Antiproliferative Activity of β-Hydroxy-β-Arylalkanoic Acids

Abstract
:1. Introduction
2. Results and Discussion
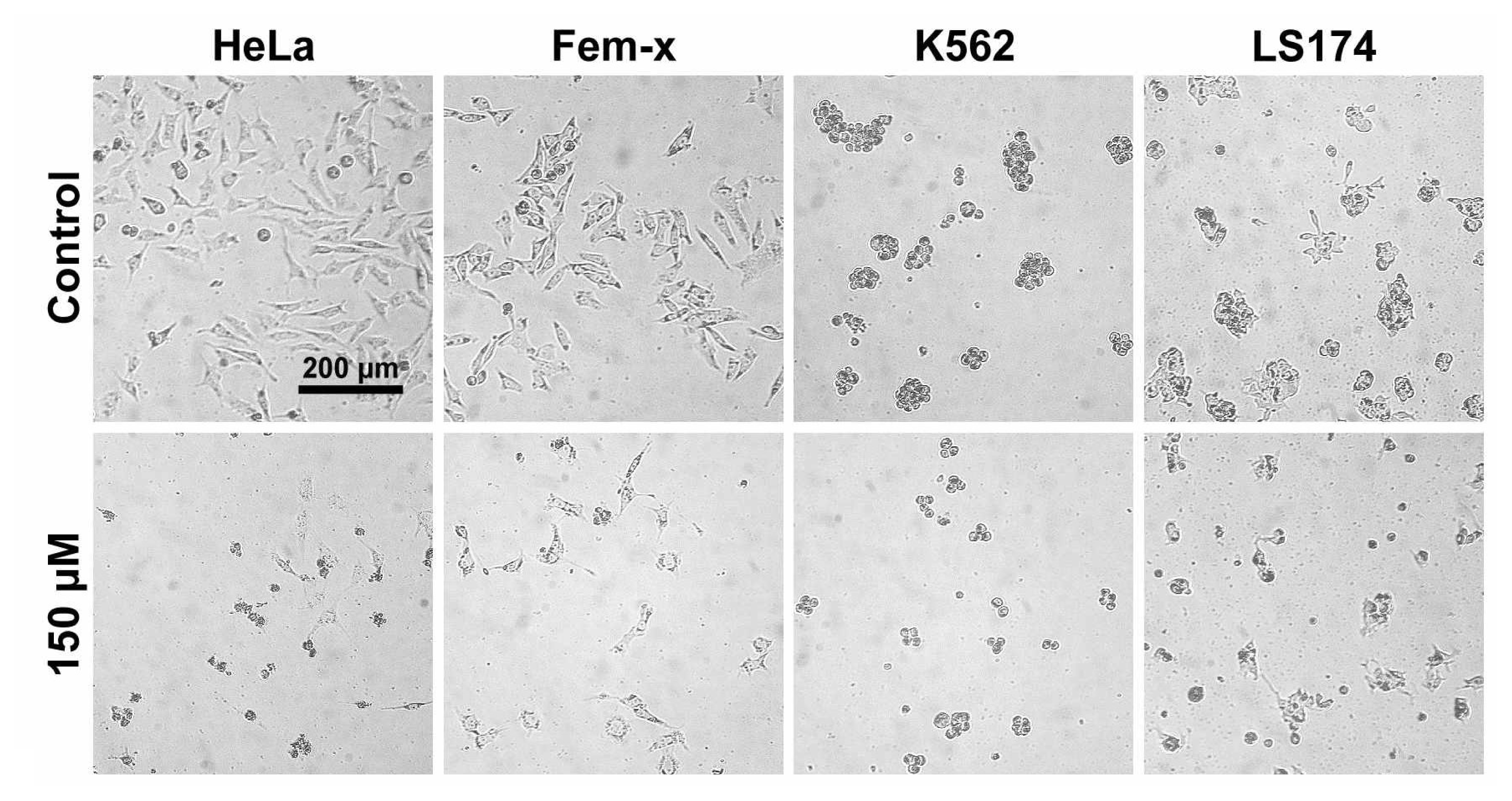
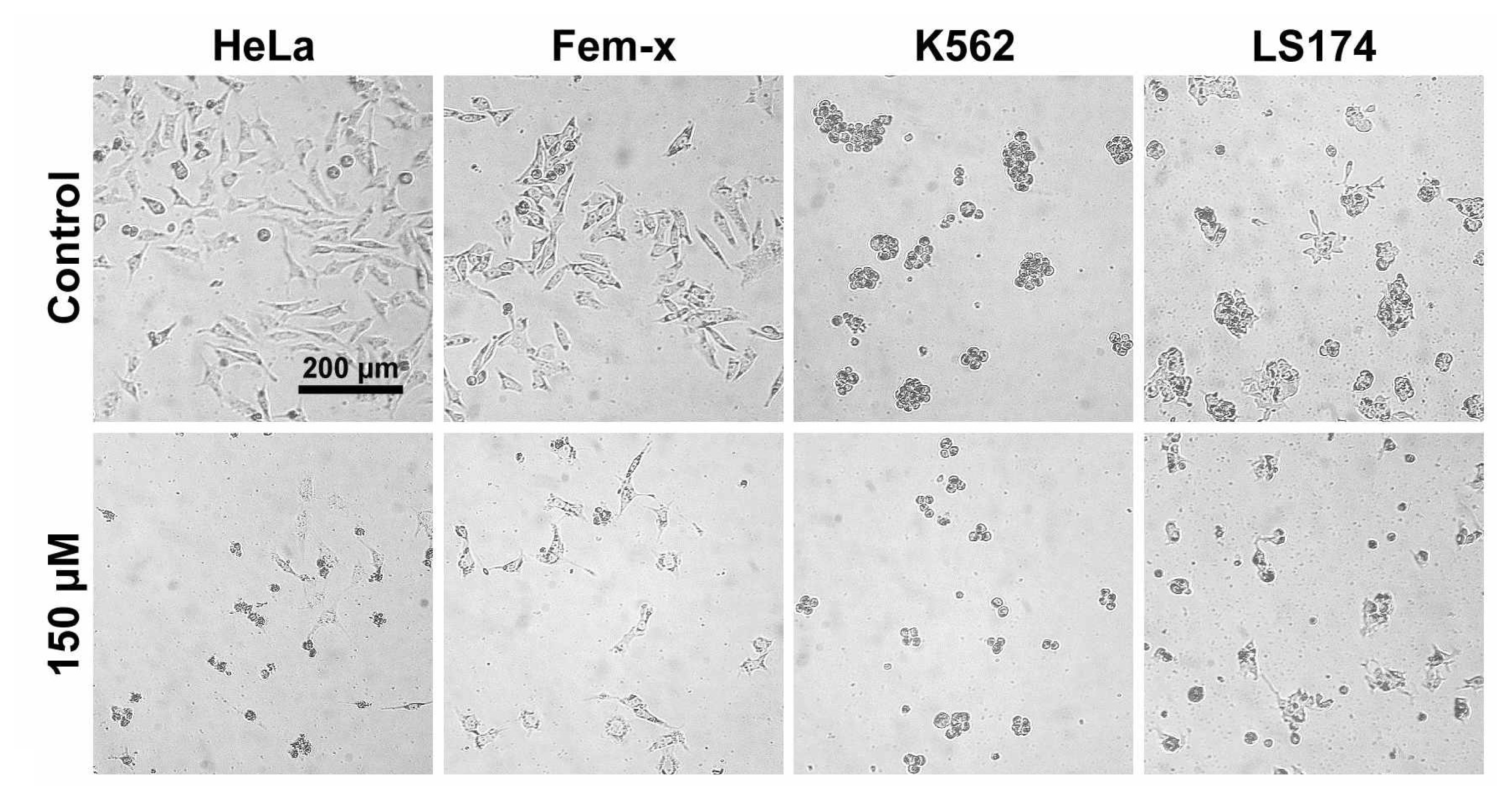
2.1. Antiproliferative activity
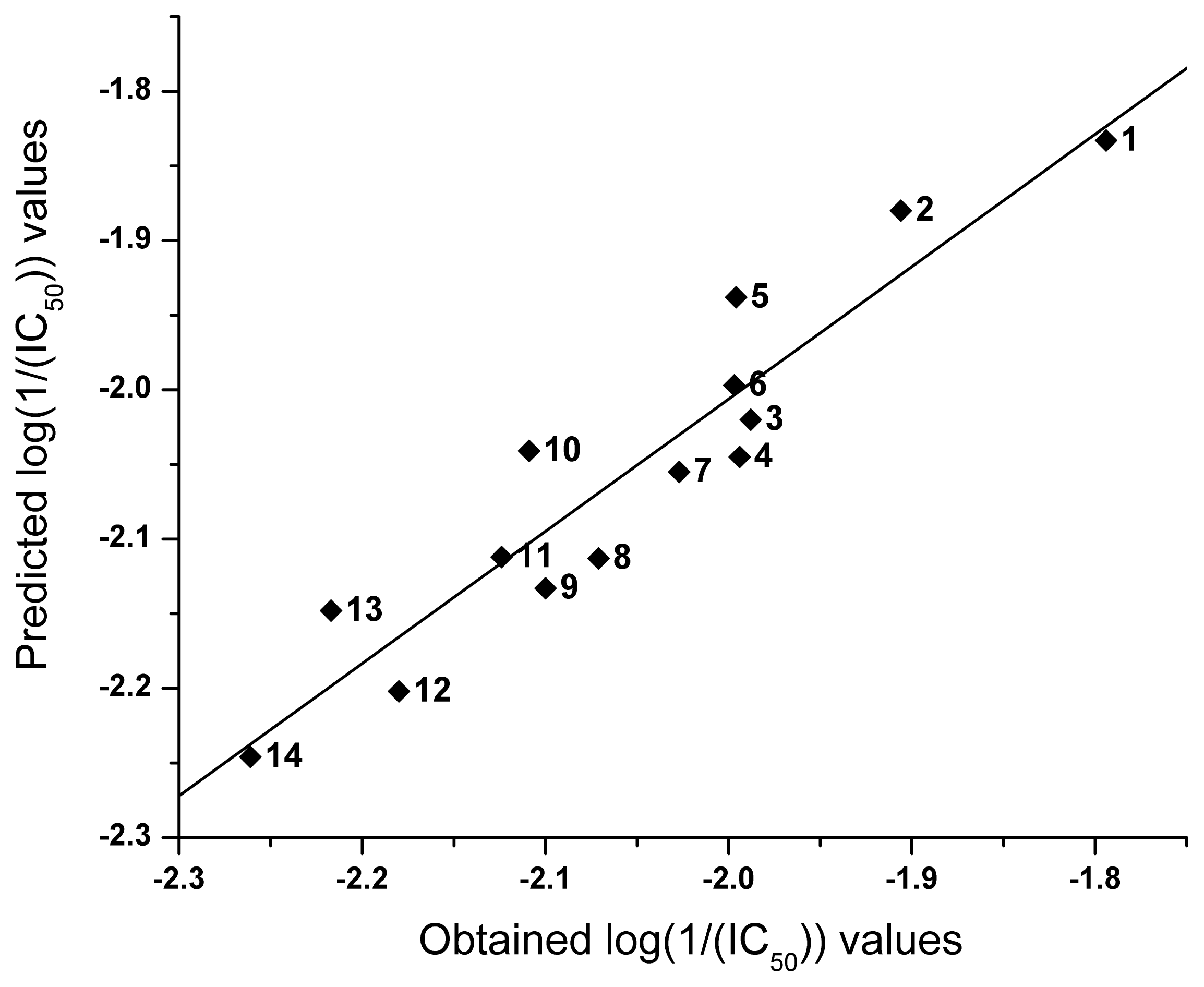
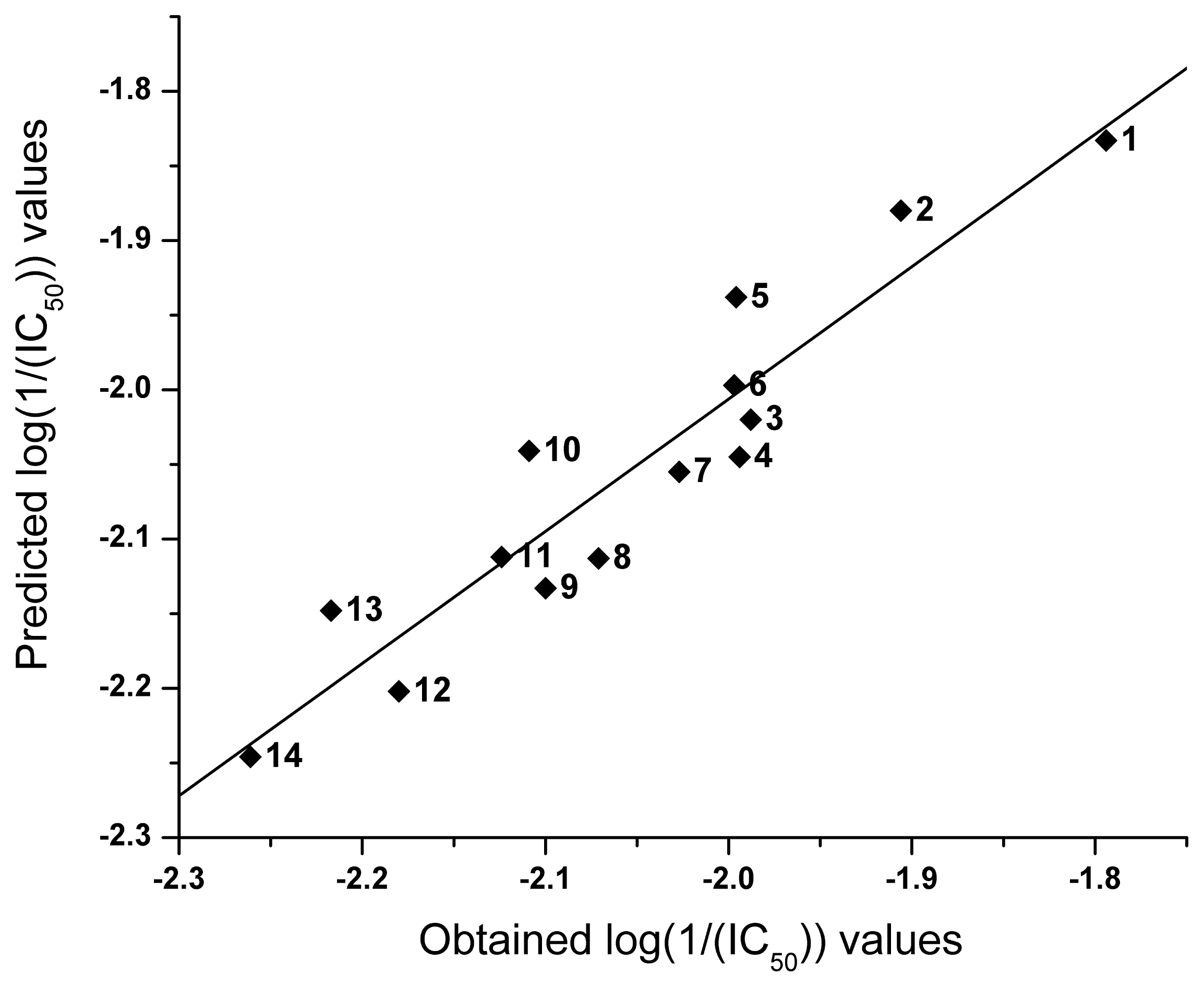
2.2. QSAR
2.3. Conclusion
