muscle Contraction. The Gordon Research Conference Muscle: Contractile Proteins, New London, New Hampshire, USA; 2005. [Google Scholar]
- Nath, S. The Rotation-Twist-Tilt Energy Storage Mechanism of Muscle Contraction. The Gordon Research Conference Muscle: Contractile Proteins, New London, New Hampshire, USA; 2005. [Google Scholar]
- Nath, S. Achievements, Ramifications and Implications of Nath’s Torsional Mechanism of Energy Transduction and ATP Synthesis and the Rotation-Uncoiling-Tilt (RUT) Energy Storage Mechanism of Muscle Contraction. The National Conference on Thermodynamics of Chemical and Biological Systems, The Indian Thermodynamics Society, Amritsar; 2005; pp. PL-4–PL-5. [Google Scholar]
- Jain, S; Murugavel, R; Hansen, LD. ATP Synthase and the Torsional Mechanism: Resolving a 50-Year-Old Mystery. Curr. Sci 2004, 87, 16–19. [Google Scholar]
- Adachi, K; Oiwa, K; Nishizaka, T; Furuike, S; Noji, H; Itoh, H; Yoshida, M; Kinosita, K. Coupling of Rotation and Catalysis in F1-ATPase Revealed by Single-Molecule Imaging and Manipulation. Cell 2007, 130, 309–321. [Google Scholar]
- Weber, J; Senior, AE. Catalytic Mechanism of F1-ATPase. Biochim. Biophys. Acta 1997, 1319, 19–58. [Google Scholar]
- Weber, J; Senior, AE. Bi-site Catalysis in F1-ATPase: Does it Exist? J. Biol. Chem 2001, 276, 35422–35428. [Google Scholar]
- Menz, RI; Walker, JE; Leslie, AGW. Structure of Bovine Mitochondrial F1-ATPase with Nucleotide Bound to All Three Catalytic Sites: Implications for the Mechanism of Rotary Catalysis. Cell 2001, 106, 331–341. [Google Scholar]
- Ross, J. Energy Transfer from Adenosine Triphosphate. J. Phys. Chem. B 2006, 110, 6987–6990. [Google Scholar]
- Senior, AE. ATP Synthase: Motoring to the Finish Line. Cell 2007, 130, 220–221. [Google Scholar]
- Dittrich, M; Hayashi, S; Schulten, K. On the Mechanism of ATP Hydrolysis in F1-ATPase. Biophys. J 2003, 85, 2253–2266. [Google Scholar]
- Gao, YQ; Yang, W; Karplus, M. A Structure-Based Model for the Synthesis and Hydrolysis of ATP by F1-ATPase. Cell 2005, 123, 195–205. [Google Scholar]
- Al-Shawi, MK; Ketchum, CJ; Nakamoto, RK. The Escherichia coli FOF1 γM23K Uncoupling Mutant has a Higher K0.5 for Pi. Transition State Analysis of this Mutant and Others Reveals that Synthesis and Hydrolysis Utilize the Same Kinetic Pathway. Biochemistry 1997, 36, 12961–12969. [Google Scholar]
- Boyer, PD. Catalytic Site Forms and Controls in ATP Synthase Catalysis. Biochim. Biophys. Acta 2000, 1458, 252–262. [Google Scholar]
- Abrahams, JP; Leslie, AGW; Lutter, R; Walker, JE. Structure at 2.8 Å Resolution of F1-ATPase from Bovine Heart Mitochondria. Nature 1994, 370, 621–628. [Google Scholar]
- Berden, JA; Hartog, AF. Analysis of the Nucleotide Binding Sites of Mitochondrial ATP Synthase Provides Evidence for a Two-Site Catalytic Mechanism. Biochim. Biophys. Acta 2000, 1458, 234–251. [Google Scholar]
- Berden, JA. Rotary Movements within the ATP Synthase Do Not Constitute an Obligatory Element of the Catalytic Mechanism. IUBMB Life 2003, 55, 473–481. [Google Scholar]
- Penefsky, HS; Cross, RL. Structure and Mechanism of FOF1-type ATP synthases and ATPases. Adv. Enzymol 1991, 64, 173–214. [Google Scholar]
- Sugi, H; Kobayashi, T; Gross, T; Noguchi, K; Karr, T; Harrington, WF. Contraction Characteristics and ATPase Activity of Skeletal Muscle Fibers in the Presence of Antibody to Myosin Subfragment 2. Proc. Natl. Acad. Sci. USA 1992, 89, 6134–6137. [Google Scholar]
