Modulatory Effects of Polyphenols on Apoptosis Induction: Relevance for Cancer Prevention

{kind=link}
{kind=link}
Abstract
:1. Introduction
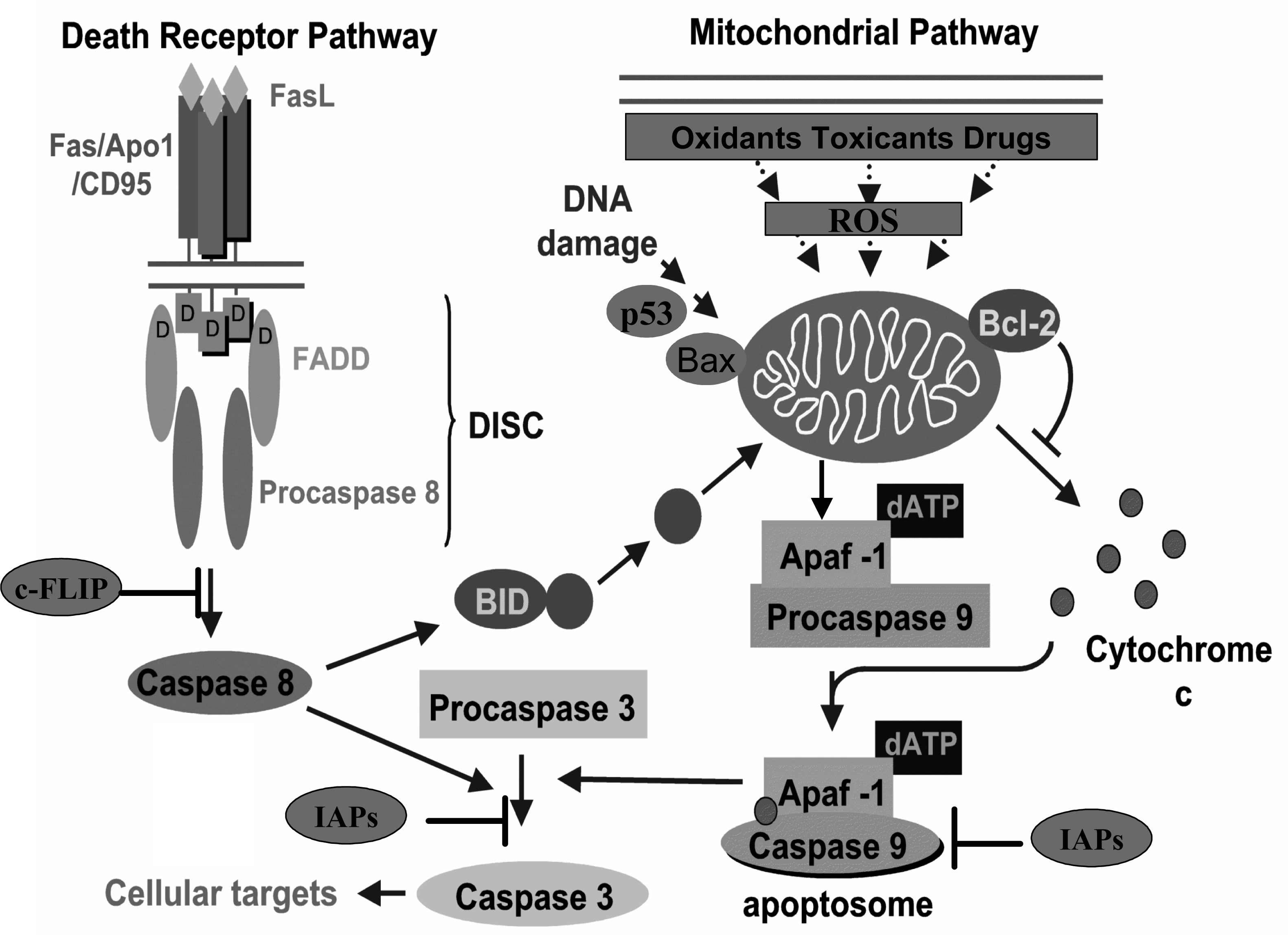
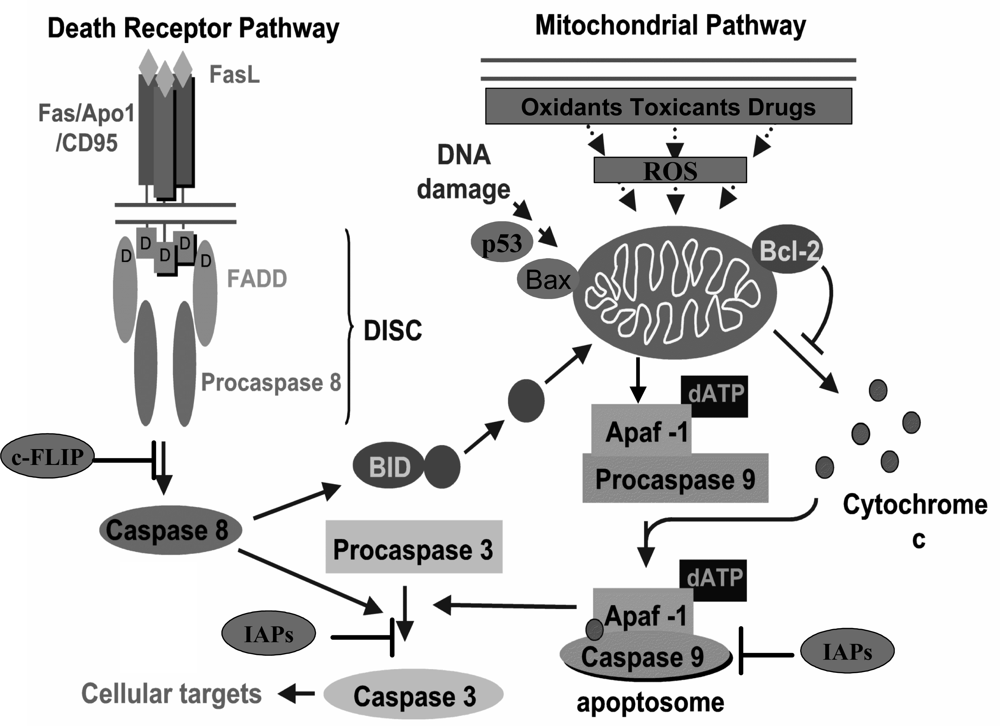
2. Apoptosis and cancer
3. Carcinogenesis and modulation of apoptosis by polyphenols
4. Conclusion
References
- Manach, C; Scalbert, A; Morand, C; Remesy, C; Jimenez, L. Polyphenols: food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79(5), 727–747. [Google Scholar]
- Benzie, IF; Strain, JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal. Biochem. 1996, 239(1), 70–76, DOI 10.1006/abio.1996.0292. [Google Scholar]
- Rice-Evans, CA; Miller, NJ; Bolwell, PG; Bramley, PM; Pridham, JB. The relative antioxidant activities of plant-derived polyphenolic flavonoids. Free Radic Res 1995, 22(4), 375–383. [Google Scholar]
- Scalbert, A; Manach, C; Morand, C; Remesy, C; Jimenez, L. Dietary polyphenols and the prevention of diseases. Crit Rev Food Sci Nutr 2005, 45(4), 287–306, DOI 10.1080/1040869059096. [Google Scholar]
- Casalini, C; Lodovici, M; Briani, C; Paganelli, G; Remy, S; Cheynier, V; Dolara, P. Effect of complex polyphenols and tannins from red wine (WCPT) on chemically induced oxidative DNA damage in the rat. Eur J Nutr 1999, 38(4), 190–195, DOI 10.1007/s003940050061. [Google Scholar]
- Giovannelli, L; Testa, G; De Filippo, C; Cheynier, V; Clifford, MN; Dolara, P. Effect of complex polyphenols and tannins from red wine on DNA oxidative damage of rat colon mucosa in vivo. Eur J Nutr 2000, 39(5), 207–212, DOI 10.1007/s003940070013. [Google Scholar]
- Takagi, A; Sai, K; Umemura, T; Hasegawa, R; Kurokawa, Y. Inhibitory effects of vitamin E and ellagic acid on 8-hydroxydeoxyguanosine formation in liver nuclear DNA of rats treated with 2-nitropropane. Cancer Lett 1995, 91(1), 139–144, DOI 10.1016/0304-3835(95)03734-E. [Google Scholar]
- Huber, WW; McDaniel, LP; Kaderlik, KR; Teitel, CH; Lang, NP; Kadlubar, FF. Chemoprotection against the formation of colon DNA adducts from the food-borne carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in the rat. Mutat. Res. 1997, 376, 115–122. [Google Scholar]
- Lodovici, M; Casalini, C; De Filippo, C; Copeland, E; Xu, X; Clifford, M; Dolara, P. Inhibition of 1,2-dimethylhydrazine-induced oxidative DNA damage in rat colon mucosa by black tea complex polyphenols. Food Chem Toxicol 2000, 38(12), 1085–1088, DOI 10.1016/S0278-5 6915(00)00109-5. [Google Scholar]
- Boyle, SP; Dobson, VL; Duthie, SJ; Kyle, JA; Collins, AR. Absorption and DNA protective effects of flavonoid glycosides from an onion meal. Eur J Nutr 2000, 39(5), 213–223, DOI 10.1007/s003940070014. [Google Scholar]
- Lampe, JW. Health effects of vegetables and fruit: assessing mechanisms of action in human experimental studies. Am. J. Clin. Nutr. 1999, 70, 475S–490S. [Google Scholar]
- Lean, ME; Noroozi, M; Kelly, I; Burns, J; Talwar, D; Sattar, N; Crozier, A. Dietary flavonols protect diabetic human lymphocytes against oxidative damage to DNA. Diabetes 1999, 48(1), 176–181, DOI 10.2337/diabetes.48.1.176. [Google Scholar]
