RNA Interference in Mammalia Cells by RNA-3’-PNA Chimeras


Abstract
:1. Introduction
2. Results and Discussion
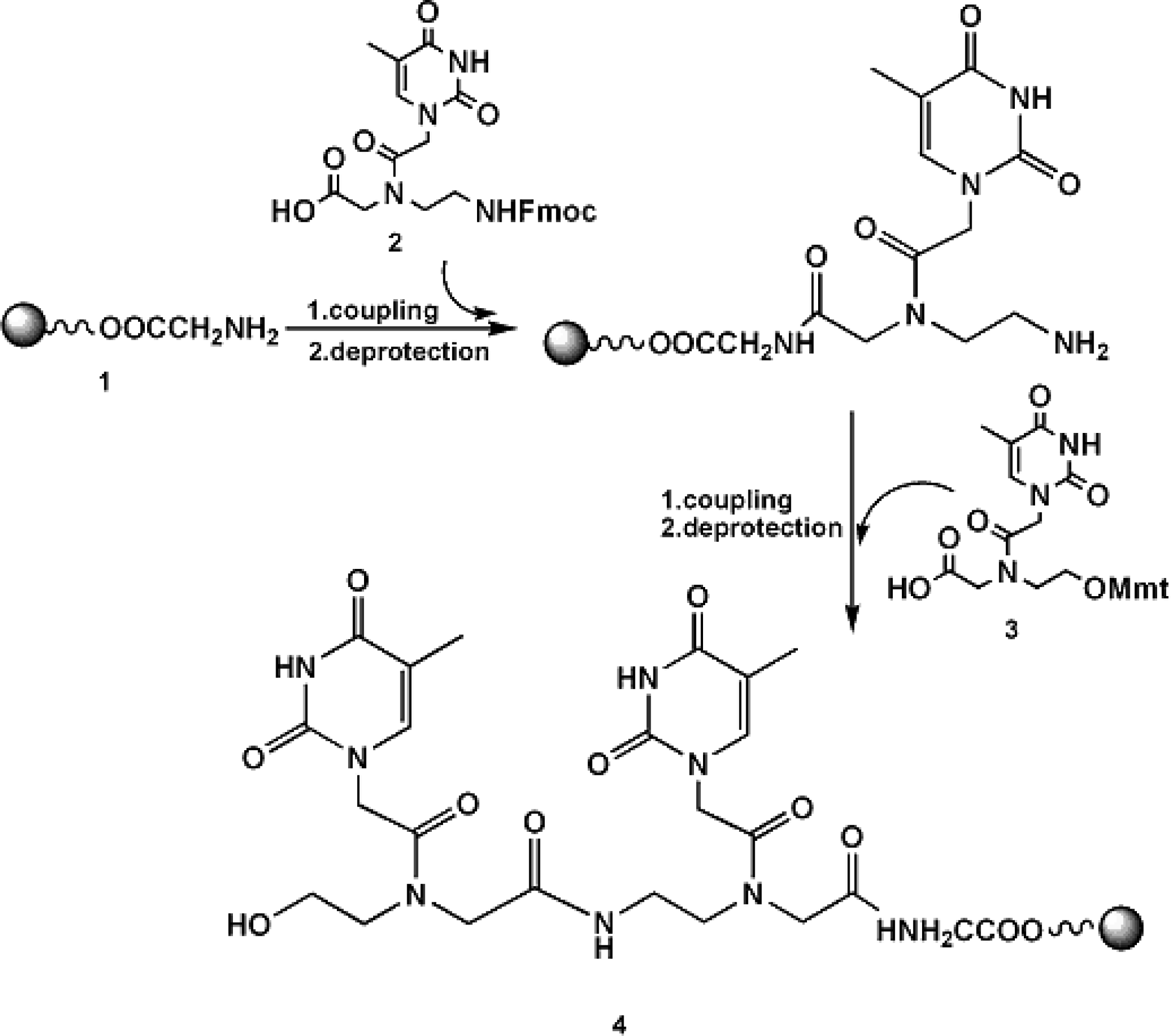
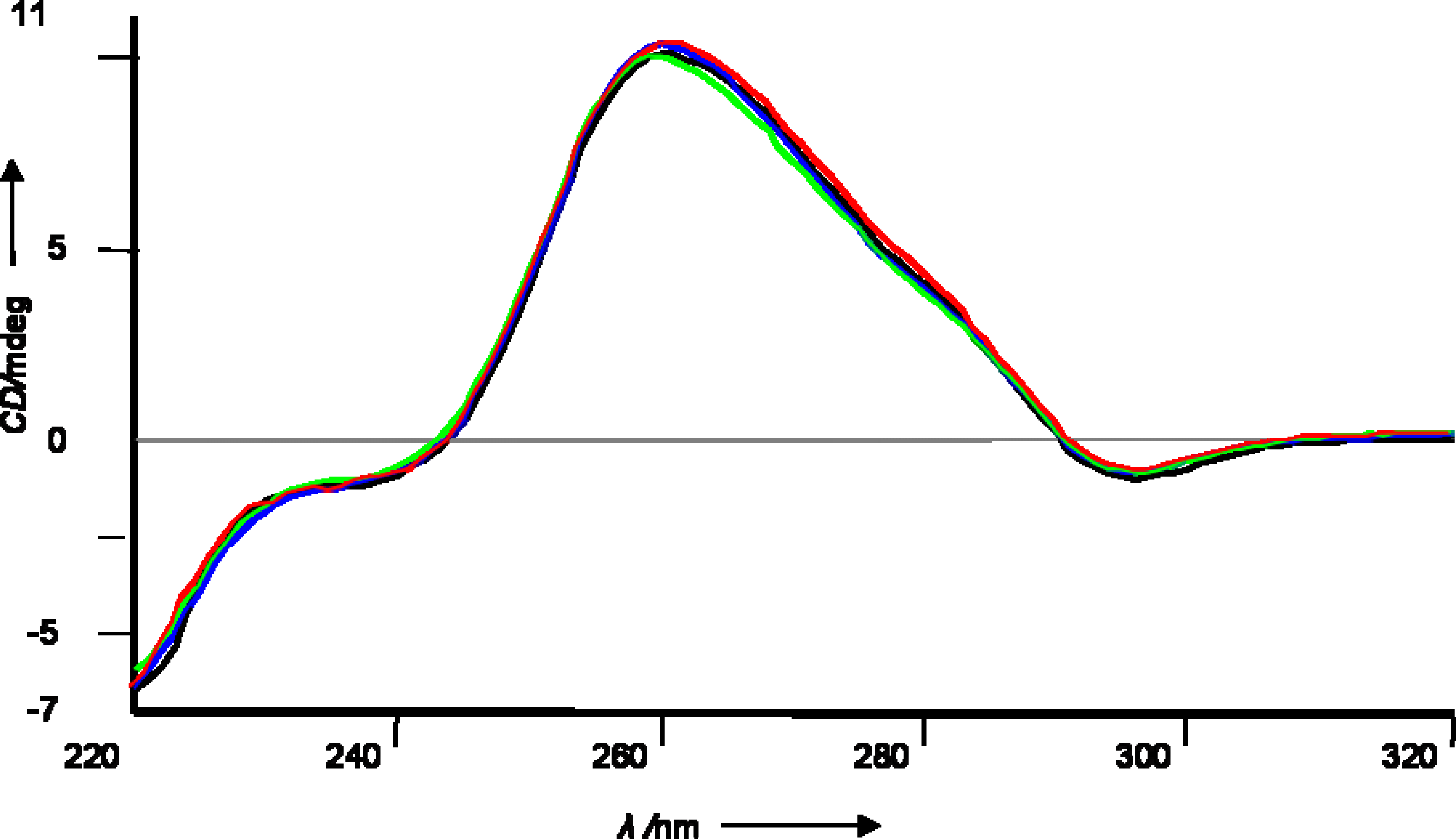
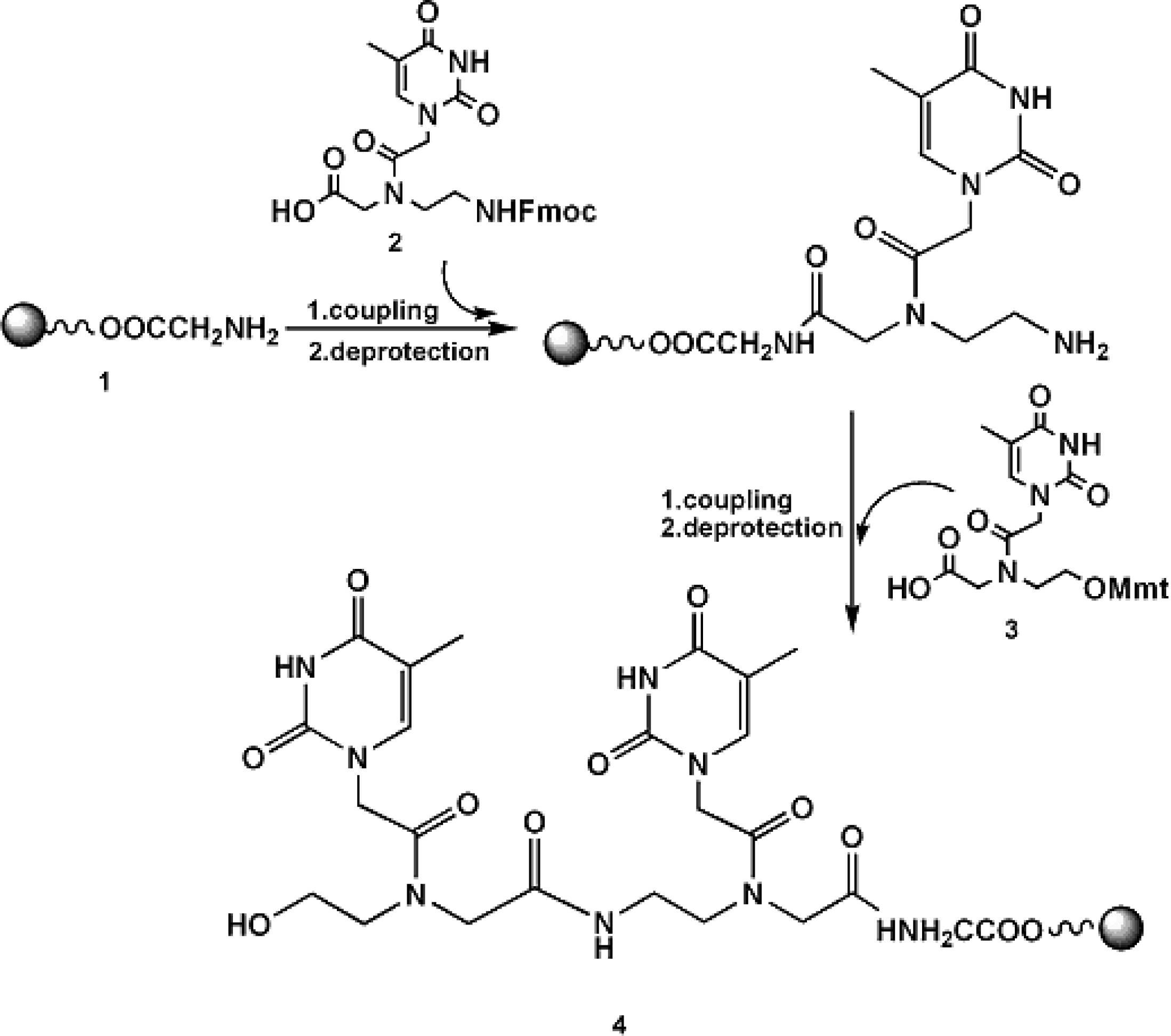
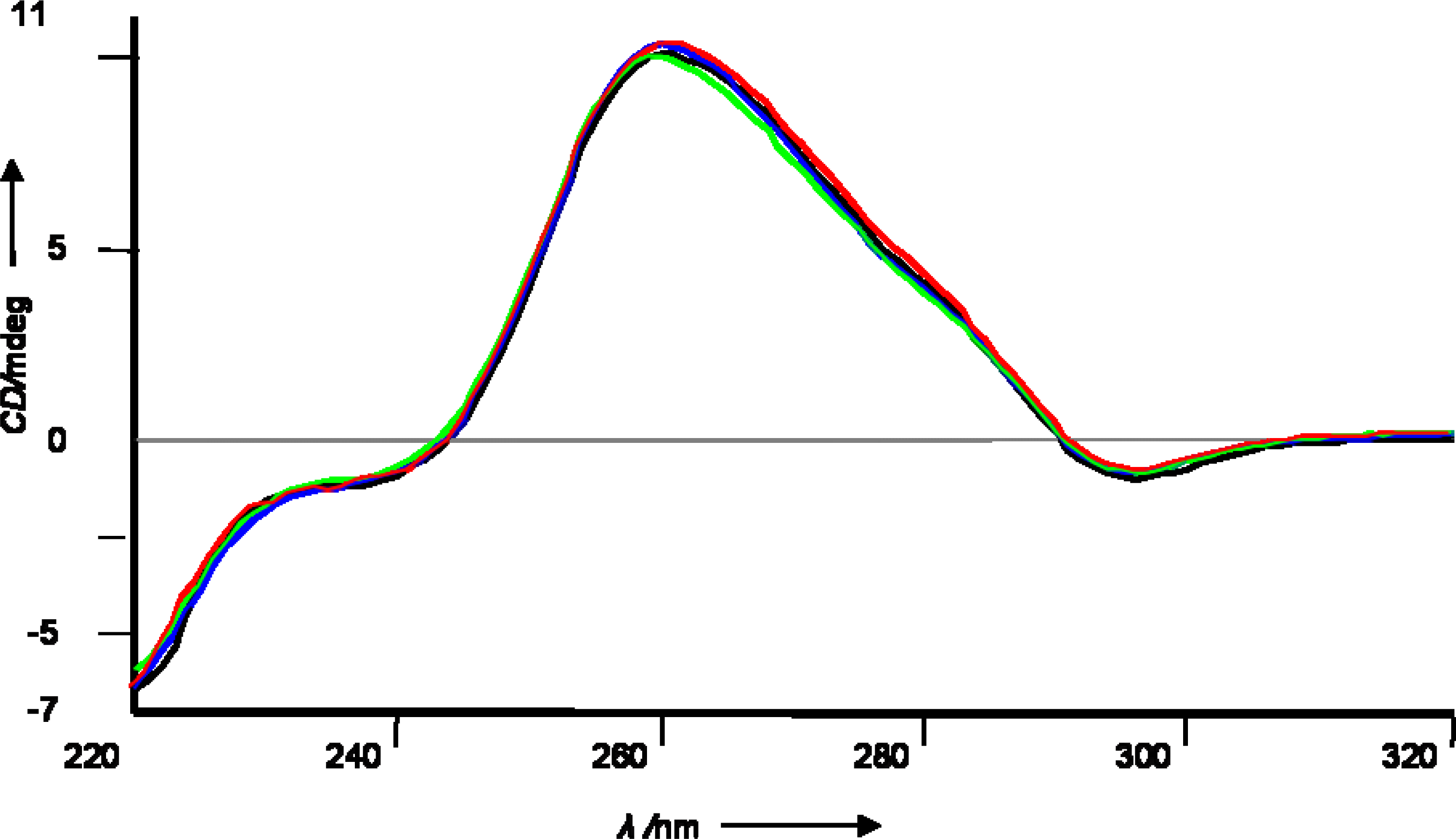
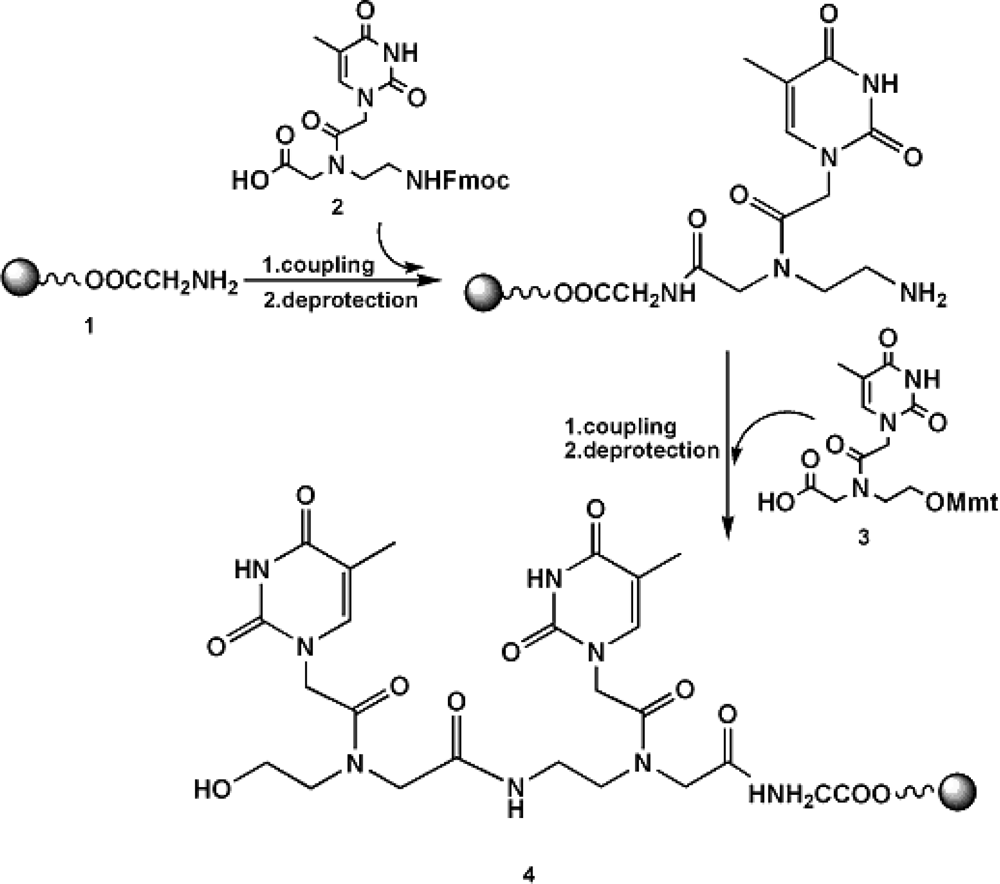
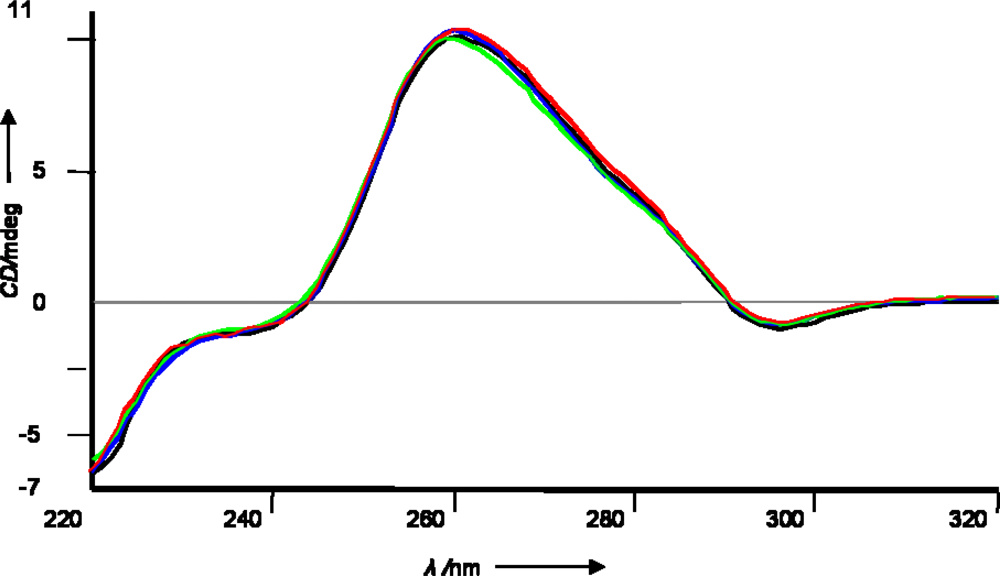
2.1 Synthetic strategy and characterization of native and chemically modified siRNAs
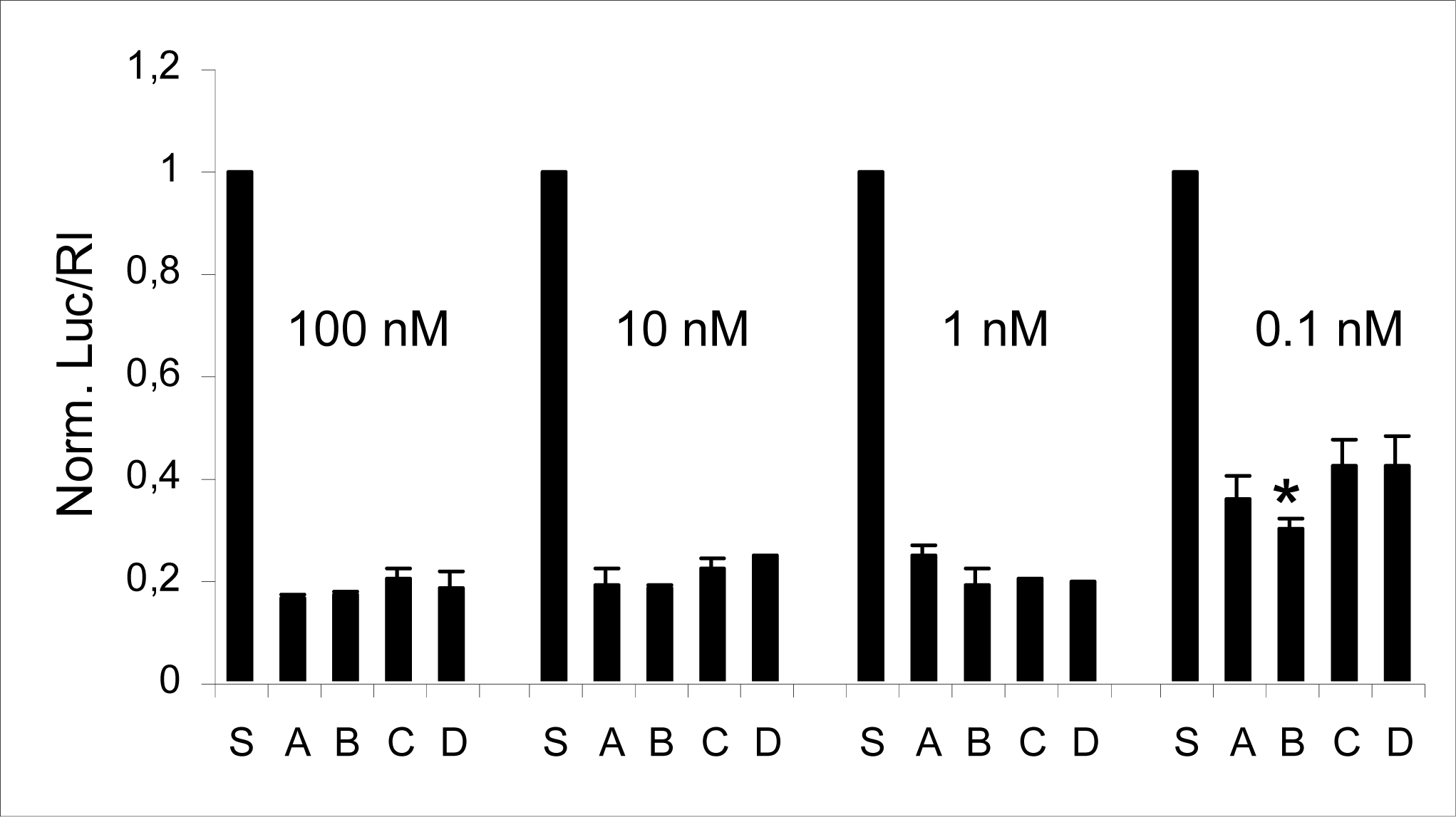
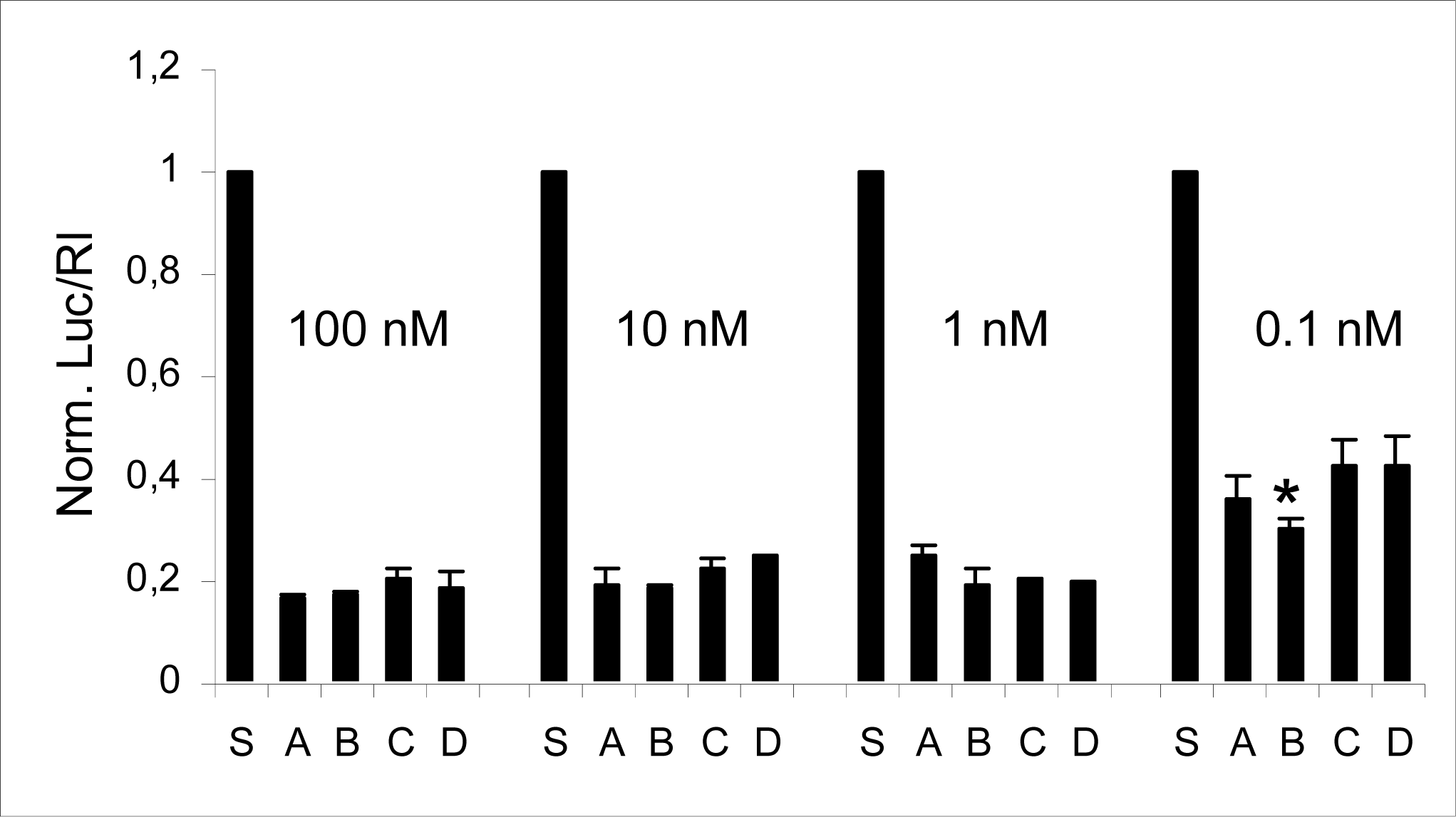
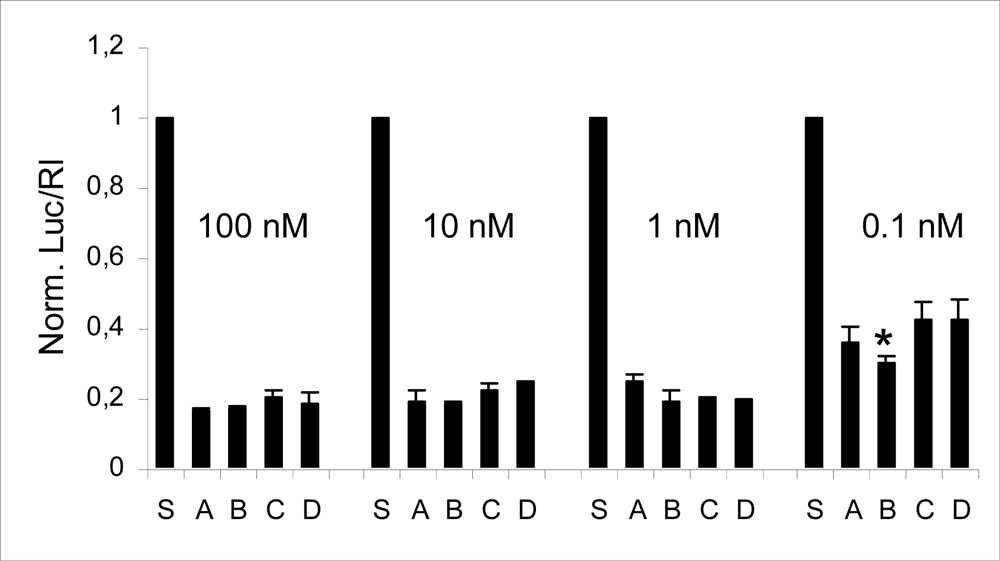
2.2 Compatibility of RNA-3’-PNA chimeras with the siRNA machinery
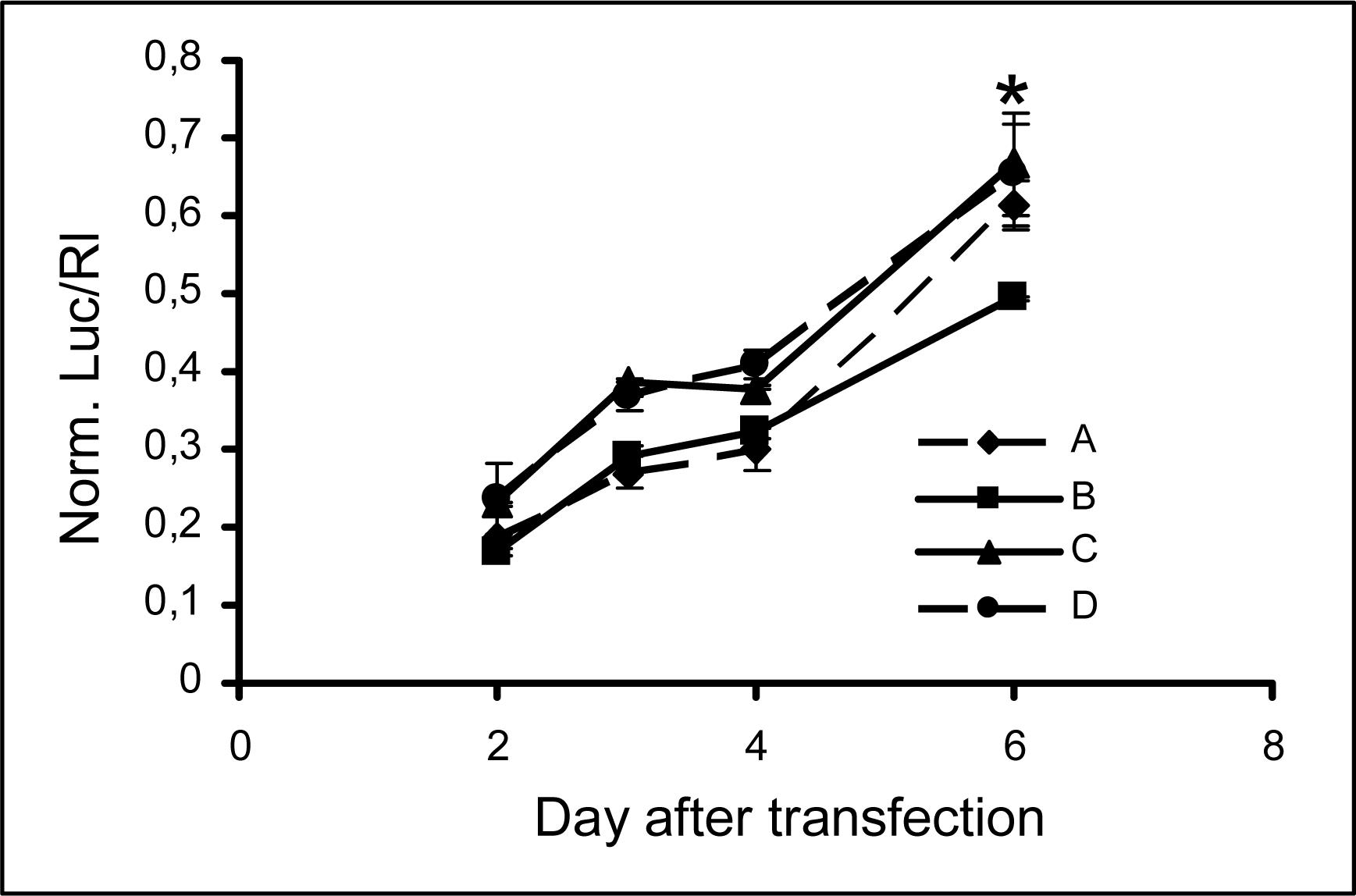
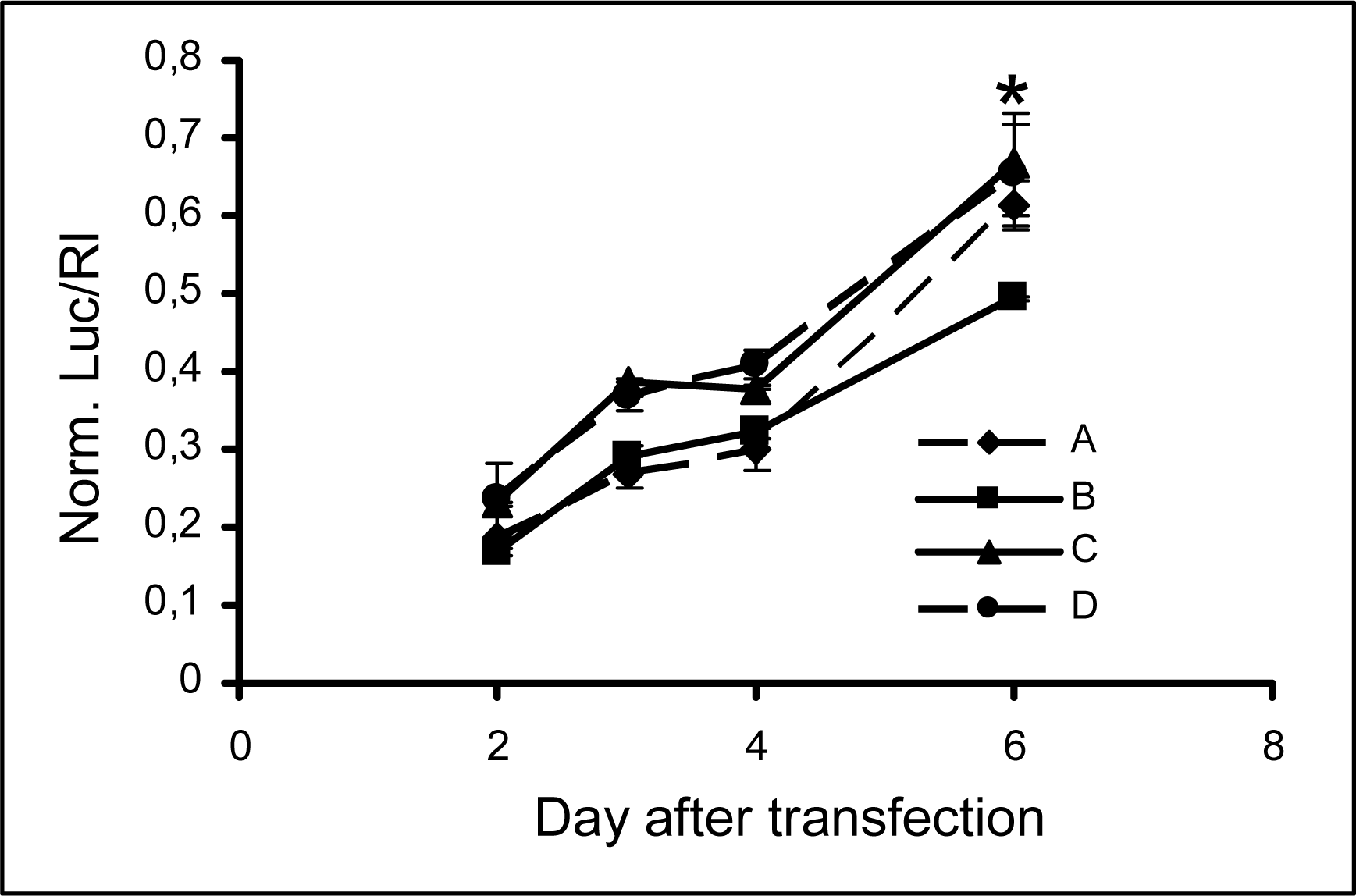
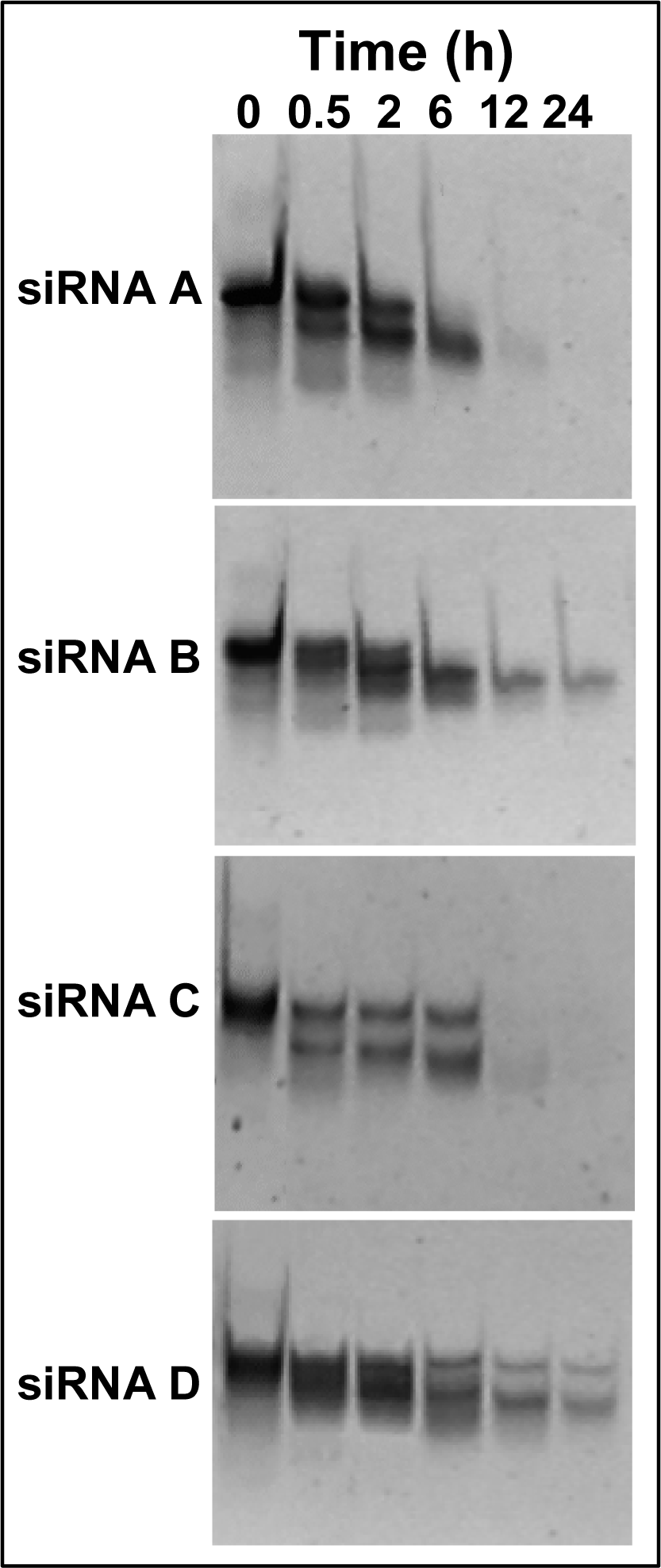
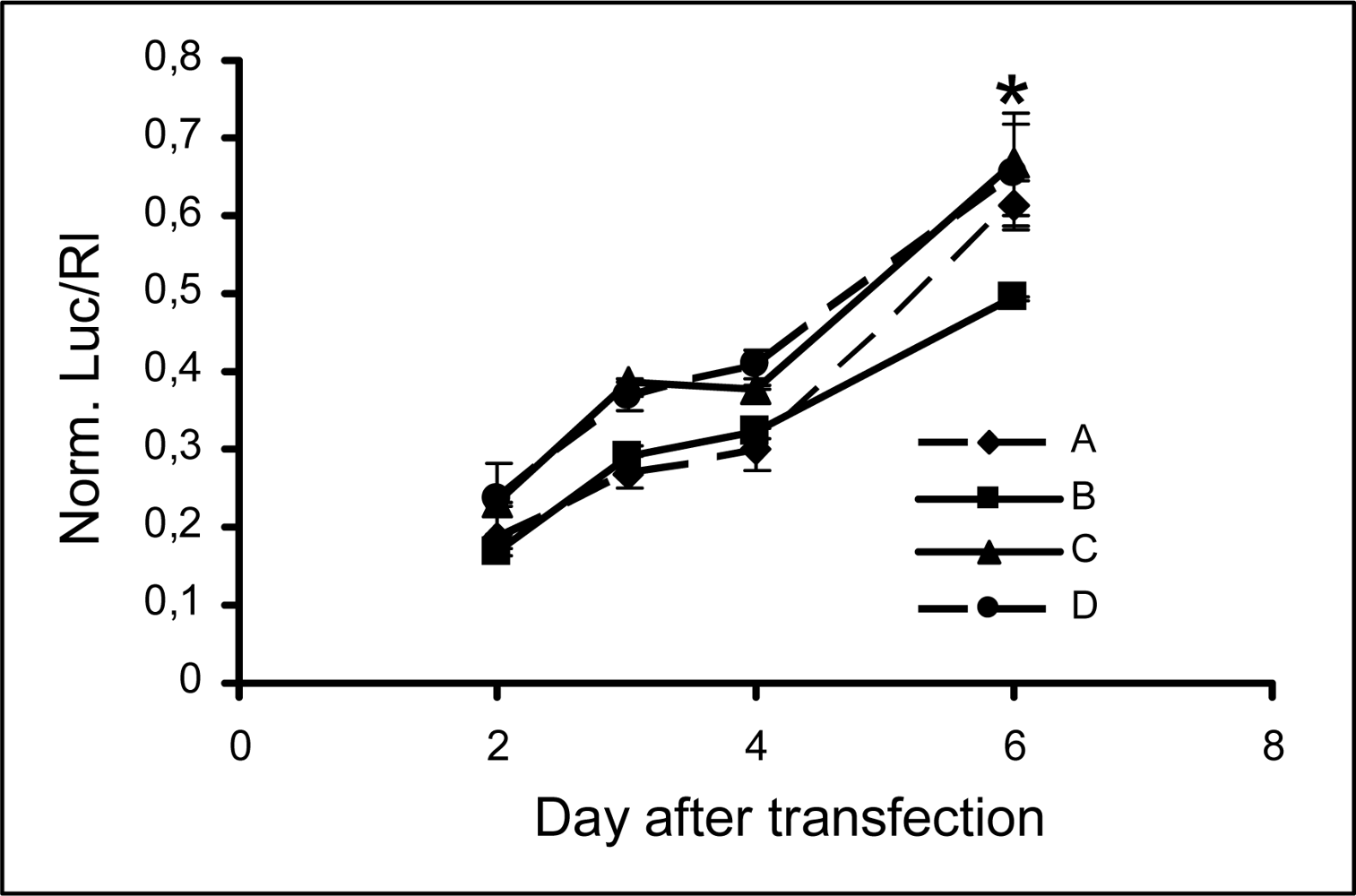
2.3 Persistence of modified siRNA-mediated silencing
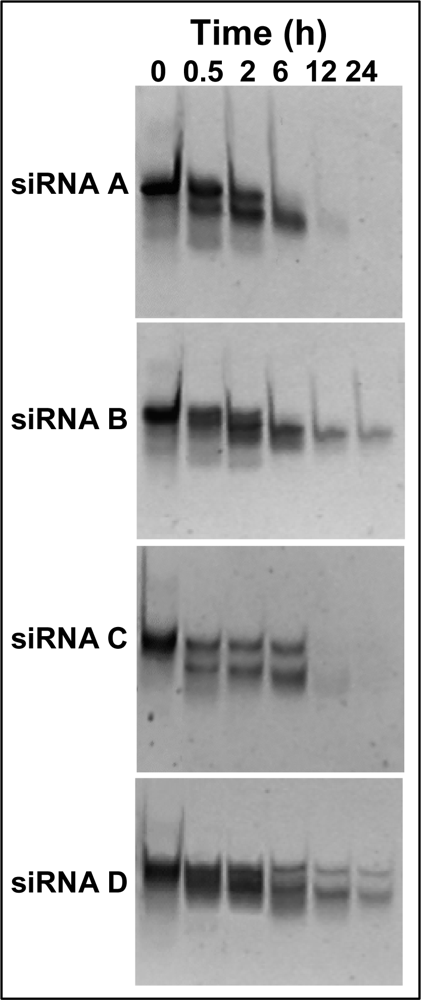
2.4 Serum stability of modified siRNA
2.5 Discussion
3. Experimental Section
3.1 Synthesis of RNA-3’-PNA chimeras
3.2 Purification and analysis of RNA-3’-PNA chimeras
3.3 siRNA preparation, Tm measurements and CD spectra
3.4 Cells cultures and transfections
3.5 Luciferase activity assay
3.6 Serum stability
Acknowledgments
References and Notes
- Fire, A; Xu, S; Montgomery, MK; Kostas, SA; Driver, SE; Mello, CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar]