- Ishijima, A; Kojima, H; Funatsu, T; Tokunaga, M; Higuchi, H; Tanaka, H; Yanagida, T. Simultaneous Observation of Individual ATPase and Mechanical Events by a Single Myosin Molecule During Interaction with Actin. Cell 1998, 92, 161–171. [Google Scholar]
- Stock, D; Leslie, AGW; Walker, JE. Molecular Architecture of the Rotary Motor in ATP Synthase. Science 1999, 286, 1700–1705. [Google Scholar]
- Hinkle, PC; Arun Kumar, M; Resetar, A; Harris, DL. Mechanistic Stoichiometry of Mitochondrial Oxidative Phosphorylation. Biochemistry 1991, 30, 3576–3582. [Google Scholar]
- Chen, C; Ko, Y; Delannoy, M; Ludtke, SJ; Chiu, W; Pedersen, PL. Mitochondrial ATP Synthasome: Three-Dimensional Structure by Electron Microscopy of the ATP Synthase in Complex Formation with Carriers for Pi and ADP/ATP. J. Biol. Chem 2004, 279, 31761–31768. [Google Scholar]
- Prebble, J. Peter Mitchell and the Ox Phos Wars. Trends Biochem. Sci 2002, 27, 209–212. [Google Scholar]
- Slater, EC. The Mechanism of the Conservation of Energy of Biological Oxidations. Eur. J. Biochem 1987, 166, 489–504. [Google Scholar]
- Reynafarje, B; Alexandre, A; Davies, P; Lehninger, AL. Proton Translocation Stoichiometry of Cytochrome Oxidase: Use of a Fast-Responding Oxygen Electrode. Proc. Natl. Acad. Sci. USA 1982, 79, 7218–7222. [Google Scholar]
- Williams, RJP. Some Unrealistic Assumptions in the Theory of Chemi-Osmosis and their Consequences. FEBS Lett 1979, 102, 126–132. [Google Scholar]
- Green, DE. A Critique of the Chemiosmotic Model of Energy Coupling. Proc. Natl. Acad. Sci. USA 1981, 78, 2240–2243. [Google Scholar]
- Battley, EH. Energetics of Microbial Growth; Wiley: New York, 1987. [Google Scholar]
- Hansen, LD; Hopkin, MS; Criddle, RS. Plant Calorimetry: A Window to Plant Physiology and Ecology. Thermochim. Acta 1997, 300, 183–197. [Google Scholar]
- Nielsen, J; Villadsen, J; Liden, G. Bioreaction Engineering Principles, 2ed Ed; Kluwer Academic/Plenum: New York, 2003. [Google Scholar]
- Lemasters, JJ. The ATP-to-Oxygen Stoichiometries of Oxidative Phosphorylation by Rat Liver Mitochondria: An Analysis of ADP-Induced Oxygen Jumps by Linear Nonequilibrium Thermodynamics. J. Biol. Chem 1984, 259, 13123–13130. [Google Scholar]
- Steinberg-Yfrach, G; Rigaud, J-L; Durantini, EN; Moore, AL; Gust, D; Moore, TA. Light-Driven Production of ATP Catalysed by F0F1-ATP Synthase in an Artificial Photosynthetic Membrane. Nature 1998, 392, 479–482. [Google Scholar]
- Tedeschi, H. Old and New Data, New Issues: The Mitochondrial Δψ. Biochim. Biophys. Acta 2005, 1709, 195–202. [Google Scholar]
- Tedeschi, H. Commentary on: Old and New Data, New Issues: The Mitochondrial Δψ. Biochim. Biophys. Acta 2005, 1710, 63–65. [Google Scholar]
- Miki, H; Setou, M; Kaneshiro, K; Hirokawa, N. All Kinesin Superfamily Protein, KIF, Genes in Mouse and Human. Proc. Natl. Acad. Sci. USA 2001, 98, 7004–7011. [Google Scholar]
- Howard, J. The Movement of Kinesin Along Microtubules. Annu. Rev. Physiol 1996, 58, 703–729. [Google Scholar]
- Howard, J. Mechanics of Motor Proteins and the Cytoskeleton; Sinauer: Sunderland, MA, USA, 2001. [Google Scholar]
- Hua, W; Chung, J; Gelles, J. Distinguishing Inchworm and Hand-Over-Hand Processive Kinesin Movement Models by Neck Rotation Measurements. Science 2002, 295, 844–848. [Google Scholar]