3. Experimental
3.1. Syntheses
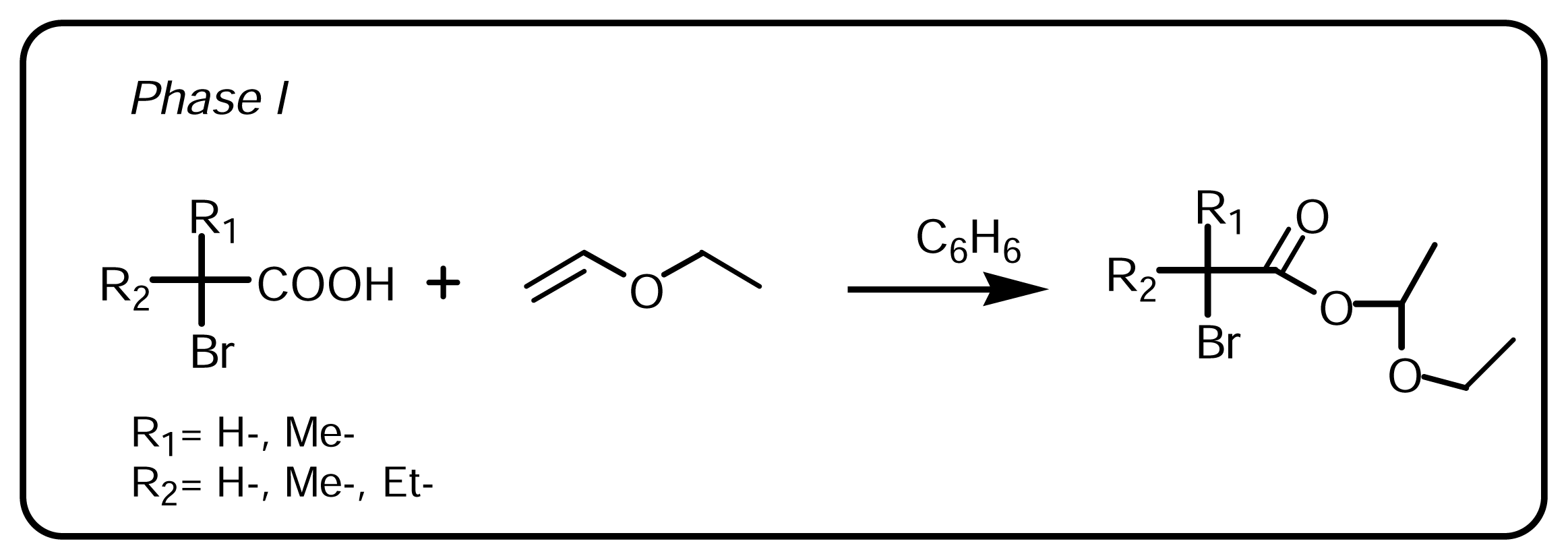
3.1.1 Synthesis of α-bromo esters-acetals (Scheme 1, Phase I)
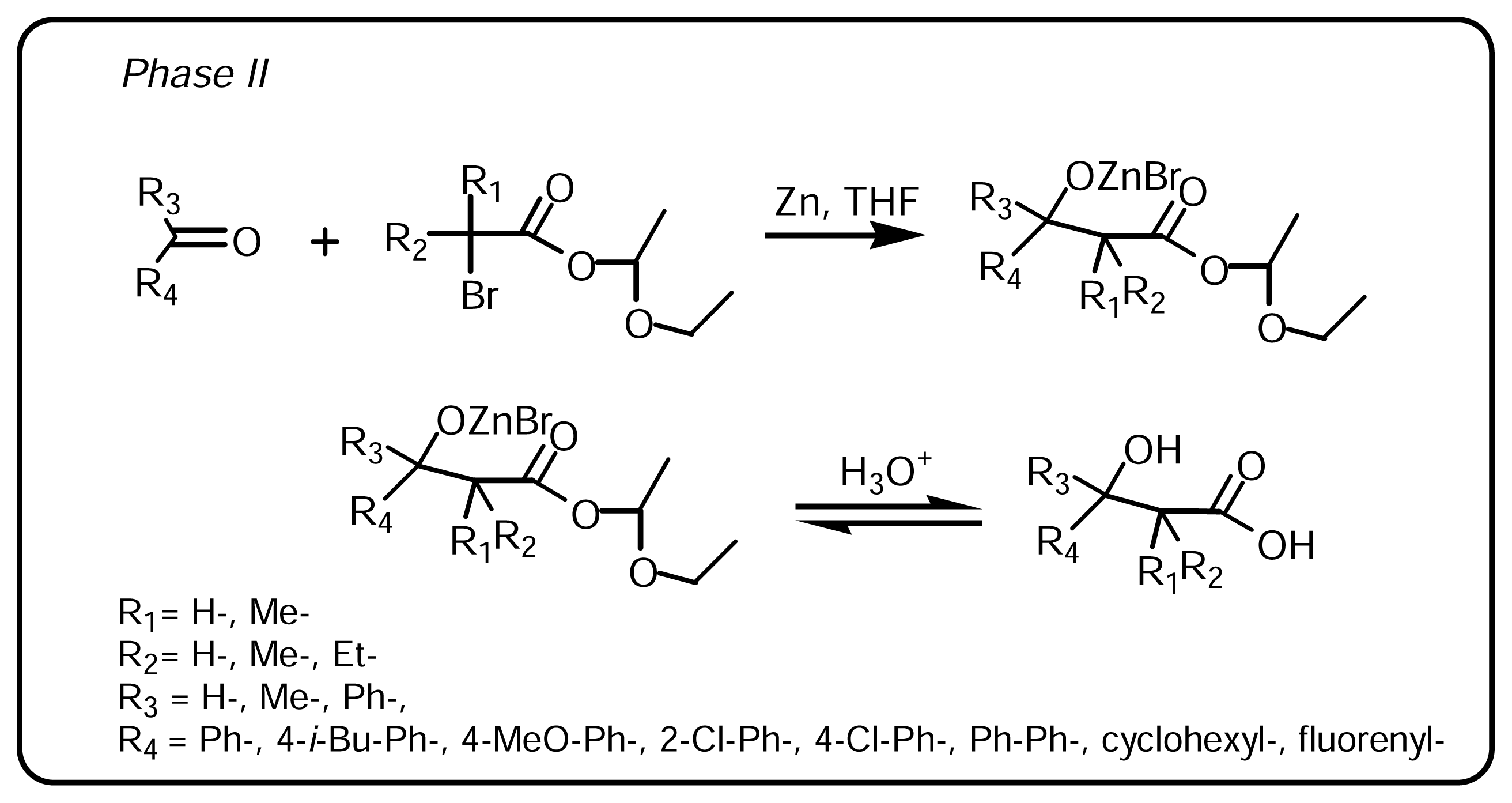
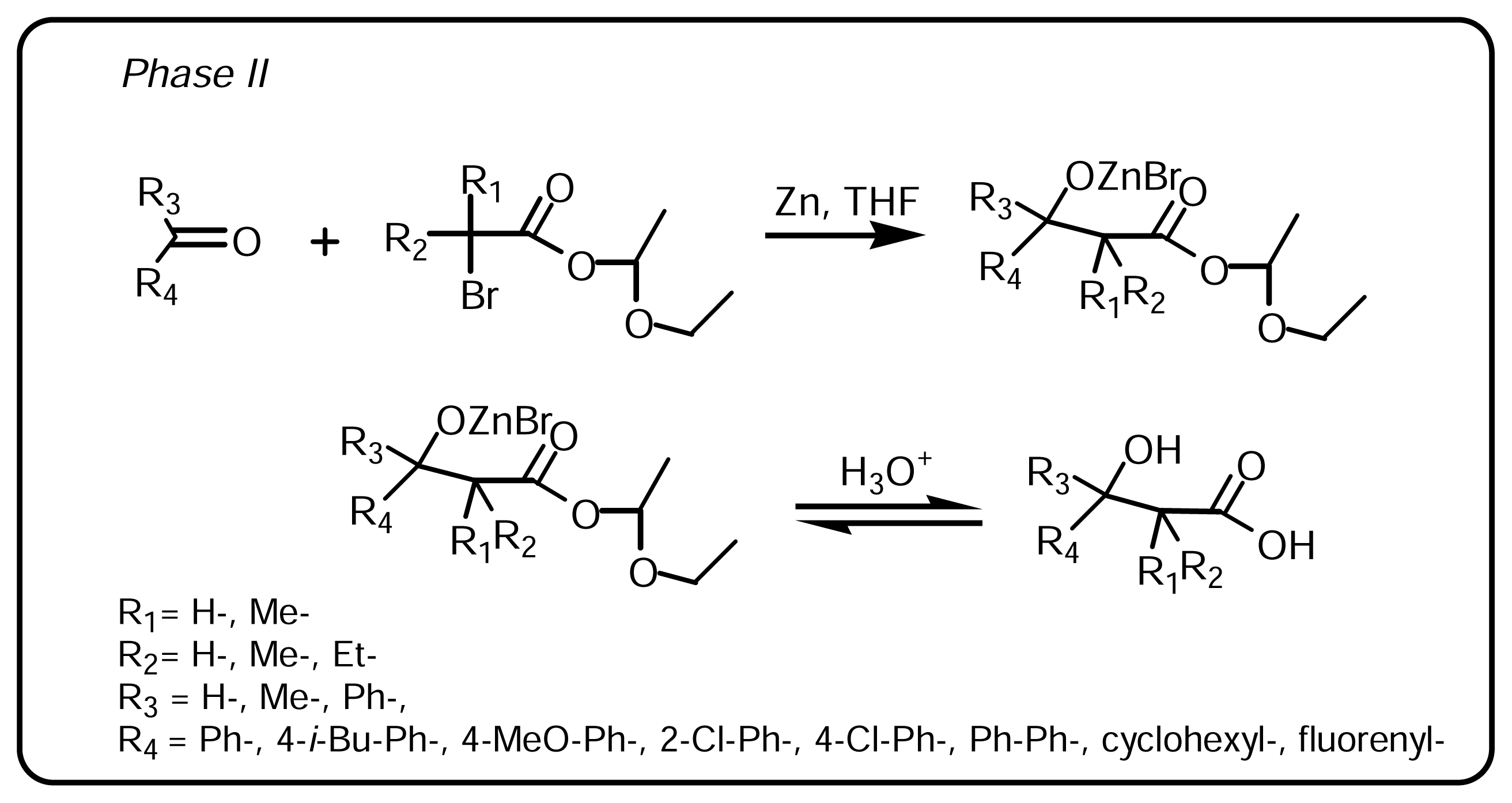
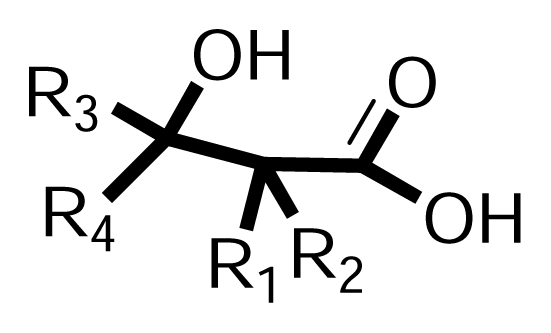
3.1.2. Synthesis of β-hydroxy-β-arylalkanoic acids (Scheme 2, Phase II)
