Prodrugs of Fluoro-Substituted Benzoates of EGC as Tumor Cellular Proteasome Inhibitors and Apoptosis Inducers

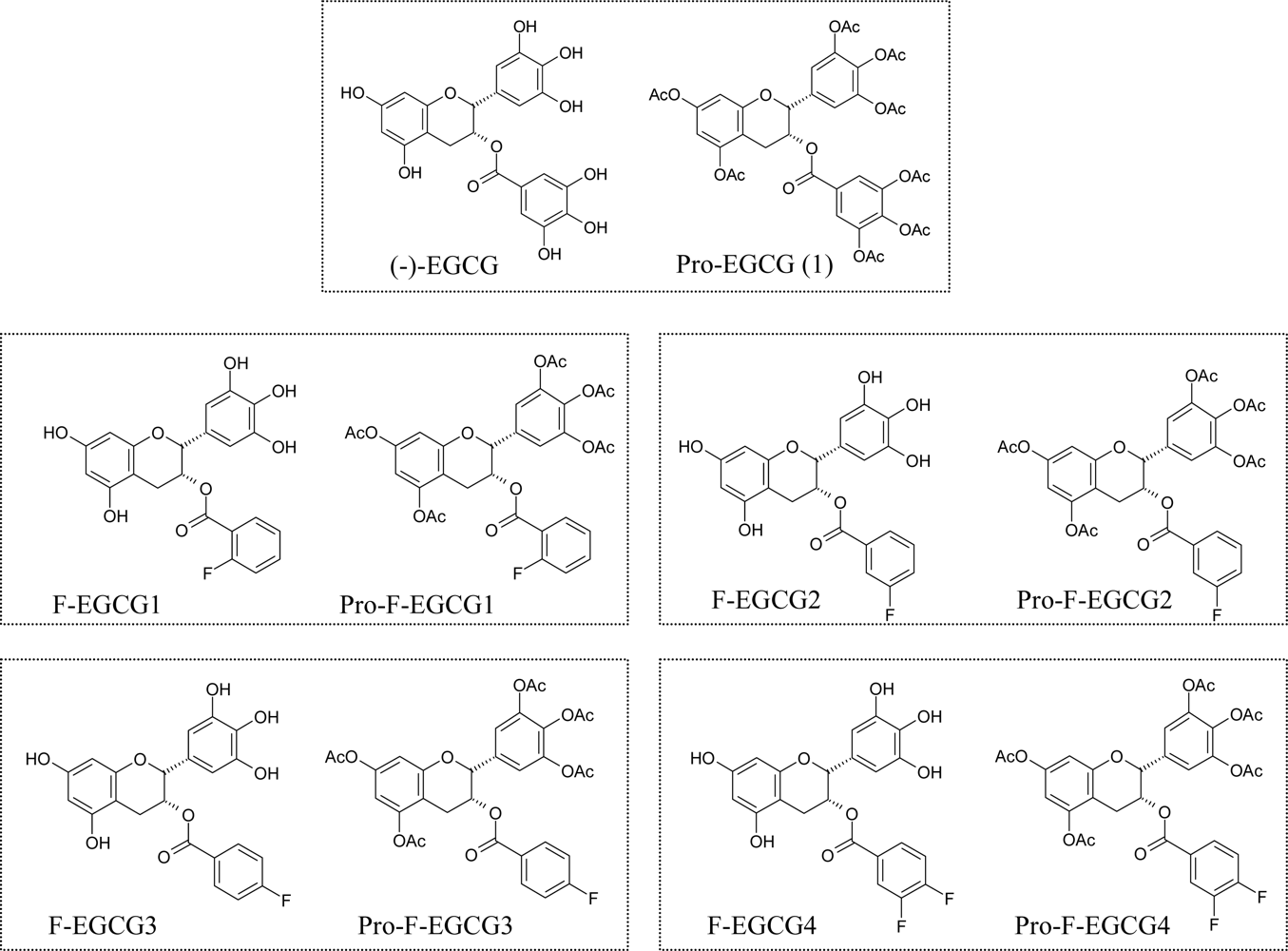{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Inhibition of the chymotrypsin-like activity of a purified 20S proteasome by synthetic F-EGCG analogs
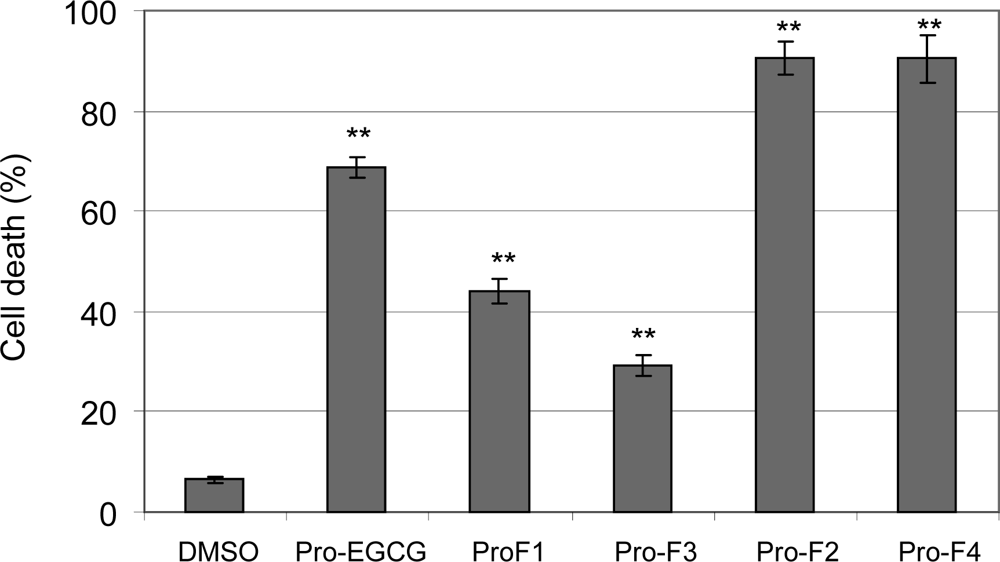
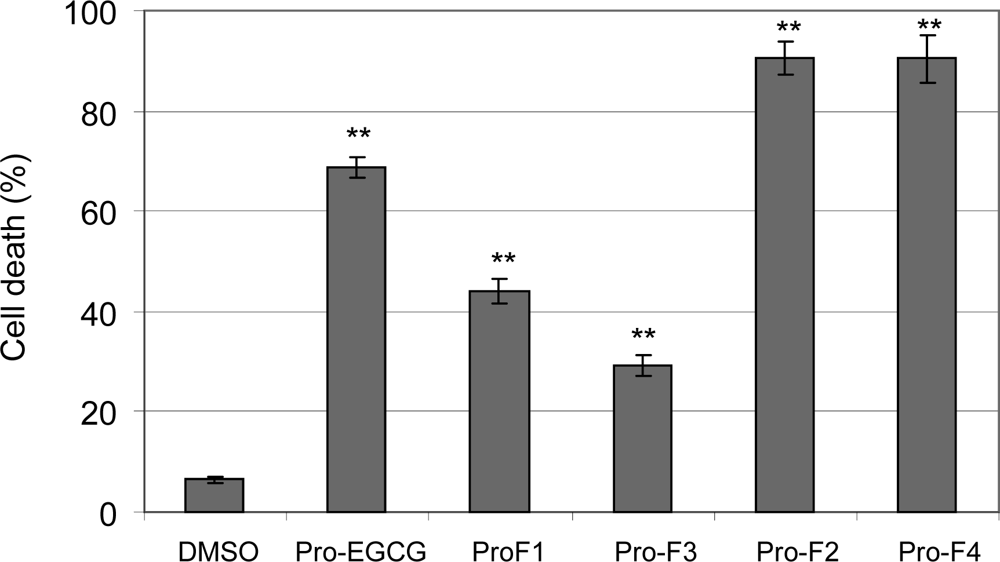
2.2. Induction of cell death in human leukemia cells by peracetates of fluoro-substituted benzoates of EGC
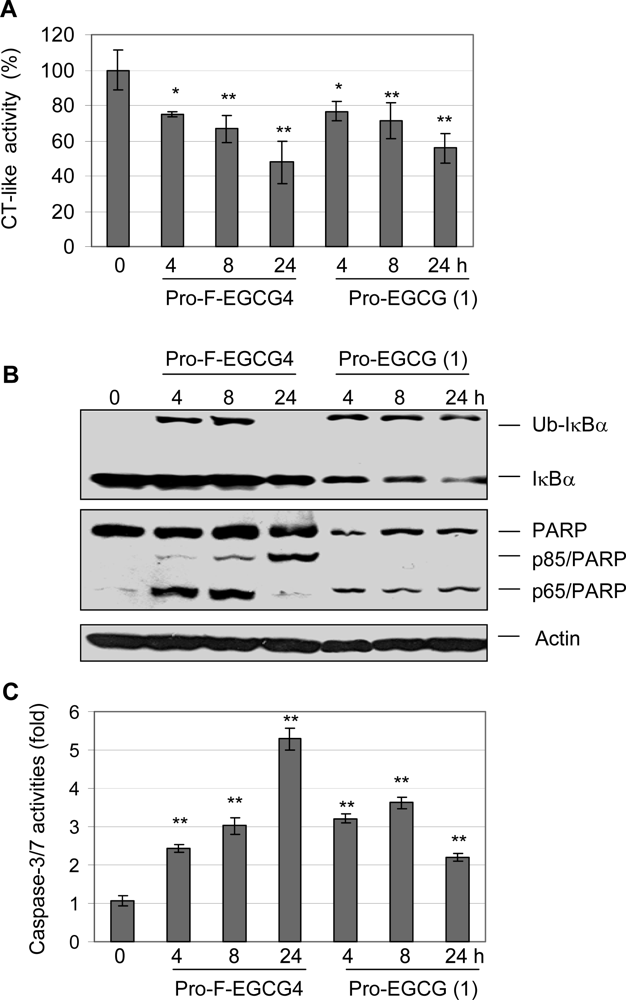
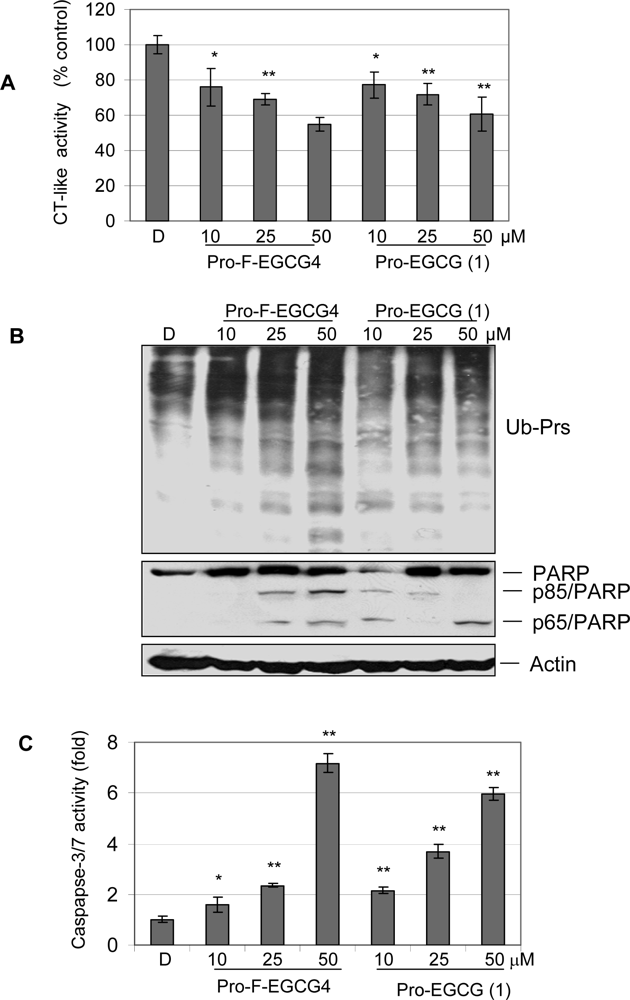
2.3 Pro-F-EGCG4 inhibits tumor cellular proteasomal activity and induces apoptosis in a time-dependent manner
2.4 Pro-F-EGCG4 inhibits tumor cellular proteasomal activity and induces apoptosis in a dose-dependent manner
3. Experimental Section
3.1. Materials
3.2. Synthesis of F-EGCG analogs and their prodrugs
3.3. Cell viability assay
3.4. Cell culture, drug treatment and protein extraction
3.5. Inhibition of cellular proteasome activity by Pro-F-EGCG analogs
3.6. Induction of caspase-3 activity by Pro-F-EGCG analogs
3.7. Western blotting
Acknowledgment
References and Notes
- Almond, JB; Cohen, GM. The proteasome: a novel target for cancer chemotherapy. Leukemia 2002, 16(4), 433–443. [Google Scholar]