- Leighton, F; Cuevas, A; Guasch, V; Perez, DD; Strobel, P; San Martin, A; Urzua, U; Diez, MS; Foncea, R; Castillo, O; Mizon, C; Espinoza, MA; Urquiaga, I; Rozowski, J; Maiz, A; Germain, A. lasma polyphenols and antioxidants, oxidative DNA damage and endothelial function in a diet and wine intervention study in humans. P. Drugs Exp Clin Res 1999, 25, 133–141. [Google Scholar]
- Masella, R; Di Benedetto, R; Vari, R; Filesi, C; Giovannini, C. Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione-related enzymes. J Nutr Biochem 2005, 16(10), 577–586, DOI 10.1016/j.jnutbio.2005.05.013. [Google Scholar]
- Bode, AM; Dong, Z. Targeting signal transduction pathways by chemopreventive agents. Mutat. Res. 2004, 555, 33–51. [Google Scholar]
- Kandaswami, C; Lee, LT; Lee, PP; Hwang, JJ; Ke, FC; Huang, YT; Lee, MT. The antitumor activities of flavonoids. In Vivo 2005, 19(5), 895–909. [Google Scholar]
- Thomasset, SC; Berry, DP; Garcea, G; Marczylo, T; Steward, WP; Gescher, AJ. Dietary polyphenolic phytochemicals--promising cancer chemopreventive agents in humans? A review of their clinical properties. Int. J. Cancer. 2007, 120(3), 451–458, DOI 10.1002/ijc.22419. [Google Scholar]
- Bode, AM; Dong, Z. Molecular and cellular targets. Mol Carcinog 2006, 45, 422–430, DOI 10.1002/mc.20222. [Google Scholar]
- Aggarwal, BB; Shishodia, S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharmacol. 2006, 71(10), 1397–1421, DOI 10.1016/j.bcp.2006.02.009. [Google Scholar]
- Na, HK; Surh, YJ. Intracellular signaling network as a prime chemopreventive target of (−)-epigallocatechin gallate. Mol. Nutr. Food Res. 2006, 50(2), 152–159, DOI 10.1002/mnfr.200500154. [Google Scholar]
- Prescott, SM; Fitzpatrick, FA. Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta 2000, 1470, M69–M78. [Google Scholar]
- Subbaramaiah, K; Dannenberg, AJ. Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol Sci 2003, 24(2), 96–102, DOI 10.1016/S0165-6147(02)00043-3. [Google Scholar]
- Turini, ME; DuBois, RN. Cyclooxygenase-2: a therapeutic target. Annu Rev Med 2002, 53, 35–57, DOI 10.1146/annurev.med.53.082901.103952. [Google Scholar]
- Oak, MH; El Bedoui, J; Schini-Kerth, VB. Antiangiogenic properties of natural polyphenols from red wine and green tea. J Nutr Biochem 2005, 16, 1–8, DOI 10.1016/j.jnutbio.2004.09.004. [Google Scholar]
- Shankar, S; Singh, G; Srivastava, RK. Chemoprevention by resveratrol: molecular mechanisms and therapeutic potential. Front Biosci 2007, 12, 4839–4854, DOI 10.2741/2432. [Google Scholar]
- Shankar, S; Ganapathy, S; Srivastava, RK. Green tea polyphenols: biology and therapeutic implications in cancer. Front Biosci 2007, 12, 4881–4899, DOI 10.2741/2435. [Google Scholar]
- Khan, N; Afaq, F; Mukhtar, H. Apoptosis by dietary factors: the suicide solution for delaying cancer growth. Carcinogenesis 2007, 28(2), 233–239, DOI 10.1093/carcin/bgl243. [Google Scholar]
- Ashkenazi, A; Dixit, VM. Death receptors: signaling and modulation. Science 1998, 281(5381), 1305–1308, DOI 10.1126/science.281.5381.1305. [Google Scholar]
- Antonsson, B; Martinou, JC. The Bcl-2 protein family. Exp Cell Res 2000, 256(1), 50–57, DOI 10.1006/excr.2000.4839. [Google Scholar]