- 3-Hydroxy-2,2-dimethyl-3-(4-biphenylyl)butanoic acid: C18H20O3; Mw = 284.35; Melting point: 142 °C; IR (KBr): ν̄max (cm−1) 3513 (ν̄,-CH-OH), 3034 (ν̄,-C(O)-OH), 2977, 1688 (ν̄, >C=O), 1238 (ν̄, -C-O); 1H NMR (CDCl3) (δ): 1.17 (s, 3 H); 1.20 (s, 3 H); 1.69 (s, 3 H); 7.31–7.63 (m, 9 H); 13C NMR (CDCl3) (δ): 21.65 (-C(CH3)2-); 25.10 (-C(OH)CH3-); 50.05 (-C(CH3)2-); 77.36 (- C(OH)CH3-); 126.07 (o-Ph); 126.98 (o′-Ph); 127.31 (p′-Ph); 127.63 (m-Ph); 128.74 (m′-Ph); 139.94 (pipso-Ph); 140.47 (Cipso′-Ph); 141.63 (Cipso-Ph); 182.91 (COOH); MS (CI): 285 (M+H)+, 267 (M-17), 197 (M-87); Yield (%): 32; Elemental analysis (%): Calc.: C, 76.03; H, 7.09. Found: C, 75.57; H, 7.28.
