Protein Folding and Misfolding on Surfaces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
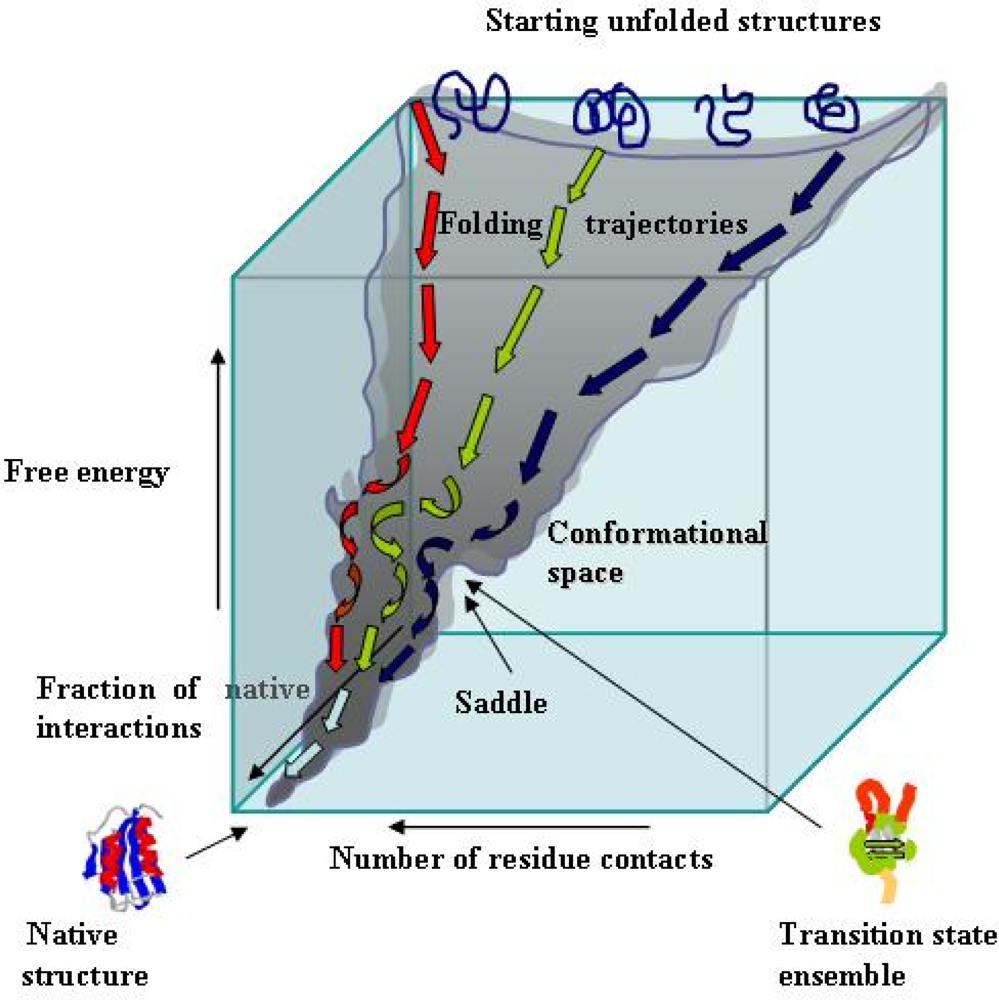
2. Essentials of protein folding
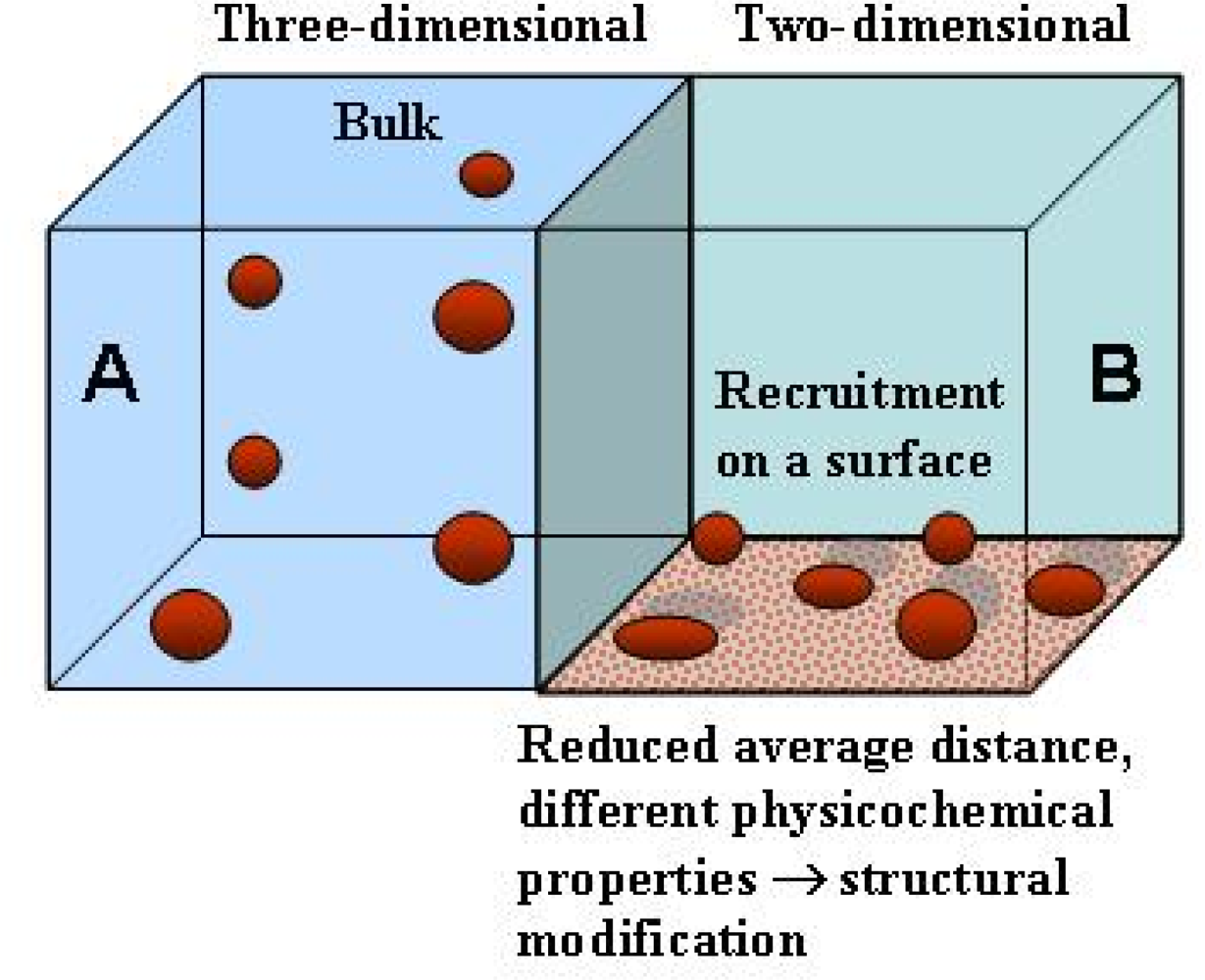
3. Protein folding on surfaces
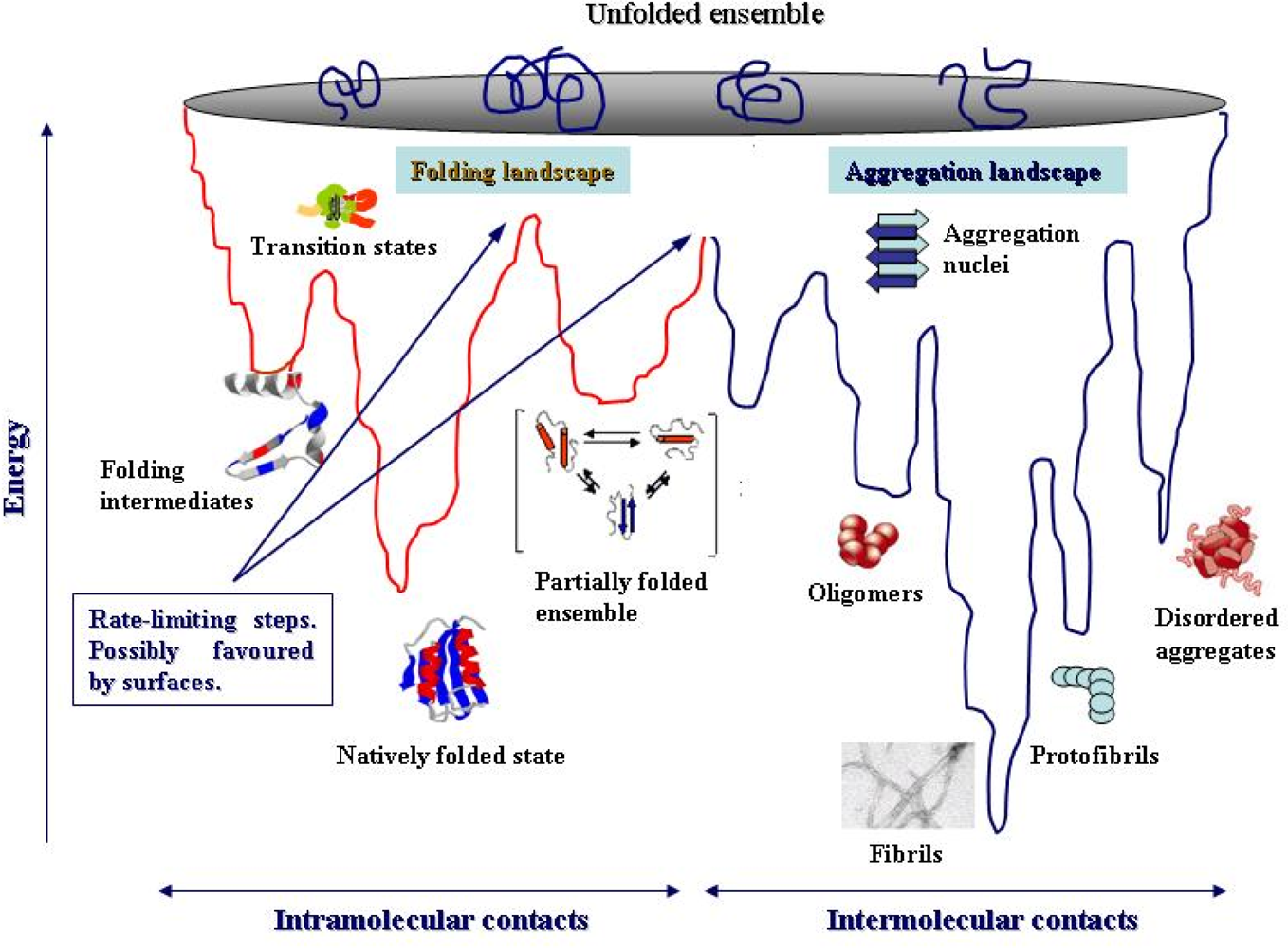
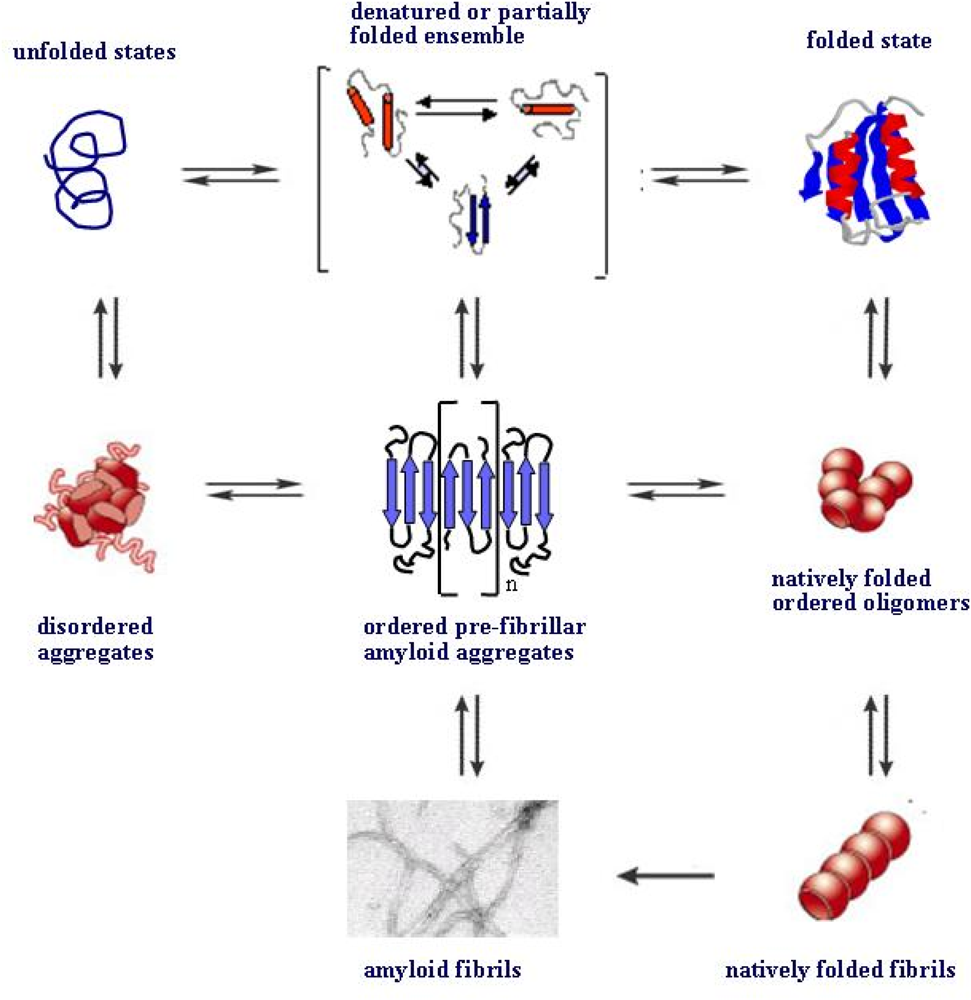
4. Protein folding and aggregation are competing pathways
5. Surfaces can favour protein unfolding/misfolding
6. Biological surfaces are primary sites of amyloid interaction and toxicity
7. Conclusions
Acknowledgments
References
- Reilly, MM. Genetically determined neuropathies. J. Neurol 1998, 245, 6–13. [Google Scholar]
- Kelly, J. Alternative conformation of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct.Biol 1998, 8, 101–106. [Google Scholar]
- Dobson, CM. The structural basis of protein folding and its links with human disease. Phil. Trans. R. Soc. Lond 2001, B 356, 133–145. [Google Scholar]
- Stefani, M; Dobson, CM. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med 2003, 81, 768–699. [Google Scholar]
- Bader, R; Bamford, R; Zurdo, J; Luisi, B; Dobson, CM. Probing the mechanism of amyloidogenesis through a tandem repeat of the PI3-SH3 domain suggests a generic model for protein aggregation and fibril formation. J. Mol. Biol 2006, 356, 189–208. [Google Scholar]
- Serpell, LC; Sunde, M; Benson, MD; Tennent, GA; Pepys, MB; Fraser, PE. The protofilament substructure of amyloid fibrils. J. Mol. Biol 2000, 300, 1033–1039. [Google Scholar]
- Sunde, M; Serpell, LC; Bartham, M; Fraser, PE; Pepys, MB; Blake, CF. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol 1997, 273, 729–739. [Google Scholar]
- Nelson, R; Sawaya, MR; Balbirnie, M; Madsen, AØ; Riekel, C; Grothe, R; Eisenberg, D. Structure of the cross-β pine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar]
- Tycko, R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr. Opin. Struct. Biol 2004, 14, 96–103. [Google Scholar]
- Fowler, DM; Koulov, AV; Alory-Jost, C; Marks, MS; Balch, WE; Kelly, JW. Functional amyloid formation within mammalian tissue. PLOS Biol 2006, 4, 1–8. [Google Scholar]
- Relini, A; Torrassa, S; Rolandi, R; Gliozzi, A; Rosano, C; Canale, C; Bolognesi, M; Plakoutsi, G; Bucciantini, M; Chiti, F; Stefani, M. Monitoring the process of HypF fibrillization and liposome permeabilization by protofibrils. J. Mol. Biol 2004, 338, 943–957. [Google Scholar]
- Lashuel, HA; Petre, BM; Wall, J; Simon, M; Nowak, RJ; Walz, T; Lansbury, PT. α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol 2002, 322, 1089–1102. [Google Scholar]
- Poirier, MA; Li, H; Macosko, J; Cail, S; Amzel, M; Ross, CA. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrillization. J. Biol. Chem 2002, 277, 41032–41037. [Google Scholar]
- Quintas, A; Vaz, DC; Cardoso, I; Saraiva, MJM; Brito, RMM. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J. Biol. Chem 2001, 276, 27207–27213. [Google Scholar]
- Hoshi, M; Sato, M; Matsumoto, S; Noguchi, A; Yasutake, K; Yoshida, N; Sato, K. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar]
- Chung, J; Yang, H; de Beus, MD; Ryu, CY; Cho, K; Colón, W. Cu/Zn superoxide dismutase can form pore-like structures. Biochem. Biophys. Res. Commun 2003, 312, 873–876. [Google Scholar]
- Dickson, DW. Correlation of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging 1995, 16, 285–298. [Google Scholar]
- Hartley, D; Walsh, DM; Ye, CP; Diehl, T; Vasquez, S; Vassilev, PM. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci 1999, 19, 8876–8884. [Google Scholar]