- Zamore, PD; Tuschl, T; Sharp, PA; Bartel, DP. RNAi: doublestranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar]
- Bernstein, E; Caudy, AA; Hammond, SM; Hannon, GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar]
- Martinez, J; Patkaniowska, A; Urlaub, H; Lührmann, R; Tuschl, T. Single-stranded antisense siRNAs target RNA cleveage in RNAi. Cell 2002, 110, 563–574. [Google Scholar]
- Meister, G; Landthaler, M; Patkaniowska, A; Dorsett, Y; Teng, G; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell. 2004, 15, 185–197. [Google Scholar]
- Liu, J; Carmell, MA; Rivas, FV; Marsden, CG; Thomson, JM; Song, JJ; Hammond, SM; Joshua-Tor, L; Hannon, GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar]
- Elbashir, SM; Harborth, J; Lendeckel, W; Yalcin, A; Weber, K; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar]
- Harborth, J; Elbashir, SM; Vandenburgh, K; Manninga, H; Scaringe, SA; Weber, K; Tuschl, T. Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev. 2003, 13, 83–105. [Google Scholar]
- Elbashir, SM; Harborth, J; Weber, K; Tuschl, T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002, 26, 199–213. [Google Scholar]
- Sullenger, BA; Gilboa, E. Emerging clinical applications of RNA. Nature 2002, 418, 252–258. [Google Scholar]