- Asbury, CL; Fehr, AN; Block, SM. Kinesin Moves by an Asymmetric Hand-Over-Hand Mechanism. Science 2003, 302, 2130–2134. [Google Scholar]
- Thoresen, T; Gelles, J. Processive Movement by a Mutant Kinesin Heterodimer with Only One Active Head. Biophys. J 2004, 86, 409a. [Google Scholar]
- Walker, RA. ncd and Kinesin Motor Domains Interact with Both α- and β-Tubulin. Proc. Natl. Acad. Sci. USA 1995, 92, 5960–5964. [Google Scholar]
- Larcher, J-C; Boucher, D; Lazereg, S; Gros, F; Denoulet, P. Interaction of Kinesin Motor Domains with α- and β-Tubulin Subunits at a Tau-Independent Binding Site. J. Biol. Chem 1996, 271, 22117–22124. [Google Scholar]
- Tucker, C; Goldstein, LSB. Probing the Kinesin-Microtubule Interaction. J. Biol. Chem 1997, 272, 9481–9488. [Google Scholar]
- Han, Y; Sablin, EP; Nogales, E; Fletterick, RJ; Downing, KH. Visualizing a New Binding Site of ncd-Motor Domain on Tubulin. J. Struct. Biol 1999, 128, 26–33. [Google Scholar]
- Yildiz, A; Tomishige, M; Vale, RD; Selvin, PR. Kinesin Walks Hand-Over-Hand. Science 2004, 303, 676–678. [Google Scholar]
- Carter, NJ; Cross, RA. Mechanics of the Kinesin Step. Nature 2005, 435, 308–312. [Google Scholar]
- Carter, NJ; Cross, RA. Kinesin’s Moonwalk. Curr. Opin. Cell Biol 2006, 18, 61–67. [Google Scholar]
- Huang, T-G; Hackney, DD. Drosophila Kinesin Minimal Motor Domain Expressed in Escherichia coli: Purification and Kinetic Characterization. J. Biol. Chem 1994, 269, 16493–16501. [Google Scholar]
- Huang, T-G; Suhan, J; Hackney, DD. Drosophila Kinesin Minimal Motor Domain Extending to Amino Acid Position 392 is Dimeric When Expressed in Escherichia coli. J. Biol. Chem 1994, 269, 16502–16507. [Google Scholar]
- Yildiz, A; Selvin, PR. Kinesin: Walking, Crawling or Sliding Along? Trends Cell Biol 2006, 15, 112–120. [Google Scholar]
- Guydosh, NR; Block, SM. Backsteps Induced by Nucleotide Analogs Suggest the Front Head of Kinesin is Gated by Strain. Proc. Natl. Acad. Sci. USA 2006, 103, 8054–8059. [Google Scholar]
- Visscher, K; Schnitzer, MJ; Block, SM. Single Kinesin Molecules Studied with a Molecular Force Clamp. Nature 1999, 400, 184–189. [Google Scholar]
- Nishiyama, M; Higuchi, H; Yanagida, T. Chemomechanical Coupling of the Forward and Backward Steps of Single Kinesin Molecules. Nature Cell Biol 2002, 4, 790–797. [Google Scholar]
- Kawaguchi, K; Ishiwata, S. Temperature Dependence of Force, Velocity, and Processivity of Single Kinesin Molecules. Biochem. Biophys. Res. Commun 2000, 272, 895–899. [Google Scholar]
- Belzacq, A-S; Vieira, HLA; Kroemer, G; Brenner, C. The Adenine Nucleotide Translocator in Apoptosis. Biochimie 2002, 84, 167–176. [Google Scholar]
- Halestrap, AP; McStay, GP; Clarke, SJ. The Permeability Transition Pore Complex: Another View. Biochimie 2002, 84, 153–166. [Google Scholar]
- Belzacq, A-S; Vieira, HLA; Verrier, F; Vandecasteele, G; Cohen, I; Prevost, M-C; Larquet, E; Pariselli, F; Petit, PX; Kahn, A; Rizzuto, R; Brenner, C; Kroemer, G. Bcl-2 and Bax Modulate Adenine Nucleotide Translocase Activity. Cancer Res 2003, 63, 541–546. [Google Scholar]
- Willis, SN; Fletcher, JI; Kaufmann, T; van Delft, MF; Chen, L; Czabotar, PE; Ierino, H; Lee, EF; Fairlie, WD; Bouillet, P; Strasser, A; Kluck, RM; Adams, JM; Huang, DCS. Apoptosis Initiated When BH3 Ligands Engage Multiple Bcl-2 Homologs, Not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar]