- Sitia, R; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar]
- Langer, T; Kaser, M; Klanner, C; Leonhard, K. AAA proteases of mitochondria: Quality control of membrane proteins and regulatory function during mitochondrial biogenesis. Biochem. Soc. Trans 2001, 29, 431–436. [Google Scholar]
- Goldberg, AL. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar]
- Schubert, U; Anton, LC; Gibbs, J; Norbury, CC; Yewdell, JW; Bennink, JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar]
- Levinthal, C. Are there pathways for protein folding? J. Chem. Phys 1968, 85, 44–45. [Google Scholar]
- Onochuc, JN; Wolynes, PG. Theory of protein folding. Curr. Opin. Struct. Biol 2004, 14, 70–75. [Google Scholar]
- Dobson, CM. Protein folding and misfolding. Nature 2003, 426, 884–891. [Google Scholar]
- Uversky, VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci 2002, 11, 739–756. [Google Scholar]
- Ellis, RJ. Macromolecular crowding: An important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol 2001, 11, 114–119. [Google Scholar]
- Bychkova, VE; Pain, RH; Ptitsyn, OB. The molten globule state is involved in the translocation of protein across membranes. FEBS Lett 1988, 238, 231–234. [Google Scholar]
- Hartl, FU; Hayer-Hartl, M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar]
- Rousseau, F; Serrano, L; Schymkowitz, JWH. How evolutionary pressure against protein aggregation shaped chaperone specificity. J. Mol. Biol 2006, 355, 1037–1047. [Google Scholar]
- Romero, P; Obradovich, Z; Kissinger, C; Villafranca, JE; Garner, E; Guilliot, S; Bunker, AK. Thousands of proteins likely to have long disordered regions. Pac. Symp. Biocomput 1998, 3, 437–448. [Google Scholar]
- Minton, AP. Influence of macromolecular crowding on intracellular association reactions: Possible role in volume regulation. In Cellular and Molecular Physiology of Cell Volume Regulation; Strange, K, Ed.; CRC Press: Boca Raton, Fl, 1994; pp. 181–190. [Google Scholar]
- Wright, PE; Dyson, HJ. Intrinsically unstructured proteins: reassessing the protein structure-function paradigm. J. Mol. Biol 1999, 293, 321–331. [Google Scholar]
- Dedmon, MM; Patel, CN; Young, GB; Pielak, GJ. FlgM gains structure in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 12681–12684. [Google Scholar]
- Gujiarro, JI; Sunde, M; Jones, JA; Campbell, ID; Dobson, CM. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA 1998, 95, 4224–4228. [Google Scholar]
- Litvinovich, SV; Brew, SA; Aota, S; Akiyama, SK; Haudenschild, C; Ingham, KC. Formation of amyloid-like fibrils by self-association of a partially unfolded fibronectin type III module. J. Mol. Biol 1998, 280, 245–258. [Google Scholar]
- Wiseman, RL; Powers, ET; Kelly, JW. Partitioning conformational intermediates between competing refolding and aggregation pathways: Insights into transthyretin amyloid disease. Biochemistry 2005, 44, 16612–16623. [Google Scholar]
- Chiti, F; Stefani, M; Taddei, N; Ramponi, G; Dobson, CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar]
- Jahn, TT; Redford, SE. Folding versus aggregation: polypeptide conformations on competing pathways. Arch. Biochem. Biophys 2008, 469, 100–117. [Google Scholar]
- Kaylor, J; Bodner, N; Edridge, S; Yamin, G; Hong, DP; Fink, AL. Characterization of oligomeric intermediates in alpha-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F alpha-synuclein. J. Mol. Biol 2005, 353, 357–372. [Google Scholar]
- Dusa, A; Kaylor, J; Edridge, S; Bodner, N; Hong, DP; Fink, AL. Characterization of oligomers during alpha-synuclein aggregation using intrinsic tryptophan fluorescence. Biochemistry 2006, 45, 2752–2760. [Google Scholar]