- Saulnier, A; Pelletier, I; Labadie, K; Colbere-Garapin, F. Complete Cure of Persistent Virus Infections by Antiviral siRNAs. Molecular Therapy 2006, 13, 142–150. [Google Scholar]
- Bitko, V; Musiyenko, A; Shulyayeva, O; Barik, S. Inhibition of Respiratory viruses by nasally administered siRNA. Nature Medicine 2005, 11, 31–36. [Google Scholar]
- McCaffrey, AP; Nakai, H; Pande, K; Huang, Z; Salazar, FH; Xu, H; Wieland, SF; Marion, PL; Kay, MA. Inhibition of hepatitis B virus in mice byRNA interference. Nature Biotechnol 2003, 21, 639–644. [Google Scholar]
- Dorsett, Y; Tuschl, T. siRNAs:applications in functional genomics and potential therapeutics. Nat. Rev. Drug Discov. 2004, 3, 318–329. [Google Scholar]
- Zhang, HY; Du, Q; Wahlestedt, C; Liang, Z. RNA Interference with Chemically Modified siRNA. Curr. Top. Med. Chem. 2006, 6, 893–900. [Google Scholar]
- Czauderna, F; Fechtner, M; Dames, S; Aygu″n, H; Klippel, A; Pronk, GJ; Giese, K; Kaufmann, J. Structural variations and stabilizing modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003, 31, 2705–2716. [Google Scholar]
- Amarzguioui, M; Holen, T; Babaie, E; Prydz, H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003, 31, 589–595. [Google Scholar]
- Chiu, YL; Rana, TM. siRNA function in RNAi: a chemical modification analysis. RNA 2003, 9, 1034–1048. [Google Scholar]