- Smith, DM; Daniel, KG; Dou, QP. Exploiting the Ubiquitin-Proteasome Pathway for Anticancer Drug Discovery: Unanswered Questions and Future Directions. Lett. Drug Des. Discov. 2005, 2, 77–84. [Google Scholar]
- Adam, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004, 5, 417–421. [Google Scholar]
- Chen, D; Daniel, KG; Kuhn, DJ; Kazi, A; Bhuiyan, M; Li, L; Wang, Z; Wan, SB; Lam, MH; Chan, TH; Dou, QP. Green tea and tea polyphenols in cancer prevention. Front Biosci. 2004, 26, 18–31. [Google Scholar]
- Nam, S; Smith, DM; Dou, QP. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. Biol. Chem. 2001, 276(16), 13322–13330. [Google Scholar]
- Haslam, E. Plant Polyphenols. Vegetable Tannins Revisited; Cambridge University Press: Cambridge, UK, 1989. [Google Scholar]
- Yang, CS; Wang, ZY. Tea and Cancer. J. Natl. Cancer Inst. 1993, 85, 1038–1049. [Google Scholar]
- Fujiki, HJ. Two stages of cancer prevention with green tea. Cancer Res. Clin. Oncol. 1999, 125, 589–597. [Google Scholar]
- Zhu, Q; Zhang, A; Tsang, D; Huang, Y; Chen, Z. Stability of green tea catechins. J Agric Food Chem. 1997, 45, 4624–4628. [Google Scholar]
- Smith, DM; Wang, Z; Kazi, A; Li, L; Chan, TH; Dou, QP. Synthetic analogs of green tea polyphenols as proteasome inhibitors. Mol. Med. 2002, 8(7), 382–392. [Google Scholar]