- Petkova, AT; Leapman, RD; Guo, Z; Yau, W-M; Mattson, MP; Tycko, R. Self-propagating, molecular level polymorphism in Alzheimer’s β-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar]
- Colon, W; Kelly, JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 1992, 31, 8654–8660. [Google Scholar]
- Chiti, F; Webster, P; Taddei, N; Clark, A; Stefani, M; Ramponi, G; Dobson, CM. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA 1999, 96, 3590–3594. [Google Scholar]
- McParland, VJ; Kad, NM; Kalverda, AP; Brown, A; Kirwin-Jones, P; Hunter, MG; Sunde, M; Radford, SE. Partially unfolded states of beta(2)-microglobulin and amyloid formation in vitro. Biochemistry 2000, 39, 8735–8746. [Google Scholar]
- Apetri, AC; Surewicz, K; Surewicz, WK. The effect of disease-associated mutations on the folding pathway of human prion protein. J. Biol. Chem 2004, 279, 18008–18014. [Google Scholar]
- Uversky, VN; Li, J; Fink, AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem 2001, 276, 10737–10744. [Google Scholar]
- Dumoulin, M; Camet, D; Last, AM; Pardon, E; Archer, S; Muyldermans, S; Wyns, L; Matagne, A; Robinson, CV; Redfield, C; Dobson, CM. Reduced global cooperativity is a common feature underlying the amyloidogenicity of pathogenic lysozyme mutations. J. Mol. Biol 2005, 346, 773–788. [Google Scholar]
- Harper, JD; Lansbury, PT. Models of amyloid seeding in Alzheimer's disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem 1997, 66, 385–407. [Google Scholar]
- Modler, AJ; Gast, K; Lutsch, G; Damaschun, G. Assembly of amyloid protofibrils via critical oligomers--A novel pathway of amyloid formation. J. Mol. Biol 2003, 325, 135–148. [Google Scholar]
- Gosal, WS; Morten, LJ; Hewitt, EW; Smith, DA; Thomson, NH; Radford, SE. Competing pathways determine fibril morphology in the self-assembly of beta2-microglobulin into amyloid. J. Mol. Biol 2005, 351, 850–864. [Google Scholar]
- Smith, AM; Jahn, TR; Ashcroft, AE; Radford, SE. Direct observation of oligomeric species formed in the early stages of amyloid fibril formation using electrospray ionisation mass spectrometry. J. Mol. Biol 2006, 364, 9–19. [Google Scholar]
- Bitan, G; Kirkitadze, MD; Lomakin, A; Volles, SS; Benedek, GB; Teplow, DB. Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar]
- Baskakov, IV; Legname, G; Baldwin, MA; Prusiner, SB; Cohen, FE. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem 2002, 277, 21140–21148. [Google Scholar]
- Necula, M; Kayed, R; Milton, S; Glabe, CG. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem 2007, 282, 10311–10324. [Google Scholar]
- Kad, NM; Myers, SL; Smith, DP; Smith, DA; Radford, SE; Thomson, NH. Hierarchical assembly of beta2-microglobulin amyloid in vitro revealed by atomic force microscopy. J. Mol. Biol 2003, 330, 785–797. [Google Scholar]
- McParland, VJ; Kalverda, AP; Homans, SW; Radford, SE. Structural properties of an amyloid precursor of beta(2)-microglobulin. Nat. Struct. Biol 2002, 9, 326–331. [Google Scholar]
- Serag, AA; Altenbach, C; Gingery, M; Hubbell, WL; Yeates, TO. Arrangement of subunits and ordering of beta-strands in an amyloid sheet. Nat. Struct. Biol 2002, 9, 734–739. [Google Scholar]
- Guo, Z; Eisenberg, D. Runaway domain swapping in amyloid-like fibrils of T7 endonuclease I. Proc. Natl. Acad. Sci. USA 2006, 103, 8042–8047. [Google Scholar]
- Rousseau, F; Wilkinson, H; Villanueva, J; Serrano, L; Schymkowitz, JW; Itzhaki, LS. Domain swapping in p13suc1 results in formation of native-like, cytotoxic aggregates. J. Mol. Biol 2006, 363, 496–505. [Google Scholar]