- Manoharan, M. RNA interference and chemically modified small interfering RNAs. Current Opinion in Chemical Biology 2004, 8, 570–579. [Google Scholar]
- Layzer, JM; McCaffrey, AP; Tanner, AK; Huang, Z; Kay, MA; Sullenger, BA. In vivo activity of nuclease-resistant siRNAs. RNA 2004, 10, 766–771. [Google Scholar]
- Parrish, S; Fleenor, J; Xu, S; Mello, C; Fire, A. Functional anatomy of a dsRNA trigger: differential requirement for the two trigger strands in RNA interference. Mol. Cell. 2000, 6, 1077–1087. [Google Scholar]
- Braasch, DA; Jensen, S; Liu, Y; Kaur, K; Arar, K; White, MA; Corey, DR. RNA interference in mammalian cells by chemically-modified RNA. Biochemistry 2003, 42, 7967–7975. [Google Scholar]
- Elmen, J; Thonberg, H; Ljungberg, K; Frieden, M; Westergaard, M; Xu, Y; Wahren, B; Liang, Z; Orum, H; Koch, T; Wahlestedt, C. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 2005, 33, 439–447. [Google Scholar]
- Hamada, M; Ohtsuka, T; Kawaida, R; Koizuma, M; Morita, K; Furukawa, H; Imanishi, T; Miyagishi, M; Taira, K. Effects on RNA interference in gene expression (RNAi) in cultured mammalian cells of mismatches and the introduction of chemical modifications at the 3’-ends of siRNAs. Antisense Nucleic Acid Drug. Dev. 2002, 12, 301–309. [Google Scholar]