- Del Principe, MI; Del Poeta, G; Venditti, A; Buccisano, F; Maurillo, L; Mazzone, C; Bruno, A; Neri, B; Irno Consalvo, M; Lo Coco, F; Amadori, S. Apoptosis and immaturity in acute myeloid leukemia. Hematology 2005, 10(1), 25–34, DOI 10.1080/10245330400020454. [Google Scholar]
- Roset, R; Ortet, L; Gil-Gomez, G. Role of Bcl-2 family members on apoptosis: what we have learned from knock-out mice. Front Biosci 2007, 12, 4722–4730, DOI 10.2741/2421. [Google Scholar]
- Baptiste, N; Friedlander, P; Chen, X; Prives, C. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 2002, 21(1), 9–21, DOI 10.1038/sj.onc.1205015. [Google Scholar]
- Bennett, M; Macdonald, K; Chan, SW; Luzio, JP; Simari, R; Weissberg, P. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science 1998, 282(5387), 290–293, DOI 10.1126/science.282.5387.290. [Google Scholar]
- Matas, D; Sigal, A; Stambolsky, P; Milyavsky, M; Weisz, L; Schwartz, D; Goldfinger, N; Rotter, V. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. Embo J 2001, 20(15), 4163–4172, DOI 10.1093/emboj/20.15.4163. [Google Scholar]
- Schuler, M; Bossy-Wetzel, E; Goldstein, JC; Fitzgerald, P; Green, DR. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J Biol Chem 2000, 275(10), 7337–7342, DOI 10.1074/jbc.275.10.7337. [Google Scholar]
- Chipuk, JE; Kuwana, T; Bouchier-Hayes, L; Droin, NM; Newmeyer, DD; Schuler, M; Green, DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303(5660), 1010–1014, DOI 10.1126/science.1092734. [Google Scholar]
- Trosko, JE. Dietary modulation of the multistage, multimechanisms of human carcinogenesis: effects on initiated stem cells and cell-cell communication. Nutr Cancer 2006, 54, 102–110, DOI 10.1207/s15327914nc5401_12. [Google Scholar]
- Porrini, M; Riso, P; Brusamolino, A; Berti, C; Guarnieri, S; Visioli, F. Daily intake of a formulated tomato drink affects carotenoid plasma and lymphocyte concentrations and improves cellular antioxidant protection. Br J Nutr 2005, 93(1), 93–99, DOI 10.1079/BJN20041315. [Google Scholar]
- Yao, LH; Jiang, YM; Shi, J; Tomas-Barberan, FA; Datta, N; Singanusong, R; Chen, SS. Flavonoids in food and their health benefits. Plant Foods Hum Nutr 2004, 59(3), 113–122, DOI 10.1007/s11130-004-0049-7. [Google Scholar]
- Rahman, I; Biswas, SK; Kirkham, PA. Regulation of inflammation and redox signaling by dietary polyphenols. Biochem Pharmacol 2006, 72, 1439–1452, DOI 10.1016/j.bcp.2006.07.004. [Google Scholar]
- Sang, S; Hou, Z; Lambert, JD; Yang, CS. Redox properties of tea polyphenols and related biological activities. Antioxid Redox Signal 2005, 7, 1704–1714, DOI 10.1089/ars.2005.7.1704. [Google Scholar]
- Ong, CS; Tran, E; Nguyen, TT; Ong, CK; Lee, SK; Lee, JJ; Ng, CP; Leong, C; Huynh, H. Quercetin-induced growth inhibition and cell death in nasopharyngeal carcinoma cells are associated with increase in Bad and hypophosphorylated retinoblastoma expressions. Oncol Rep 2004, 11(3), 727–733. [Google Scholar]
- Shimizu, M; Deguchi, A; Lim, JT; Moriwaki, H; Kopelovich, L; Weinstein, IB. (−)-Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor-2 signaling pathways in human colon cancer cells. Clin Cancer Res 2005, 11, 2735–2746. [Google Scholar]