- Lomas, DA; Carrell, RW. Serpinopathies and the conformational dementias. Nat. Rev. Genet 2002, 3, 759–768. [Google Scholar]
- Bousset, L; Thomson, NH; Radford, SE; Melki, R. The yeast prion Ure2p retains its native alpha-helical conformation upon assembly into protein fibrils in vitro. EMBO J 2002, 21, 2903–2911. [Google Scholar]
- Plakoutsi, G; Bemporad, F; Calamai, M; Taddei, N; Dobson, CM; Chiti, F. Evidence for a Mechanism of Amyloid Formation Involving Molecular Reorganisation within Native-like Precursor Aggregates. J. Mol. Biol 2005, 351, 910–922. [Google Scholar]
- Bokvist, M; Lindström, F; Watts, A; Gröbner, G. Two types of Alzheimer’s β-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol 2004, 335, 1039–1049. [Google Scholar]
- Yip, CM; Elton, EA; Darabie, AA; Morrison, MR; McLaurin, J. Cholesterol, a modulator of membrane-associated Aβ-fibrillogenesis and neurotoxicity. J. Mol. Biol 2001, 311, 723–734. [Google Scholar]
- Kazlauskaite, J; Senghera, N; Sylvester, I; Vénien-Bryan, C; Pinheiro, TJT. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 2003, 42, 3295–3304. [Google Scholar]
- Porat, Y; Kolusheva, S; Jelinek, R; Gazit, E. The human islet amyloid polypeptide forms transient membrane-active prefibrillar assemblies. Biochemistry 2003, 42, 10971–10977. [Google Scholar]
- Linse, S; Cabaleiro-Lago, C; Xue, W-F; Lynch, I; Lindman, S; Thulin, E; Radford, SE; Dawson, KA. Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 8691–8696. [Google Scholar]
- Zhu, M; Souillac, PO; Ionesco-Zanetti, C; Carter, SA; Fink, AL. Surface-catalyzed amyloid fibril formation. J. Biol. Chem 2002, 277, 50914–50922. [Google Scholar]
- Necula, M; Chirita, C; Kuret, J. Rapid anionic micelle-mediated α-synuclein fibrillization in vitro. J. Biol. Chem 2003, 278, 46674–46680. [Google Scholar]
- Zhao, H; Jutila, A; Nurminen, T; Wickström, SA; Keski-Oja, J; Kinnunen, PKJ. Binding of endostatin to phosphatidylserine-containing membranes and formation of amyloid-like fibers. Biochemistry 2005, 44, 2857–2863. [Google Scholar]
- Zhao, H; Tuominen, EKJ; Kinnunen, PKJ. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry 2004, 43, 10302–10307. [Google Scholar]
- Arispe, N; Rojas, E; Pollard, HD. Alzheimer’s disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminium. Proc. Natl Acad. Sci. USA 1993, 89, 10940–10944. [Google Scholar]
- Mirzabekov, TA; Lin, MC; Kagan, BL. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem 1996, 271, 1988–1992. [Google Scholar]
- Lin, MC; Mirzabekov, T; Kagan, BL. Channel formation by a neurotoxic prion protein fragment. J. Biol. Chem 1997, 272, 44–47. [Google Scholar]
- Kourie, JI. Synthetic C-type mammalian natriuretic peptide forms large cation selective channels. FEBS Lett 1999, 445, 57–62. [Google Scholar]
- Lin, M-cA; Kagan, B. Electrophysiologic properties of channels induced by Aβ25-35 in planar lipid bilayers. Peptides 2002, 23, 1215–1228. [Google Scholar]
- Volles, MJ; Lansbury, PT. Vesicle permeabilization by protofibrillar α-synuclein: comparison of wild-type with Parkinson’s disease linked mutants and insights in the mechanisms. Biochemistry 2001, 40, 7812–7819. [Google Scholar]
- Ding, TT; Lee, S-J; Rochet, J-C; Lansbury, PT. Annular α-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 2002, 41, 10209–10217. [Google Scholar]
- Hirakura, Y; Carreras, I; Sipe, JD; Kagan, BL. Channel formation by serum amyloid A: A potential mechanism for amyloid pathogenesis and host defense. Amyloid 2002, 9, 13–23. [Google Scholar]