- 3-Hydroxy-2,2-dimethyl-3,3-diphenylpropanoic acid; C17H18O3; Mw = 270.32; Melting point: 162 °C; IR (KBr): ν̄max (cm−1) 3570 (ν̄,-CH-OH), 3520 (ν̄,-C(O)-OH), 1678 (ν̄, >C=O), 1160 (ν̄, - C-O); 1H NMR ( CDCl3) (δ): 1.35 (s, 6H); 7.13–7.47 (m, 10 H, after the subtraction of CHCl3 signal at 7.263 δ); 13C NMR (CDCl3) (δ): 23.93 (-C(CH3)2-); 48.71 (-C(CH3)2-); 82.44 (C-OH); 127.25 (o-Ph); 127.39 (p-Ph); 128.63 (m-Ph); 144.62 (Cipso-Ph); 185.24 (COOH); MS (CI): 253 (M-17), 183 (M-87); Yield (%): 40; Elemental analysis (%): Calcd.: C, 75.53; H, 6,71. Found: C, 75.22; H, 6,74.
- 3-Hydroxy-2-methyl-3-(2-chlorophenyl)propanoic acid; C10H11ClO3; Mw = 214.65; Melting point: 84 0C; IR (KBr):ν̄max (cm−1) 3325, 3420 (ν̄,-CH-OH), 3057, 2992 (ν̄,-C(O)-OH), 1707 (ν̄, >C=O), 1232 (ν̄, -C-O-); 1H NMR (CDCl3) (δ): 1.09 (d, J 7.2 Hz, 3 H, after the subtraction of CHCl3 signal at 7.263 δ); 1.16 (d, J 7.2 Hz, 3 H); 2.88–3.11 (m, 1 H); 5.31(d, J 8 Hz, 1 H); 5.62 (d, J 2.2 Hz, 1 H); 7.18–7.63 (m, 4 H); 13C NMR (CDCl3) (δ): 9.14 (14.15) (-CH(CH3)-); 42.55 (46.45) (-CH(CH3)-); 69.68 (71.99) (-CH(OH)-); 126.74 (127.34) (m-Ph); 127.91 (128.23) (m-Ph); 128.73 (p-Ph); 129.16 (129.44) (o-Ph); 129.52 (131.29) (C-Cl); 138.14 (Cipso-Ph); 181.15 (181.66) (COOH); (MS (EI): 214 (M+),142 (M-73), 77; Yield (%): 25; Elemental analysis (%): Calcd.: C, 55.96; H, 5.17. Found: C, 55.41; H, 5.02.
- 3-Hydroxy-3-(4-biphenylyl)butanoic acid; C16H16O3; Mw = 256.30; Melting point: 136 °C; IR (KBr):ν̄max (cm−1) 3521 (ν̄,-CH-OH), 2977 (ν̄,-C(O)-OH), 1689 (ν̄, >C=O), 1238 (ν̄, -C-O-); 1H NMR (CDCl3) (δ): 1.58 (s, 1 H); 2.96 (dd, J1J2 16.4 Hz; 2 H); 5.63 (s, OH); 7.30–7.60 (m, 9 H); 13C NMR (CDCl3) (δ): 30.57 (CH3); 47.92 (-CH2-); 72.70 (C-OH); 124.85 (o-Ph); 127.10 (o′-Ph); 127.14 (o- and p′-Ph); 127.32 (m′-Ph); 128.78 (m-Ph); 139.98 (pipso-Ph); 140.58 (Cipso′-Ph); 145.40 (Cipso-Ph); MS (EI): 256 (M+), 238 (M-18), 197 (M-59), 153; Yield (%): 27; Elemental analysis (%): Calcd.: C, 74.98; H, 6.29. Found: C, 74.70; H, 6.48.
- 2-[9-(9-Hydroxyfluorenyl)]-2-methylpropanoic acid; C17H16O3; Mw = 268.31; Melting point: 138 °C; IR (KBr):ν̄max (cm−1) 3387 (ν̄, -CH-OH), 2984 (ν̄,-C(O)-OH), 1725 (ν̄, >C=O), 1156 (ν̄, - C-O); 1H NMR (CDCl3) (δ): 1.07 (s, 6H); 7.19–7.61 (m, 8 H); 13C NMR (CDCl3) (δ): 20.98 (- C(CH3)2-); 48.72 (-C(CH3)2-); 85.39 (C-OH); 119.97 (m-Ph); 124.14 (m-Ph); 127.80 (p-Ph); 129.45 (o- Ph); 140.43 (Cipso-Ar); 145.69 (Cipso); 180.65 (COOH); ( MS (CI): 268 (M+), 251 (M-17), 223 (M-45), 181 (M-87); Yield (%): 20; Elemental analysis (%): Calcd.: C, 76.10; H, 6.01. Found: C, 76.19; H, 6.19.
- 3-Hydroxy-2-methyl-3,3-diphenylpropanoic acid; C16H16O3; Mw = 256.30; Melting point: 180 °C; IR (KBr):ν̄max (cm−1) 3525 (ν̄, -CH-OH), 2991 (ν̄, -C(O)-OH), 1677 (ν̄, >C=O), 1225 (ν̄, - C-O); 1H NMR (CDCl3) (δ) : 1.19 (d, J 7.2 Hz, 3 H); 3.65 (q, J 7.2 Hz, 1 H); 7.13–7.56 (m, 10 H); 13C NMR (CDCl3) (δ) 13.04 (CH3); 46.41 (-CH(CH3)-); 77.91 (C-OH); 125.30 (o- and o′-Ph); 126.71 (m- Ph); 127.12 (m′-Ph); 128.18 (p- and p′-Ph); 128.47 (o- and o′-Ph); 143.80 (Cipso); 147.15 (Cipso′); 182.04 (COOH); MS (CI):. 257 (M+H)+, 239 (M-17), 183 (M-73); Yield (%): 35; Elemental analysis (%): Calcd.: C, 74.98; H, 6.29. Found: C, 75.08; H, 6.11.