- Selvendiran, K; Koga, H; Ueno, T; Yoshida, T; Maeyama, M; Torimura, T; Yano, H; Kojiro, M; Sata, M. Luteolin promotes degradation in signal transducer and activator of transcription 3 in human hepatoma cells: an implication for the antitumor potential of flavonoids. Cancer Res 2006, 66(9), 4826–4834, DOI 10.1158/0008-5472.CAN-05-4062. [Google Scholar]
- Michels, G; Watjen, W; Niering, P; Steffan, B; Thi, QH; Chovolou, Y; Kampkotter, A; Bast, A; Proksch, P; Kahl, R. Pro-apoptotic effects of the flavonoid luteolin in rat H4IIE cells. Toxicology 2005, 206(3), 337–348, DOI 10.1016/j.tox.2004.07.022. [Google Scholar]
- Kuo, PL; Lin, CC. Green tea constituent (−)-epigallocatechin-3-gallate inhibits Hep G2 cell proliferation and induces apoptosis through p53-dependent and Fas-mediated pathways. J Biomed Sci 2003, 10(2), 219–227. [Google Scholar]
- Lee, HJ; Wang, CJ; Kuo, HC; Chou, FP; Jean, LF; Tseng, TH. Induction apoptosis of luteolin in human hepatoma HepG2 cells involving mitochondria translocation of Bax/Bak and activation of JNK. Toxicol Appl Pharmacol 2005, 203(2), 124–131, DOI 10.1016/j.taap.2004.08.004. [Google Scholar]
- Gong, L; Li, Y; Nedeljkovic-Kurepa, A; Sarkar, FH. Inactivation of NF-kappaB by genistein is mediated via Akt signaling pathway in breast cancer cells. Oncogene 2003, 22(30), 4702–4709, DOI 10.1038/sj.onc.1206583. [Google Scholar]
- Wenzel, E; Somoza, V. Metabolism and bioavailability of trans-resveratrol. Mol Nutr Food Res 2005, 49(5), 472–481, DOI 10.1002/mnfr.200500010. [Google Scholar]
- Gusman, J; Malonne, H; Atassi, G. A reappraisal of the potential chemopreventive and chemotherapeutic properties of resveratrol. Carcinogenesis 2001, 22, 1111–1117, DOI 10.1093/carcin/22.8.1111. [Google Scholar]
- Jang, M; Cai, L; Udeani, GO; Slowing, KV; Thomas, CF; Beecher, CW; Fong, HH; Farnsworth, NR; Kinghorn, AD; Mehta, RG; Moon, RC; Pezzuto, JM. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar]
- Kelloff, GJ; Crowell, JA; Steele, VE; Lubet, RA; Malone, WA; Boone, CW; Kopelovich, L; Hawk, ET; Lieberman, R; Lawrence, JA; Ali, I; Viner, JL; Sigman, CC. Progress in cancer chemoprevention: development of diet-derived chemopreventive agents. J Nutr 2000, 130, 467S–471S. [Google Scholar]
- Wolter, F; Akoglu, B; Clausnitzer, A; Stein, J. Downregulation of the cyclin D1/Cdk4 complex occurs during resveratrol-induced cell cycle arrest in colon cancer cell lines. J Nutr 2001, 131(8), 2197–2203. [Google Scholar]
- Kim, YA; Lee, WH; Choi, TH; Rhee, SH; Park, KY; Choi, YH. Involvement of p21WAF1/CIP1, pRB, Bax and NF-kappaB in induction of growth arrest and apoptosis by resveratrol in human lung carcinoma A549 cells. Int J Oncol 2003, 23(4), 1143–1149. [Google Scholar]
- Gao, X; Xu, YX; Divine, G; Janakiraman, N; Chapman, RA; Gautam, SC. Disparate in vitro and in vivo antileukemic effects of resveratrol, a natural polyphenolic compound found in grapes. J Nutr 2002, 132(7), 2076–2081. [Google Scholar]