- Hou, X; Richardson, SJ; Aguilar, MI; Small, DH. Binding of amyloidogenic transthyretin to the plasma membrane alters membrane fluidity and induces neurotoxicity. Biochemistry 2005, 44, 11618–11627. [Google Scholar]
- Mattson, MP. Impairment of membrane transport and signal transduction systems by amyloidogenic proteins. Methods Enzymol 1999, 309, 733–768. [Google Scholar]
- Stefani, M. Generic cell dysfunction in neurodegenerative disorders: role of surfaces in early protein misfolding, aggregation, and aggregate cytotoxicity. Neuroscientist 2007, 13, 519–531. [Google Scholar]
- Alsenbrey, C; Borowik, T; Byström, R; Bokvist, M; Lindström, F; Misiak, H; Sani, M-A; Gröbner, G. How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur. Biophys. J 2008, 37, 247–255. [Google Scholar]
- Sethuraman, A; Belfort, G. Protein structural perturbation and aggregation on homogeneous surfaces. Biophys. J 2005, 88, 1322–1333. [Google Scholar]
- Kourie, JI; Henry, CL. Ion channel formation and membrane-linked pathologies of misfolded hydrophobic proteins: the role of dangerous unchaperoned molecules. Clin. Exp. Pharmacol. Physiol 2002, 29, 741–753. [Google Scholar]
- Engel, MFM; Yiggitop, HA; Eigersma, RC; Rijkers, DTS; Liskamp, RMJ; de Kruijff, B; Höppener, JWM; Killian, A. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol 2006, 356, 783–789. [Google Scholar]
- Homma, N. Collagen-binding affinity of B2-microglobulin, a preprotein of hemodialysis-associated amyloidosis. Nephron 1998, 53, 37–40. [Google Scholar]
- Giorgetti, S; Rossi, A; Mangione, P; Raimondi, S; Marini, S; Stoppini, M; Corazza, A; Viglino, P; Esposito, G; Cetta, G; Merlini, G; Bellotti, V. Beta2-microglobulin isoforms display an heterogeneous affinity for type I collagen. Protein Sci 2005, 14, 696–702. [Google Scholar]
- Relini, A; Canale, C; De Stefano, S; Rolandi, R; Giorgetti, S; Stoppini, M; Rossi, A; Fogolari, F; Corazza, A; Esposito, G; Gliozzi, A; Bellotti, V. Collagen plays an active role in the aggregation of beta2-microglobulin under physiopathological conditions of dialysis-related amyloidosis. J. Biol. Chem 2006, 281, 16521–16529. [Google Scholar]
- Suk, JY; Zhang, F; Balch, WE; Linhardt, RJ; Kelly, JW. Heparin accelerates gelsolin amyloidogenesis. Biochemistry 2006, 45, 2234–2242. [Google Scholar]
- Calamai, M; Kumita, JR; Mifsud, J; Parrini, C; Ramazzotti, M; Ramponi, G; Taddei, N; Chiti, F; Dobson, CM. Nature and significance of the interactions between amyloid fibrils and biological polyelectrolytes. Biochemistry 2006, 45, 12806–12815. [Google Scholar]
- Nandi, PK; Nicole, J-C. Nucleic acid and prion protein interaction produces spherical amyloids which can function in vivo as coats of spongiform encephalopathy agent. J. Mol. Biol 2004, 344, 827–837. [Google Scholar]
- Selkoe, DJ. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev 2001, 81, 741–766. [Google Scholar]
- Peng, S; Glennert, J; Westermark, P. Medin-amyloid: A recently characterized age-associated arterial amyloid form affects mainly arteries in the upper part of the body. Amyloid 2005, 12, 96–102. [Google Scholar]
- Huff, ME; Balch, WE; Kelly, JW. Pathological and functional amyloid formation orchestrated by the secretory pathway. Curr. Opin. Struct. Biol 2003, 13, 674–682. [Google Scholar]
- Koudinov, AL; Koudinova, NV. Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. J. Neurol. Sci 2005, 229–230, 233–240. [Google Scholar]