- Dowler, T; Bergeron, D; Tedeschi, A; Paquet, L; Ferrari, N; Damha, MJ. Improvements in siRNA properties mediated by 2’-deoxy-2’-fluoro-β-D-arabinonucleic acid (FANA). Nucleic Acids Res. 2006, 34, 1669–1675. [Google Scholar]
- Dande, P; Prakash, TP; Sioufi, N; Gaus, H; Jarres, R; Berdeja, A; Eric, E; Swayze, EE; Griffey, RH; Bhat, B. Improving RNA interference in mammalian cells by 4’thio-modified small interfering RNA (siRNA): effect on siRNA activity and nuclease stability when used in combination with 2’-O-alkyl modifications. J. Med. Chem. 2006, 49, 1624–1634. [Google Scholar]
- Nykanen, A; Haley, B; Zamore, PD. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell. 2001, 107, 309–321. [Google Scholar]
- Chiu, YL; Ali, A; Chu, C; Cao, H; Rana, TM. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem. Biol. 2004, 11, 1165–1175. [Google Scholar]
- Lorenz, C; Hadwiger, P; John, M; Vornlocher, HP; Unverzagt, C. Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells. Bioorg. Med. Chem. Lett. 2004, 14, 4975–4977. [Google Scholar]
- Egholm, M; Nielsen, PE; Buchardt, O; Berg, RH. Recognition of guanine and adenine in DNA by cytosine and thymine containing peptide nucleic acids (PNA). J. Am. Chem. Soc. 1992, 114, 9677–9678. [Google Scholar]
- Hyrup, B; Nielsen, PE. Peptide nucleic acid (PNA): synthesis, properties and potential applications. Bioorg. Med. Chem. 1969, 4, 5–23. [Google Scholar]