- Billard, C; Izard, JC; Roman, V; Kern, C; Mathiot, C; Mentz, F; Kolb, JP. Comparative antiproliferative and apoptotic effects of resveratrol, epsilon-viniferin and vine-shots derived polyphenols (vineatrols) on chronic B lymphocytic leukemia cells and normal human lymphocytes. Leuk Lymphoma 2002, 43(10), 1991–2002, DOI 10.1080/1042819021000015952. [Google Scholar]
- Joe, AK; Liu, H; Suzui, M; Vural, ME; Xiao, D; Weinstein, IB. Resveratrol induces growth inhibition, S-phase arrest, apoptosis, and changes in biomarker expression in several human cancer cell lines. Clin Cancer Res 2002, 8(3), 893–903. [Google Scholar]
- Clement, MV; Hirpara, JL; Chawdhury, SH; Pervaiz, S. Chemopreventive agent resveratrol, a natural product derived from grapes, triggers CD95 signaling-dependent apoptosis in human tumor cells. Blood 1998, 92(3), 996–1002. [Google Scholar]
- Surh, YJ; Hurh, YJ; Kang, JY; Lee, E; Kong, G; Lee, SJ. Resveratrol, an antioxidant present in red wine, induces apoptosis in human promyelocytic leukemia (HL-60) cells. Cancer Lett 1999, 140(1–2), 1–10. [Google Scholar]
- Kim, YA; Choi, BT; Lee, YT; Park, DI; Rhee, SH; Park, KY; Choi, YH. Resveratrol inhibits cell proliferation and induces apoptosis of human breast carcinoma MCF-7 cells. Oncol Rep 2004, 11(2), 441–446. [Google Scholar]
- Alkhalaf, M. Resveratrol-Induced Apoptosis Is Associated with Activation of p53 and Inhibition of Protein Translation in T47D Human Breast Cancer Cells. Pharmacology 2007, 80(2–3), 134–143. [Google Scholar]
- Shih, A; Davis, FB; Lin, HY; Davis, PJ. Resveratrol induces apoptosis in thyroid cancer cell lines via a MAPK- and p53-dependent mechanism. J Clin Endocrinol Metab 2002, 87(3), 1223–1232, DOI 10.1210/jc.87.3.1223. [Google Scholar]
- Signorelli, P; Ghidoni, R. Resveratrol as an anticancer nutrient: molecular basis, open questions and promises. J Nutr Biochem 2005, 16, 449–466, DOI 10.1016/j.jnutbio.2005.01.017. [Google Scholar]
- Lambert, JD; Hong, J; Yang, GY; Liao, J; Yang, CS. Inhibition of carcinogenesis by polyphenols: evidence from laboratory investigations. Am J Clin Nutr 2005, 81, 284S–291S. [Google Scholar]
- Chen, C; Shen, G; Hebbar, V; Hu, R; Owuor, ED; Kong, AN. Epigallocatechin-3-gallate-induced stress signals in HT-29 human colon adenocarcinoma cells. Carcinogenesis 2003, 24(8), 1369–1378, DOI 10.1093/carcin/bgg091. [Google Scholar]
- Hastak, K; Gupta, S; Ahmad, N; Agarwal, MK; Agarwal, ML; Mukhtar, H. Role of p53 and NF-kappaB in epigallocatechin-3-gallate-induced apoptosis of LNCaP cells. Oncogene 2003, 22(31), 4851–4859, DOI 10.1038/sj.onc.1206708. [Google Scholar]
- Aura, AM; Martin-Lopez, P; O'Leary, KA; Williamson, G; Oksman-Caldentey, KM; Poutanen, K; Santos-Buelga, C. In vitro metabolism of anthocyanins by human gut microflora. Eur J Nutr 2005, 44(3), 133–142, DOI 10.1007/s00394-004-0502-2. [Google Scholar]
- Masella, R; Cantafora, A; Modesti, D; Cardilli, A; Gennaro, L; Bocca, A; Coni, E. Antioxidant activity of 3,4-DHPEA-EA and protocatechuic acid: a comparative assessment with other olive oil biophenols. Redox Rep 1999, 4(3), 113–121, DOI 10.1179/135100099101534792. [Google Scholar]
- Hudson, EA; Dinh, PA; Kokubun, T; Simmonds, MS; Gescher, A. Characterization of potentially chemopreventive phenols in extracts of brown rice that inhibit the growth of human breast and colon cancer cells. Cancer Epidemiol Biomarkers Prev 2000, 9(11), 1163–1170. [Google Scholar]