- Sjögren, M; Mielke, M; Gustafson, D; Zandi, P; Skoog, I. Cholesterol and Alzheimer’s disease –is there a relation? Mech. Ageing Develop 2006, 127, 138–147. [Google Scholar]
- Stefani, M; Liguri, G. Cholesterol in Alzheimer’s disease: Unresolved questions. Curr. Alz. Res 2009, 6. in press. [Google Scholar]
- Arispe, N; Doh, M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease Aβ(1–40) and (1–42) peptides. FASEB J 2002, 16, 1526–1536. [Google Scholar]
- Mason, RP; Shoemaker, WJ; Shajenko, L; Chambers, TE; Herbette, LG. Evidence for changes in the Alzheimer’s disease brain cortical membrane structure mediated by cholesterol. Neurobiol. Aging 1992, 13, 413–419. [Google Scholar]
- Allen, JA; Halverson-Tamboli, RA; Rasenick, MM. Lipid raft microdomains and neurotransmitter signalling. Nature Rev 2007, 8, 128–140. [Google Scholar]
- Kojro, E; Gimpl, G; Lammich, S; März, W; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar]
- Abad-Rodriguez, J; Ledesma, MD; Craessaerts, K; Perga, S; Medina, M; Delacourte, A; Dingwall, C; De Strooper, B; Dotti, CG. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell. Biol 2004, 167, 953–960. [Google Scholar]
- Crameri, A; Biondi, E; Kuehnle, K; Lutjohann, D; Thelen, KM; Perga, S; Dotti, CG; Nitsch, RM; Ledesma, MD; Mohajeri, MH. The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J 2006, 25, 432–443. [Google Scholar]
- Gharibyan, AL; Zamotin, V; Yanamandra, K; Moskaleva, OS; Margulis, BA; Kostanyan, IA; Morozova-Roche, LA. Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J. Mol. Biol 2007, 365, 1337–13349. [Google Scholar]
- Novitskaya, V; Bocharova, OV; Bronstein, I; Baskakov, IV. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J. Biol. Chem 2006, 281, 13828–13836. [Google Scholar]
- Hotze, EM; Heuck, AP; Czajkowsky, M; Shao, Z; Johnson, AE; Tweten, R. Monomer-monomer interactions drive the prepore to pore conversion of a β-barrel-forming cholesterol-dependent cytolysin. J. Biol. Chem 2002, 277, 11597–115605. [Google Scholar]
- Chung, J; Yang, H; de Beus, MD; Ryu, CY; Cho, K; Colón, W. Cu/Zn superoxide dismutase can form pore-like structures. Biochem. Biophys. Res. Commun 2003, 312, 873–876. [Google Scholar]
- Azimov, R; Azimova, R; Hirakura, Y; Kagan, BL. Ion channels with different selectivity formed by transthyretin. Biophys. J 2001, 80, 129a. [Google Scholar]
- Lashuel, HA; Hartley, D; Petre, BM; Walz, T; Lansbury, PT. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar]
- Hirakura, Y; Kagan, BL. Pore formation by beta-2-microglobulin: A mechanism for the pathogenesis of dialysis-associated amyloidosis. Amyloid 2001, 8, 94–100. [Google Scholar]
- Cecchi, C; Baglioni, S; Fiorillo, C; Pensalfini, A; Liguri, G; Nosi, D; Rigacci, S; Bucciantini, M; Stefani, M. Insights into the molecular basis of the differing susceptibility of varying cell types to the toxicity of amyloid aggregates. J. Cell Sci 2005, 118, 3459–3470. [Google Scholar]
- Cecchi, C; Pensalfini, A; Stefani, M; Baglioni, S; Fiorillo, C; Cappadona, S; Caporale, R; Nosi, D; Ruggiero, M; Liguri, G. Vulnerability to amyloid toxicity depends on the cell-cycle phase in neuroblastoma replicating cells. J. Mol. Med 2007, 86, 197–209. [Google Scholar]
- Cecchi, C; Rosati, F; Pensalfini, A; Formigli, L; Nosi, D; Liguri, G; Dichiara, F; Morello, M; Danza, G; Pieraccini, G; Peri, A; Serio, M; Stefani, M. Seladin-1/dhcr24 protects neuroblastoma cells against Aβ toxicity by increasing membrane cholesterol content. J. Cell. Mol. Med, 2008; Epub ahead of print. [Google Scholar]