- 3-Hydroxy-2,2-dimethyl-3-(4-chlorophenyl)propanoic acid; C11H13ClO3; Mw = 228.67; Melting point: 142 °C; IR (KBr):ν̄max (cm−1) 3501 (ν̄, -CH-OH), 2989 (ν̄, -C(O)-OH), 1683 (ν̄, >C=O), 1167 (ν̄, -C-O); 1H NMR (CDCl3) (δ) : 1.13 (s, 3 H); 1.15 (s, 3 H); 4.94 (s, 3 H); 7.25–7.36 (m, 4 H); 13C NMR (CDCl3) (δ) 18.34 (CH3); 23.05 (CH3); 47.50 (-C(CH3)2-); 77.64 (-CH(OH)-); 128.07 (m-Ph); 129.07 (o-Ph); 137.96 (Cipso); 183.08 (COOH); MS (CI) 229 (M+H)+, 211 (M-18), 141; Yield (%): 40; Elemental analysis (%): Calcd.: C, 57.78; H, 5.73. Found: C, 57.73; H, 5.75.
- 3-Hydroxy-3-(2-chlorophenyl)propanoic acid; C9H9ClO3; Mw = 200.62; Melting point: 92 °C; IR (KBr):ν̄max (cm−1) 3476, 3311 (ν̄, -C(O)-OH), 1709 (ν̄, >C=O), 1164 (ν̄, -C-O); 1H NMR (CDCl3) (δ): 2.71 (dd, J 9.8 Hz, J 16.8 Hz, 1H); 2.97 (dd, J 2.6 Hz, J 16.8 Hz, 1H); 5.53 (dd, J 2.2 Hz, J 8 Hz, 1H); 6.51 (s, OH) 7.19–7.64 (m, 4 H); 13C NMR (CDCl3) (δ): 41.29 (-CH2-); 66.91 (-CH(OH)- ); 126.96 (m-Ph); 127.32 (p-Ph); 128.98 (m-Ph); 129.49 (o-Ph); 131.38 (C-Cl); 139.45 (Cipso); 177.57 (COOH); MS (CI) 201 (M+), 183 (M-18), 141 (M-59); Yield (%): 20; Elemental analysis (%): Calcd.: C, 53.88; H, 4.52. Found: C, 53.65; H, 4.63.
- 3-Hydroxy-2,2-dimethyl-(4-methoxyphenyl)butanoic acid; C13H18O4; Mw = 238.28; Melting point: 120 °C; IR (KBr):ν̄max (cm−1) 3420 (ν̄, -C(O)-OH), 2996 (ν̄, -C(O)-OH), 1728 (ν̄, >C=O),1251 (ν̄, -C-O); 1H NMR (CDCl3) (δ): 1.12 (s, 3 H); 1.15 (s, 3 H); 1.64 (s, 3 H); 3.79 (s, 3 H); 6.81–7.37 (m, 4 H); 13C NMR (CDCl3) (δ): 21.56 (-C(CH3)2-); 25.13 (-C(OH)CH3-); 50.11 (-C(CH3)2- ); 55.13 (CH3-O-Ar); 77.18 (-C(OH)CH3-); 112.70 (m-Ar); 128.32 (o-Ar); 134.72 (C-OCH3); 158.59 (Cipso); 182.67 (COOH); MS (CI) 239 (M+H)+, 221 (M-18), 151 (M-87); Yield (%): 45; Elemental analysis (%): Calcd.: C, 65.53; H, 7.61. Found: C, 65.76; H, 7.94.
- 2-Hethyl-2-(1-(1-hydroxycyclohexyl))propanoic acid; C10H18O3; Mw = 186.25; Melting point: 89 °C; IR (KBr): ν̄max (cm−1) 3497 (ν̄, -CH-OH), 3411 (ν̄, -C(O)-OH), 2970 (ν̄, -C(O)-OH), 1686 (ν̄, >C=O), 1144 (ν̄, -C-O); 1H NMR (CDCl3) (δ) : 1.25 (s, 6 H); 1.21–1.69 (m, 10 H); 13C NMR (CDCl3) (δ): 20.74 (-C(CH3)2-); 21.41 ( 2′ (4′)-CH2-); 25.53 (4′-CH2-); 31.30 (2′ (6′)-CH2-); 50.11 (-C(CH3)2-); 74.63 (C-OH); 183.09 (COOH); MS (CI) 187 (M+H)+, 169 (M-17); Yield (%): 28; Elemental analysis (%): Calcd.: C, 64.49; H, 9.74. Found: C, 64.20; H, 10.02.
- 3-Hydroxy-2,2-dimethyl-3-phenylbutanoic acid; C12H16O3; Mw = 208.26; Melting point: 89 °C; IR (KBr): ν̄max (cm−1) 3230 (ν̄, -CH-OH), 3205, (ν̄, -C(O)-OH), 2989 (ν̄, -C(O)-OH), 1723 (ν̄, >C=O), 1159 (ν̄, -C-O); 1H NMR (CDCl3) (δ): 1.13 (s, 3 H); 1.16 (s, 3 H); 1.66 (s, 3 H); 7.25–7.46 (m, 5 H); 13C NMR (DMSO) (δ) 21.54 (-C(CH3)2-); 21.59 (-C(CH3)2-); 25.00 (-C(OH)CH3-); 49.98 (- C(CH3)2-); 77.38 (-C(OH)CH3-); 127.12 (o-Ph); 127.23 (p-Ph); 127.43 (m-Ph); 142.52 (Cipso-Ph); 182.98 (COOH); MS (CI) 208 (M+), 191 (M-17), 121 (M-87); Yield (%): 33; Elemental analysis (%): Calcd.: C, 69.21; H, 7.77. Found: C, 68.97; H, 7.74.