- Yen, GC; Hsieh, CL. Reactive oxygen species scavenging activity of Du-zhong (Eucommia ulmoides oliv.) and its active compounds. J Agric Food Chem 2000, 48, 3431–3436. [Google Scholar]
- Lin, HH; Chen, JH; Huang, CC; Wang, CJ. Apoptotic effect of 3,4-dihydroxybenzoic acid on human gastric carcinoma cells involving JNK/p38 MAPK signaling activation. Int J Cancer 2007, 120(11), 2306–2316, DOI 10.1002/ijc.22571. [Google Scholar]
- Watabe, M; Hishikawa, K; Takayanagi, A; Shimizu, N; Nakaki, T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J Biol Chem 2004, 279(7), 6017–6026, DOI 10.1074/jbc.M306040200. [Google Scholar]
- Kurata, R; Adachi, M; Yamakawa, O; Yoshimoto, M. Growth suppression of human cancer cells by polyphenolics from sweetpotato (Ipomoea batatas L.) leaves. J Agric Food Chem 2007, 55, 185–190, DOI 10.1021/jf0620259. [Google Scholar]
- Lee, YJ; Kuo, HC; Chu, CY; Wang, CJ; Lin, WC; Tseng, TH. Involvement of tumor suppressor protein p53 and p38 MAPK in caffeic acid phenethyl ester-induced apoptosis of C6 glioma cells. Biochem Pharmacol 2003, 66(12), 2281–2289, DOI 10.1016/j.bcp.2003.07.014. [Google Scholar]
- Thatte, U; Bagadey, S; Dahanukar, S. Modulation of programmed cell death by medicinal plants. Cell Mol Biol (Noisy-le-grand) 2000, 46(1), 199–214. [Google Scholar]
- Keum, YS; Kim, J; Lee, KH; Park, KK; Surh, YJ; Lee, JM; Lee, SS; Yoon, JH; Joo, SY; Cha, IH; Yook, JI. Induction of apoptosis and caspase-3 activation by chemopreventive [6]-paradol and structurally related compounds in KB cells. Cancer Lett 2002, 177(1), 41–47, DOI 10.1016/S0304-3835(01)00781-9. [Google Scholar]
- Miyoshi, N; Nakamura, Y; Ueda, Y; Abe, M; Ozawa, Y; Uchida, K; Osawa, T. Dietary ginger constituents, galanals A and B, are potent apoptosis inducers in Human T lymphoma Jurkat cells. Cancer Lett 2003, 199(2), 113–119, DOI 10.1016/S0304-3835(03)00381-1. [Google Scholar]
- Shishodia, S; Amin, HM; Lai, R; Aggarwal, BB. Curcumin (diferuloylmethane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem Pharmacol 2005, 70(5), 700–713, DOI 10.1016/j.bcp.2005.04.043. [Google Scholar]
- Bharti, AC; Donato, N; Singh, S; Aggarwal, BB. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 2003, 101(3), 1053–1062, DOI 10.1182/blood-2002-05-1320. [Google Scholar]
- Shankar, S; Srivastava, RK. Involvement of Bcl-2 family members, phosphatidylinositol 3′-kinase/AKT and mitochondrial p53 in curcumin (diferulolylmethane)-induced apoptosis in prostate cancer. Int J Oncol 2007, 30(4), 905–918. [Google Scholar]
- Loo, G. Redox-sensitive mechanisms of phytochemical-mediated inhibition of cancer cell proliferation (review). J Nutr Biochem 2003, 14(2), 64–73, DOI 10.1016/S0955-2863(02)00251-6. [Google Scholar]
- Sun, Y; Oberley, LW. Redox regulation of transcriptional activators. Free Radic Biol Med 1996, 21(3), 335–348, DOI 10.1016/0891-5849(96)00109-8. [Google Scholar]
- Martin, KR. Targeting apoptosis with dietary bioactive agents. Exp Biol Med (Maywood) 2006, 231(2), 117–129. [Google Scholar]