- Wan, SB; Landis-Piwowar, KR; Kuhn, DJ; Chen, D; Dou, QP; Chan, TH. Structure-activity study of epi-gallocatechin gallate (EGCG) analogs as proteasome inhibitors. Bioorg. Med. Chem. 2005, 13(6), 2177–2185. [Google Scholar]
- Landis-Piwowar, KR; Huo, CD; Chen, D; Cui, QC; Minic, V; Shi, GQ; Chan, TH; Dou, QP. A Novel Pro-drug of the Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate as a Potential Anti-Cancer Agent. Cancer Res. 2007, 67, 4303–4310. [Google Scholar]
- Chen, D; Chen, MS; Cui, QZC; Yang, HJ; Dou, QP. Structure-proteasome-inhibitory activity relationships of dietary flavonoids in human cancer cells. Front. Biosci. 2007, 12, 1935–1345. [Google Scholar]
- Pink, JJ; Wuerzberger-Davis, S; Tagliarino, C; Planchon, SM; Yang, X; Froelich, CJ; Boothman, DA. Activation of a cysteine protease in MCF-7 and T47D breast cancer cells during ß-Lapachone-mediated apoptosis. Exp. Cell Res. 2000, 255, 144–155. [Google Scholar]
- Lu, H; Meng, X; Yang, CS. Enzymology of methylation of tea catechins and inhibition of catechol-O-methyltransferase by (-)-epigallocatechin gallate. Drug Metab. Dispos. 2003, 31, 572–579. [Google Scholar]
- Lambert, J; Sang, S; Hong, J; et al. Peracetylation as a means of enhancing in vitro bioactivity and bioavailability of epigallocatechin-3-gallate. Drug Metab. Dispos. 2006, 34, 2111–2116. [Google Scholar]