- Morellet, N; Bouaziz, S; Petitjean, P; Roques, BP. NMR Structure of the HIV-1 Regulatory Protein VPR. J. Mol. Biol 2003, 327, 215–227. [Google Scholar]
- Bruns, K; Fossen, T; Wray, V; Henklein, P; Tessmer, U; Schubert, U. Structural Characterization of the HIV-1 Vpr N Terminus. J. Biol. Chem 2003, 278, 43188–43201. [Google Scholar]
- Bruns, K; Studtrucker, N; Sharma, A; Fossen, T; Mitzner, D; Eissman, A; Tessmer, U; Roder, R; Henklein, P; Wray, V; Schubert, U. Structural Characterization and Oligomerization of PB1-F2, a Proapoptotic Influenza A Virus Protein. J. Biol. Chem 2007, 282, 353–363. [Google Scholar]
- Tripathy, BC; Mohanty, P. Zinc Inhibition of Electron Transport in Isolated Chloroplasts. Plant Physiol 1980, 66, 1174–1178. [Google Scholar]
- Arnon, DI. Copper Enzymes in Isolated Chloroplasts. Plant Physiol 1949, 24, 1–15. [Google Scholar]
- Jagendorf, AT; Uribe, E. ATP Formation Caused by Acid-Base Transition of Spinach Chloroplasts. Proc. Natl. Acad. Sci. USA 1966, 55, 170–177. [Google Scholar]
- Taussky, HH; Shorr, E. A Microcolorimetric Method for the Determination of Inorganic Phosphorus. J. Biol. Chem 1953, 202, 675–685. [Google Scholar]
- Schägger, H; Pfeiffer, K. The Ratio of Oxidative Phosphorylation Complexex I-V in Bovine Heart Mitochondria and the Composition of Respiratory Chain Supercomplexes. J. Biol. Chem 2001, 276, 37861–37867. [Google Scholar]
- Wittig, I; Carrozzo, R; Santorelli, FM; Schägger, H. Supercomplexes and Subcomplexes of Mitochondrial Oxidative Phosphorylation. Biochim. Biophys. Acta 2006, 1757, 1066–1072. [Google Scholar]
- Lancaster, CRD; Sauer, US; Groβ, R; Haas, AH; Graf, J; Schwalbe, H; Mäntele, W; Simon, J; Madej, MG. Experimental Support for the “E Pathway Hypothesis” of Coupled Transmembrane e− and H+ Transfer in Dihemic Quinol: Fumarate Reductase. Proc. Natl. Acad. Sci. USA 2005, 102, 18860–18865. [Google Scholar]
- MacMillan, F; Budiman, K; Angerer, H; Michel, H. The Role of Tryptophan 272 in the Paracoccus denitrificans Cytochrome c Oxidase. FEBS Lett 2006, 580, 1345–1349. [Google Scholar]
- Belevich, I; Bloch, DA; Belevich, N; Wikström, M; Verkhovsky, MI. Exploring the Proton Pump Mechanism of Cytochrome c Oxidase in Real Time. Proc. Natl. Acad. Sci. USA 2007, 104, 2685–2690. [Google Scholar]
- Kim, YC; Wikström, M; Hummer, G. Kinetic Models of Redox-Coupled Proton Pumping. Proc. Natl. Acad. Sci. USA 2007, 104, 2169–2174. [Google Scholar]

© 2008 by MDPI This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nath, S. The New Unified Theory of ATP Synthesis/Hydrolysis and Muscle Contraction, Its Manifold Fundamental Consequences and Mechanistic Implications and Its Applications in Health and Disease. Int. J. Mol. Sci. 2008, 9, 1784-1840. https://doi.org/10.3390/ijms9091784
Nath S. The New Unified Theory of ATP Synthesis/Hydrolysis and Muscle Contraction, Its Manifold Fundamental Consequences and Mechanistic Implications and Its Applications in Health and Disease. International Journal of Molecular Sciences. 2008; 9(9):1784-1840. https://doi.org/10.3390/ijms9091784
Chicago/Turabian StyleNath, Sunil. 2008. "The New Unified Theory of ATP Synthesis/Hydrolysis and Muscle Contraction, Its Manifold Fundamental Consequences and Mechanistic Implications and Its Applications in Health and Disease" International Journal of Molecular Sciences 9, no. 9: 1784-1840. https://doi.org/10.3390/ijms9091784