- Cowan, KJ; Storey, KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol 2003, 206(7), 1107–1115. [Google Scholar]
- Cross, TG; Scheel-Toellner, D; Henriquez, NV; Deacon, E; Salmon, M; Lord, JM. Serine/threonine protein kinases and apoptosis. Exp Cell Res 2000, 256(1), 34–41, DOI 10.1006/excr.2000.4836. [Google Scholar]
- Ramos, S. Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. J Nutr Biochem 2007, 18(7), 427–442, DOI 10.1016/j.jnutbio.2006.11.004. [Google Scholar]
- Manna, SK; Mukhopadhyay, A; Aggarwal, BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol 2000, 164(12), 6509–6519. [Google Scholar]
- Yu, R; Hebbar, V; Kim, DW; Mandlekar, S; Pezzuto, JM; Kong, AN. Resveratrol inhibits phorbol ester and UV-induced activator protein 1 activation by interfering with mitogen-activated protein kinase pathways. Mol Pharmacol 2001, 60(1), 217–224. [Google Scholar]
- Katiyar, SK; Afaq, F; Azizuddin, K; Mukhtar, H. Inhibition of UVB-induced oxidative stress-mediated phosphorylation of mitogen-activated protein kinase signaling pathways in cultured human epidermal keratinocytes by green tea polyphenol (minus;)-epigallocatechin-3-gallate. Toxicol Appl Pharmacol 2001, 176(2), 110–107, DOI 10.1006/taap.2001.9276. [Google Scholar]
- Yin, F; Giuliano, AE; Van Herle, AJ. Signal pathways involved in apigenin inhibition of growth and induction of apoptosis of human anaplastic thyroid cancer cells (ARO). Anticancer Res 1999, 19, 4297–4303. [Google Scholar]
- Long, LH; Clement, MV; Halliwell, B. Artifacts in cell culture: rapid generation of hydrogen peroxide on addition of (−)-epigallocatechin(−)-epigallocatechin gallate(+)-catechin, and quercetin to commonly used cell culture media. Biochem Biophys Res Commun 2000, 273(1), 50–53, DOI 10.1006/bbrc.2000.2895. [Google Scholar]
- Yang, GY; Liao, J; Li, C; Chung, J; Yurkow, EJ; Ho, CT; Yang, CS. Effect of black and green tea polyphenols on c-jun phosphorylation and H(2)O(2) production in transformed and non-transformed human bronchial cell lines: possible mechanisms of cell growth inhibition and apoptosis induction. Carcinogenesis 2000, 21(11), 2035–2039, DOI 10.1093/carcin/21.11.2035. [Google Scholar]
- Sakagami, H; Arakawa, H; Maeda, M; Satoh, K; Kadofuku, T; Fukuchi, K; Gomi, K. Production of hydrogen peroxide and methionine sulfoxide by epigallocatechin gallate and antioxidants. Anticancer Res 2001, 21, 2633–2641. [Google Scholar]
- Chen, C; Yu, R; Owuor, ED; Kong, AN. Activation of antioxidant-response element (ARE), mitogen-activated protein kinases (MAPKs) and caspases by major green tea polyphenol components during cell survival and death. Arch Pharm Res 2000, 23(6), 605–612. [Google Scholar]
- Chung, JH; Han, JH; Hwang, EJ; Seo, JY; Cho, KH; Kim, KH; Youn, JI; Eun, HC. Dual mechanisms of green tea extract (EGCG)-induced cell survival in human epidermal keratinocytes. Faseb J 2003, 17, 1913–1915. [Google Scholar]
- Brusselmans, K; De Schrijver, E; Heyns, W; Verhoeven, G; Swinnen, JV. Epigallocatechin-3-gallate is a potent natural inhibitor of fatty acid synthase in intact cells and selectively induces apoptosis in prostate cancer cells. Int J Cancer 2003, 106(6), 856–862, DOI 10.1002/ijc.11317. [Google Scholar]