- Komiyama, M; Ye, S; Liang, X; Yamamoto, Y; Tomita, T; Zhou, JM; Aburatani, H. PNA for one-base differentiating protection of DANN from nuclease and ist use for SNP detection. J. Am. Chem. Soc. 2003, 125, 3758–3762. [Google Scholar]
- Uhlmann, E; Peyman, A; Breipohl, G; Will, DW. PNA: synthetic polyamide nucleic acids with unusual binding properties. Angew. Chem., Int. Ed. 1998, 37, 2796–2823. [Google Scholar]
- Uhlmann, E. Peptide nucleic acids (PNA) and PNA-DNA chimeras: from high binding affinity towards biological function. Biol. Chem. 1998, 379, 1045–1052. [Google Scholar]
- Capasso, D; De Napoli, L; Di Fabio, G; Messere, A; Montesarchio, D; Pedone, C; Piccialli, G; Saviano, M. Solid phase synthesis of DNA-3’-PNA chimeras by using Bhoc/Fmoc PNA monomers. Tetrahedron 2001, 57(46), 9481–9486. [Google Scholar]
- Esposito, V; Randazzo, A; Messere, A; Galeone, A; Petraccone, L; Giancola, C; Piccialli, G; Mayol, L. Synthesis and structural characterization of PNA-DNA quadruplex forming chimeras. Eur. J. Org. Chem. 2003, 17, 3364–3371. [Google Scholar]
- Petraccone, L; Erra, E; Messere, A; Montesarchio, D; Piccialli, G; De Napoli, L; Barone, G; Giancola, C. Targeting duplex DNA with DNA-PNA chimeras? Physico-chemical characterisation of a triplex DNA-PNA/DNA/DNA. Biopolymers 2004, 73, 434–442. [Google Scholar]