- 3-Hydroxy-3(4-isobutylphenyl)butanoic acid;C14H20O3; Mw = 236.31; Melting point: 90 °C; IR (KBr):ν̄max (cm−1) 3515, 3400 (ν̄, -C(O)-OH), 2957 (ν̄, -C(O)-OH), 1683 (ν̄, >C=O), 1244 (ν̄, - C-O); 1H NMR (CDCl3) (δ): 0.87 (d, J 6.6 Hz, 3H); 0.90 (d, J 6.6 Hz, 3H); 1.55 (s, 3H); 1.85 (h, 1 H); 2.45 (d, J 7.2 Hz, 2 H); 2.91 (dd, J 16 Hz, J 39.2 Hz, 2 H); 5.76 (b, OH); 7.10 (d, J 8 Hz) 7.30 (d, J 8 Hz); 13C NMR (CDCl3) (δ): 22.34 (-CH(CH3)2); 30.10 (-CH(CH3)2); 30.39 (CH3-C(OH)-); 44.88 (Ar- CH2-); 46.08 (-CH2-COOH); 72.74 (CH3-C(OH)-); 124.06 (o-Ph); 129.09 (m-Ph); 140.51 (Cipso-i-Bu); 143.57 (Cipso); 176.98 (COOH); MS (CI) 236 (M+), 218 (M-18), 177 (M-59); Yield (%): 42; Elemental analysis (%): Calcd.: C, 70.11; H, 8.53; Found: C, 70.08; H, 8.58. (with 0.2 mol H2O; in 1H NMR spectra, presence of water is observed)
- 3-Hydroxy-2,2-dimethyl-3-phenylpropanoic acid; C11H14O3; Mw = 194.23; Melting point: 132 °C; IR (KBr):ν̄max (cm−1) 3439, 3380 (ν̄, -OH), 2984 (ν̄, -C(O)-OH), 1707 (ν̄, >C=O), 1134 (ν̄, -C-O); 1H NMR (CDCl3) (δ) 1.17 (s, 6 H); 4.96 (s, 1 H); 7.32–7.36 (m, 5 H); 13C NMR (CDCl3) (δ) 18.50 (-C(CH3)2-); 23.29 (-C(CH3)2-); 47.49 (-C(CH3)2-); 78.47 (HO-CH(Ph)-); 127.72 (o-Ph); 127.96 (m-Ph); 128.07 (p-Ph); 139.47 (Cipso-Ph); 182.80 (COOH); MS (CI). 194 (M+), 176 (M-18), 107 (M-87); Yield (%): 25; Elemental analysis (%): Calcd.: C, 68.02; H, 7.26; Found: C, 67.71; H, 7.55.
- 3-Hydroxy-3,3-diphenylpropanoic acid; C15H14O3; Mw = 242.09; Melting point: 217 °C; IR (KBr):ν̄max (cm−1) 3478 (ν̄, -OH), 2908 (ν̄, -C(O)-OH), 1688 (ν̄, >C=O), 1232 (ν̄, -C-O); 1H NMR (DMSO) (δ) 3.28 (s, 2 H); 7.12–7.48 (m, 10 H); 13C NMR (DMSO) (δ) 45.33 (-CH2-COOH); 75.71 (HO-C(Ph)2-); 125.69 (o-Ph); 126.60 (p-Ph); 128.07 (m-Ph); 147.54 (Cipso-Ph); 173.35 (COOH); MS (CI). 242 (M+) 183 (M-59); Yield (%): 40; Elemental analysis (%): Calcd.: C, 74.36; H,. 5.82; Found: C, 74.22; H, 6.01.
- 2-(1′-(1′-Hydroxycyclohexyl))butanoic acid; C10H18O3; Mw = 186.25; Melting point: 86 °C; IR (KBr):ν̄max (cm−1) 3384 (ν̄,-OH), 2936 (ν̄, -C(O)-OH), 1710 (ν̄, >C=O), 1193 (ν̄, -C-O); 1HNMR (CDCl3) (δ) 0.97 (t, J 7.3 Hz. 3H); 1.22–1.81 (m, 12H); 2.38 (t, J 7.5 Hz, 3H); 13C NMR (CDCl3) (δ) 12.32 (-CH2-CH3); 19.41(-CH2-CH3) ; 21.59 (3′ (5′)-CH2); 21.79 (5′ (3′)-CH2); 25.51 (4′-CH2); 34.19 (2′ (6′)-CH2); 37.11 (6′ (2′)-CH2); 56.48 (-CH(Et)COOH); 71.96 (1′-COH); 181.00 (COOH); (MS (CI) 186 (M+), 169 (M-17), 100 (M-87); Yield (%): 40; Elemental analysis (%): Calcd.: C, 64.49; H, 9.74; Found: C, 64.23; H, 9.86.
3.2. Cell culture
3.2.1. Investigated compounds
3.2.2. Preparation of peripheral blood mononuclear cells
3.2.3. Treatment of LS174, HeLa, Fem-x and K562 cells
3.2.4. Treatment of PBMC
3.2.5. Determination of cell survival


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
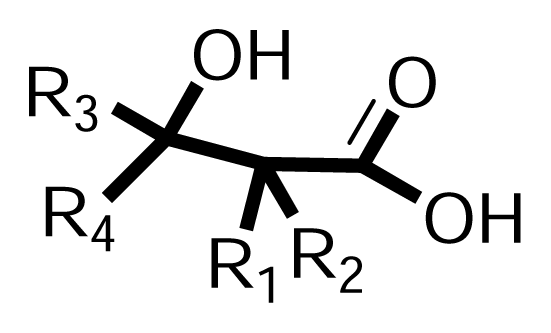 | ||||||||
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | ||||||||
| NO | R1- | R2- | R3- | R4- | HeLa | Fem-X | K562 | LS174 |
| 1 | Me- | Me- | Me- | Ph-Ph- | 62.20 | 205 | 141 | 154 |
| 2 | Me- | Me- | Ph- | Ph- | 80.56 | / | / | / |
| 3 | Me- | H- | H- | 2-Cl-Ph- | 97.20 | 89 | 92 | 93 |
| 4 | H- | H- | Me- | Ph-Ph- | 98.70 | >200 | >200 | >200 |
| 5 | Me- | Me- | fluorenyl- | 99.07 | / | / | / | |
| 6 | Me- | H- | Ph- | Ph- | 99.41 | / | / | / |
| 7 | Me- | Me- | H- | 4-Cl-Ph- | 106.30 | >200 | >200 | >200 |
| 8 | H- | H- | H- | 2-Cl-Ph- | 117.80 | >200 | >200 | >200 |
| 9 | Me- | Me- | Me- | 4-MeO-Ph- | 125.80 | 136 | >200 | >200 |
| 10 | Me- | Me- | cyclohexyl- | 128.40 | >200 | >200 | >200 | |
| 11 | Me- | Me- | Me- | Ph- | 133.17 | / | / | / |
| 12 | H- | H- | Me- | 4-i-Bu-Ph- | 151.40 | >200 | >200 | >200 |
| 13 | Me- | Me- | H- | Ph- | 164.88 | / | / | / |
| 14 | H- | H- | Ph- | Ph- | 182.55 | / | / | / |
| 15 | H- | Et- | cyclohexyl- | >200 | >200 | / | / | |
| Cis-DDP | / | 4.31 | 4.70 | 5.77 | / | |||
| NO | logP | I | Obtained log(1/(IC50)) | Predicted log(1/(IC50)) |
|---|---|---|---|---|
| 1 | 4.16 | 0 | −1.794 | −1.833 |
| 2 | 3.88 | 0 | −1.906 | −1.880 |
| 3 | 2.12 | −1 | −1.988 | −2.020 |
| 4 | 2.89 | 0 | −1.994 | −2.045 |
| 5 | 3.53 | 0 | −1.996 | −1.938 |
| 6 | 3.18 | 0 | −1.997 | −1.997 |
| 7 | 2.83 | 0 | −2.027 | −2.055 |
| 8 | 1.56 | −1 | −2.071 | −2.113 |
| 9 | 2.36 | 0 | −2.100 | −2.133 |
| 10 | 1.99 | −1 | −2.109 | −2.041 |
| 11 | 2.49 | 0 | −2.124 | −2.112 |
| 12 | 2.87 | 1 | −2.180 | −2.202 |
| 13 | 2.27 | 0 | −2.217 | −2.148 |
| 14 | 2.61 | 1 | −2.261 | −2.246 |
Acknowledgements
References and Notes
- Botting, R. M. Cyclooxygenase: Past, present and future. A tribute to John R. Vane (1927–2004). Journal of Thermal Biology 2006, 31, 208–219. [Google Scholar]
- Kiefer, W.; Dannhardt, G. Novel Insights and Therapeutical Applications in the Field of Inhibitors of COX-2. Cur. Med. Chem. 2004, 11, 3147–3161. [Google Scholar]
- Goodsell, D. S. The Molecular Perspective: Cyclooxygenase-2; Oncologist 2000, 5, 169–171. [Google Scholar]
- Gobec, S.; Brožič, P.; Lanišnik-Rižner, T. Nonsteroidal drugs and their analogues as inhibitors of aldo-keto reductase AKR1C3; New lead compounds for the development of anticancer agents. Bioorg. Med. Chem. Lett. 2005, 15( 23), 5170–5175. [Google Scholar]
- The Merck Index on CD; 1997; Chapman and Hall EPD; Edition 12:2; entry 4924.