- Chen, ZP; Schell, JB; Ho, CT; Chen, KY. Green tea epigallocatechin gallate shows a pronounced growth inhibitory effect on cancerous cells but not on their normal counterparts. Cancer Lett 1998, 129(2), 173–179, DOI 10.1016/S0304-3835(98)00108-6. [Google Scholar]
- Kawai, K; Tsuno, NH; Kitayama, J; Okaji, Y; Yazawa, K; Asakage, M; Sasaki, S; Watanabe, T; Takahashi, K; Nagawa, H. Epigallocatechin gallate induces apoptosis of monocytes. J Allergy Clin Immunol 2005, 115(1), 186–191, DOI 10.1016/j.jaci.2004.10.005. [Google Scholar]
- Ahmad, N; Feyes, DK; Nieminen, AL; Agarwal, R; Mukhtar, H. Green tea constituent epigallocatechin-3-gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. J Natl Cancer Inst 1997, 89(24), 1881–1886, DOI 10.1093/jnci/89.24.1881. [Google Scholar]
- Hsu, S; Bollag, WB; Lewis, J; Huang, Q; Singh, B; Sharawy, M; Yamamoto, T; Schuster, G. Green tea polyphenols induce differentiation and proliferation in epidermal keratinocytes. J Pharmacol Exp Ther 2003, 306(1), 29–34, DOI 10.1124/jpet.103.049734. [Google Scholar]
- Tan, X; Hu, D; Li, S; Han, Y; Zhang, Y; Zhou, D. Differences of four catechins in cell cycle arrest and induction of apoptosis in LoVo cells. Cancer Lett 2000, 158, 1–6. [Google Scholar]
- Pantuck, AJ; Leppert, JT; Zomorodian, N; Aronson, W; Hong, J; Barnard, RJ; Seeram, N; Liker, H; Wang, H; Elashoff, R; Heber, D; Aviram, M; Ignarro, L; Belldegrun, A. Phase II study of pomegranate juice for men with rising prostate-specific antigen following surgery or radiation for prostate cancer. Clin Cancer Res 2006, 12, 4018–4026, DOI 10.1158/1078-0432.CCR-05-2290. [Google Scholar]
- Seeram, NP; Aronson, WJ; Zhang, Y; Henning, SM; Moro, A; Lee, RP; Sartippour, M; Harris, DM; Rettig, M; Suchard, MA; Pantuck, AJ; Belldegrun, A; Heber, D. Pomegranate ellagitannin-derived metabolites inhibit prostate cancer growth and localize to the mouse prostate gland. J Agric Food Chem 2007, 55, 7732–7737. [Google Scholar]
- Siddiqui, IA; Saleem, M; Adhami, VM; Asim, M; Mukhtar, H. Tea beverage in chemoprevention and chemotherapy of prostate cancer. Acta Pharmacol Sin 2007, 28, 1392–1408, DOI 10.1111/j.1745-7254.2007.00693.x. [Google Scholar]
Share and Cite
D’Archivio, M.; Santangelo, C.; Scazzocchio, B.; Varì, R.; Filesi, C.; Masella, R.; Giovannini, C. Modulatory Effects of Polyphenols on Apoptosis Induction: Relevance for Cancer Prevention. Int. J. Mol. Sci. 2008, 9, 213-228. https://doi.org/10.3390/ijms9030213
D’Archivio M, Santangelo C, Scazzocchio B, Varì R, Filesi C, Masella R, Giovannini C. Modulatory Effects of Polyphenols on Apoptosis Induction: Relevance for Cancer Prevention. International Journal of Molecular Sciences. 2008; 9(3):213-228. https://doi.org/10.3390/ijms9030213
Chicago/Turabian StyleD’Archivio, Massimo, Carmela Santangelo, Beatrice Scazzocchio, Rosaria Varì, Carmela Filesi, Roberta Masella, and Claudio Giovannini. 2008. "Modulatory Effects of Polyphenols on Apoptosis Induction: Relevance for Cancer Prevention" International Journal of Molecular Sciences 9, no. 3: 213-228. https://doi.org/10.3390/ijms9030213