- Greiner, B; Breipohl, G; Uhlmann, E. (2’-O-Methyl-RNA)-3’-PNA Chimeras: A New Class of Mixed Backbone Oligonucleotide Analogues with High Binding Affinity to RNA. Helv. Chim. Acta 2002, 85, 2619–2626. [Google Scholar]
- Turner, JJ; Jones, S; Fabani, MM; Ivanova, G; Arzumanov, AA; Gait, MJ. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells, Molec. and Dis. 2007, 38, 1–7. [Google Scholar]
- Van der Laan, C; Havenaar, P; Oosting, RS; Kuyl-Yheskiely, E; Uhlmann, E; van Boom, JH. Bioorg. Med. Lett. 1998, 8, 663–668.
- Breipohl, G; Will, DW; Peyman, A; Uhlmann, E. Novel synthetic routes to PNA monomers and PNA-DNA linker molecules. Tetrahedron 1997, 53(43), 14671–14686. [Google Scholar]
- O’Toole, AS; Miller, S; Serra, MJ. Stability of 3′ double nucleotide overhangs that model the 3′ ends of siRNA. RNA 2005, 11(4), 512–516. [Google Scholar]
- O’Toole, AS; Miller, S; Haines, N; Zink, MC; Serra, MJ. Comprehensive thermodynamic analysis of 3′ double-nucleotide overhangs neighboring Watson-Crick terminal base pairs. Nucleic Acids Res. 2006, 34(11), 3338–3344. [Google Scholar]
- Lingel, A; Simon, B; Izaurralde, E; Sattler, M. Nucleic acid 3′-end recognition by the Argonaute 2 PAZ domain. Nat. Struct. Mol. Biol. 2004, 11, 576–577. [Google Scholar]
- Ma, JB; Ye, K; Patel, DJ. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature 2004, 429, 318–322. [Google Scholar]