- Landis-Piwowar, KR; Wan, SB; Wiegand, RA; Kuhn, DJ; Chan, TH; Dou, QP. Methylation Suppresses the Proteasome-Inhibitory Function of Green Tea Polyphenols. J. Cell. Physiol. 2007, 213(1), 252–260. [Google Scholar]
- An, B; Goldfarb, RH; Siman, R; Dou, QP. Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ. 1998, 5, 1062–1075. [Google Scholar]

Share and Cite
Yu, Z.; Qin, X.L.; Gu, Y.Y.; Chen, D.; Cui, Q.C.; Jiang, T.; Wan, S.B.; Dou, Q.P. Prodrugs of Fluoro-Substituted Benzoates of EGC as Tumor Cellular Proteasome Inhibitors and Apoptosis Inducers. Int. J. Mol. Sci. 2008, 9, 951-961. https://doi.org/10.3390/ijms9060951
Yu Z, Qin XL, Gu YY, Chen D, Cui QC, Jiang T, Wan SB, Dou QP. Prodrugs of Fluoro-Substituted Benzoates of EGC as Tumor Cellular Proteasome Inhibitors and Apoptosis Inducers. International Journal of Molecular Sciences. 2008; 9(6):951-961. https://doi.org/10.3390/ijms9060951
Chicago/Turabian StyleYu, Zhiyong, Xu Long Qin, Yan Yan Gu, Di Chen, Qiuzhi Cindy Cui, Tao Jiang, Sheng Biao Wan, and Q. Ping Dou. 2008. "Prodrugs of Fluoro-Substituted Benzoates of EGC as Tumor Cellular Proteasome Inhibitors and Apoptosis Inducers" International Journal of Molecular Sciences 9, no. 6: 951-961. https://doi.org/10.3390/ijms9060951