- Shadek, GM; Kummer, MP; Lu, DC; Galvan, V; Bredesen, DE; Koo, EH. Aβ induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597-624). FASEB J 2006, 20, 1254–1266. [Google Scholar]
- He, P; Zhong, Z; Lindholm, K; Berning, L; Lee, W; Lemere, C; Staufenbiel, M; Li, R; Shen, Y. Tumor necrosis factor death receptor signaling cascade Is required for amyloid-β protein-induced neuron death. J. Neurosci 2004, 24, 1760–1771. [Google Scholar]
- Yan, SD; Chen, X; Fu, J; Chen, M; Zhu, H; Roher, A; Slattery, T; Zhan, M; Nagashima, J; Morser, A; Migheli, P; Nawroth, D; Stern, AM; Schmidt, AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 1996, 382, 685–691. [Google Scholar]
- Yan, SD; Zhu, H; Zhu, A; Golabek, A; Du, H; Roher, A; Yu, J; Soto, C; Schmidt, AM; Stern, D; Kindy, M. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nature Med 2000, 6, 643–651. [Google Scholar]
- Yan, SD; Stern, D; Kane, MD; Kuo, YM; Lampert, HC; Roher, AE. RAGE-Abeta interactions in the pathophysiology of Alzheimer's disease. Resto. Neurol. Neurosci 1998, 12, 167–173. [Google Scholar]
- Sasaki, N; Takeuchi, M; Chowei, H; Kikuchi, S; Hayashi, N; Nakano, H; Ikeda, S; Yamagishi, T; Kitamoto, T; Saito, T; Makita, Z. Advanced glycation end products (AGE) and their receptor (RAGE) in the brain of patients with Creutzfeldt-Jakob disease with prion plaques. Neurosci. Lett 2002, 326, 117–120. [Google Scholar]
- Monteiro, FA; Cardoso, I; Mendes Sousa, M; Saraiva, MJ. In vitro inhibition of transthyretin aggregate-induced cytotoxicity by full and peptide derived forms of the soluble receptor for advanced glycation end products (RAGE). FEBS Lett 2006, 580, 3451–3456. [Google Scholar]
- Geroldi, D; Falcone, C; Emanuele, E. Soluble receptor for advanced glycation end products: from disease marker to potential therapeutic target. Curr. Med. Chem 2006, 13, 1971–1978. [Google Scholar]
- Hou, X; Parkington, HC; Coleman, HA; Mechler, A; Martin, LL; Aguilar, M-I; Small, DH. Transthyretin oligomers induce calcium influx via voltage-gated calcium channels. J. Neurochem 2007, 100, 446–457. [Google Scholar]
- Hsieh, H; Boehm, J; Sato, C; Iwatsubo, T; Tomita, T; Sisodia, S; Malinow, R. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar]
- De Felice, FG; Velasco, PT; Lambert, MP; Viola, K; Fernandez, SJ; Ferreira, ST; Klein, WL. Aβ oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem 2007, 282, 11590–11601. [Google Scholar]
- Pellistri, F; Bucciantini, M; Relini, A; Gliozzi, A; Robello, M; Stefani, M. Generic interaction of pre-fibrillar amyloid aggregates with NMDA and AMPA receptors results in free Ca2+ increase in primary neuronal cells. J. Biol. Chem, 2008; Epub ahead of print. [Google Scholar]
- Kranenburg, O; Bouma, B; Kroon-Batenburg, LMJ; Reijerkerk, A; Wu, Y-P; Voest, EE; Gebbink, MFBG. Tissue-type plasminogen activator is a multiligand cross-beta structure receptor. Current Biol 2002, 12, 1833–1839. [Google Scholar]
- Lacor, PN; Buniel, MC; Chang, L; Fernandez, SJ; Gong, Y; Viola, KL; Lambert, M; Velasco, PT; Bigio, EH; Finch, CE; Krafft, G; Klein, WI. Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J. Neurosci 2004, 24, 10191–10200. [Google Scholar]
- Lee, G; Pollard, HB; Arispe, N. Annexin 5 and apolipoprotein E2 protect against Alzheimer’s amyloid-β-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides 2002, 23, 1249–1263. [Google Scholar]
- Bhatia, R; Lin, H; Lal, R. Fresh and nonfibrillar amyloid β protein(1–42) induces rapid cellular degeneration in aged human fibroblasts: evidence for AβP-channel-mediated cellular toxicity. FASEB J 2000, 14, 1233–1243. [Google Scholar]