- Muller, S; Wolf, J; Ivanov, SA. Current strategies for the synthesis of RNA. Curr. Org. Synth. 2004, 1, 293–307. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | 5’UCGAAGUAUUCCGCGUACGTT(sense) |
| 3’TT AGCUUCAUAAGGCGCAUGC(antisense) | |
| B | 5’ CGAAGUAUUCCGCGUACGttGly (sense) |
| 3’TT AGCUUCAUAAGGCGCAUGC(antisense) | |
| C | 5’ UCGAAGUAUUCCGCGUACGTT(sense) |
| 3’Glytt AGCUUCAUAAGGCGCAUGC(antisense) | |
| D | 5’ UCGAAGUAUUCCGCGUACGttGly(sense) |
| 3’Glytt AGCUUCAUAAGGCGCAUGC(antisense) |
Share and Cite
Potenza, N.; Moggio, L.; Milano, G.; Salvatore, V.; Di Blasio, B.; Russo, A.; Messere, A. RNA Interference in Mammalia Cells by RNA-3’-PNA Chimeras. Int. J. Mol. Sci. 2008, 9, 299-315. https://doi.org/10.3390/ijms9030299
Potenza N, Moggio L, Milano G, Salvatore V, Di Blasio B, Russo A, Messere A. RNA Interference in Mammalia Cells by RNA-3’-PNA Chimeras. International Journal of Molecular Sciences. 2008; 9(3):299-315. https://doi.org/10.3390/ijms9030299
Chicago/Turabian StylePotenza, Nicoletta, Loredana Moggio, Giovanna Milano, Vincenzo Salvatore, Benedetto Di Blasio, Aniello Russo, and Anna Messere. 2008. "RNA Interference in Mammalia Cells by RNA-3’-PNA Chimeras" International Journal of Molecular Sciences 9, no. 3: 299-315. https://doi.org/10.3390/ijms9030299