- Caughey, B; Lansbury, PT. Protofibrils, Pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci 2003, 26, 267–298. [Google Scholar]
- Watt, JA; Pike, CJ; Walencewicz-Wasserman, AJ; Cotman, CW. Ultrastructural analysis of beta-amyloid-induced apoptosis in cultured hippocampal neurons. Brain Res 1994, 661, 147–156. [Google Scholar]
- Morishima, Y; Gotoh, Y; Zieg, J; Barrett, T; Takano, H; Flavell, R; Davis, RJ; Shirasaki, Y; Greenberg, ME. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci 2001, 21, 7551–7560. [Google Scholar]
- Velez-Pardo, C; Arroyave, ST; Lopera, F; Castano, AD; Jimenez Del Rio, M. Ultrastructure evidence of necrotic neural cell death in familial Alzheimer's disease brains bearing presenilin-1 E280A mutation. J. Alzheimer Dis 2001, 3, 409–415. [Google Scholar]
- Ross, CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 2002, 35, 819–822. [Google Scholar]
- Bucciantini, M; Calloni, G; Chiti, F; Formigli, L; Nosi, D; Dobson, CM; Stefani, M. Pre-fibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem 2004, 279, 31374–31382. [Google Scholar]
- Sirangelo, I; Malmo, C; Iannuzzi, C; Mezzogiorno, A; Bianco, MR; Papa, M; Irace, G. Fibrillogenesis and cytotoxic activity of the amyloid-forming apomyoglobin mutant W7FW14F. J. Biol. Chem 2004, 279, 13183–13189. [Google Scholar]
- Zhu, YJ; Lin, H; Lal, R. Fresh and nonfibrillar amyloid β protein (1–40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AβP-channel-mediated cellular toxicity. FASEB J 2000, 14, 1244–1254. [Google Scholar]
- Kourie, JI. Mechanisms of amyloid β protein-induced modification in ion transport systems: Implications for neurodegenerative diseases. Cell. Mol. Neurobiol 2001, 21, 173–213. [Google Scholar]
- Butterfield, AD; Drake, J; Pocernich, C; Castegna, A. Evidence of oxidative damage in Alzeimer’s disease brain: central role for amyloid β-peptide. Trends Mol. Med 2001, 7, 548–554. [Google Scholar]
- Milhavet, O; Lehmann, S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res. Rev 2002, 38, 328–339. [Google Scholar]
- Hyun, D-H; Lee, MH; Hattori, N; Kubo, S-I; Mizuno, Y; Halliwell, B; Jenner, P. Effect of wild-type or mutant parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J. Biol. Chem 2002, 277, 28572–28577. [Google Scholar]
- Gong, Y; Chang, L; Viola, KL; Lacor, PN; Lambert, MP; Finch, CE; Krafft, GA; Klein, WL. Alzheimer’s disease-affected brain: presence of oligomeric Aβ ligands (ADDSLs) suggest a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar]
- Varadarajan, S; Yatin, S; Aksenova, M; Butterfield, DA. Alzheimer’s amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol 2000, 130, 184–208. [Google Scholar]
- Squier, TC. Oxidative stress and protein aggregation during biological aging. Exp. Gerontol 2001, 36, 1539–1550. [Google Scholar]
- Kawahara, M; Kuroda, Y; Arispe, N; Rojas, E. Alzheimer’s β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevation by a common mechanism in a hypopthalamic GnRH neuronal cell line. J. Biol. Chem 2000, 275, 14077–14083. [Google Scholar]
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stefani, M. Protein Folding and Misfolding on Surfaces. Int. J. Mol. Sci. 2008, 9, 2515-2542. https://doi.org/10.3390/ijms9122515
Stefani M. Protein Folding and Misfolding on Surfaces. International Journal of Molecular Sciences. 2008; 9(12):2515-2542. https://doi.org/10.3390/ijms9122515
Chicago/Turabian StyleStefani, Massimo. 2008. "Protein Folding and Misfolding on Surfaces" International Journal of Molecular Sciences 9, no. 12: 2515-2542. https://doi.org/10.3390/ijms9122515