- The Merck Index on CD; 1997; Chapman and Hall EPD; Edition 12:2; entry 3989.
- The Merck Index on CD; 1997; Chapman and Hall EPD; Edition 12:2; entry 4925.Am. J. Med. 1984, 77(1A), 1–125.
- The Merck Index on CD; 1997; Chapman and Hall EPD; Edition 12:2; entry 6504.Drugs 1979, 18, 241–277.
- The Merck Index on CD; 1997; Chapman and Hall EPD; Edition 12:2; entry 4003.Drugs 1981, 21, 1–22.
- Cornforth, D. A.; Opara, A. E.; Read, G. General Synthetic Routes to β-Hydroxy-acids from t- Butyl Esters and Reformatskii Reaction; J. Chem. Soc 1969, 2799–2805, references cited therein. [Google Scholar]
- Horeau, A. Synthese des β-hydroxy-acides par une reaction de Reformatsky utilisant les α-bromoesters de trimethylsilyle. Tetrahedron Lett. 1971, 34, 3227–3228. [Google Scholar]
- Bogavac, M.; Arsenijeviċ, L,; Arsenijeviċ, V. Rėaction de Reformatsky à froid avec des α- bromoesters-acėtals. I. Une mėthode gėnėrale pour la synthėse des β-hydroxyacides à partir des α- bromoesters de tėtrahydropyrannyle ; Bull. Soc. Chim. Fr 1980, II-145. [Google Scholar]
- Varoli, L.; Burneli, S.; Guarnieri, A.; Bonazzi, D.; Scapini, G.; Sarret, M. Fantuz, Nichtsteriodale Entzűndungshemmer 19.Mitteilung: Synthese und Aktivität von stereomeren Phenylessigsäure- Arylhomologen. Pharmazie 1988, 43, 764–767. [Google Scholar]
- Bellassoued, M.; Cuoffignal, R.; Gaudemar, M. Synthése directe de β-hydroxyacides par réaction de Réformatsky. J. Organomet. Chem. 1973, 61, 9–18. [Google Scholar]
- Pavlov, S.; Bogavac, M.; Arsenijeviċ, L.; Klajn, E.; Arsenijeviċ, V. Preparation of some choleretic drugs by modified Reformatsky reaction. Direct synthesis of β-hydroxy acids. Acta Pharm. Jugosl. 1986, 36, 47–51. [Google Scholar]
- Arsenijeviċ, L.; Bogavac, M.; Pavlov, S. A Simple Synthesis of Some Ssubstituted Phenylalkanoic Acids with Potential Activity. (Printed in Serbian abstract in English). Arh. farm 1992, 42, 127–130. [Google Scholar]
- Bogavac, M.; Arsenijeviċ, L.; Pavlov, S.; Arsenijeviċ, V. Reformatsky Reaction with α-Bromo Esters-Acetals in the Cold. II. A General Method for the Preparation of β-Hydroxy Acids by Using 1-Alkoxyalkyl Esters of α-Bromo Acids. (Printed in Serbian abstract in English). Arh. farm. 1988, 38(3), 93–97. [Google Scholar]
- Urano, F.; Nakahata, M.; Tonoyamacho, H.; Fuije, H.; Oono, K. Resist Material and Process for Forming Pattern Using the Same. EU pat, 0 476 865 A1 2003. CA 117:P101049n. [Google Scholar]
- Clothier, R.H. The FRAME cytotoxicity test (Kenacid Blue). Methods in Molecular Biology 1995, 43, 109–118. [Google Scholar]
- Dilber, S. P.; Juranić, Z. D.; Stanojković, T. P.; Drakulić, B. J.; Juranić, I. O. Antiproliferative Action of β-Hydroxy-β-Arylakanoic Acids Toward Human HeLa Cells. A QSAR study. Abstracts of the 43rd Symposium of Serbian Chemical Society, Belgrade, Serbia and Montenegro; 2005. [Google Scholar]
- Dilber, S. P.; Juranić, Z. D.; Žižak, S. Ž.; Drakulić, B. J.; Juranić, I. O. β-Hydroxy-β- Arylalkanoic Acids Selectively Suppress Proliferation of Four Human Tumor Cell Lines in vitro. Abstracts of the Sixth European Meeting on Environmental Chemistry (EMEC 6th), Belgrade, Serbia and Montenegro; 2005. [Google Scholar]
- Ghose, A. K.; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional- Structure-Directed Quantitative Structure-Activity Relationships. 2. Modeling Dispersive and Hydrophobic Interactions. J. Chem. Inf. Comput. Sci 1987, 27, 21–35. [Google Scholar]
- Free, S. M.; Wilson, J. W. A mathematical contribution to structure-activity studies. J. Med. Chem. 1971, 7(4), 395–399. [Google Scholar]
- Pedretti, A.L.; Villa, L.; Vistoli, G. VEGA - An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Com. Aided Mol. Des. 2004, 18, 167–173. ; Vega ZZ 2.0.5, http://www.ddl.unimi.it. [Google Scholar]
- For superimposition of the molecules the arbitrary chosen, but samestereochemistry (Sor S,R) of enantiomers or diastereomers are used. For the superimposition of cycloalkyl-substituted compound 10 the C2 and C6 of cyclohexyl ring were superimposed on the corresponding carbons of phenyl ring of the 1 For the 3,3-diphenyl compound, one of phenyl rings is arbitrarily chosen.
- Du, G. J.; Lin, H. H.; Xu, Q. T.; Wang, M.W. Bcl-2 switches the type of demise from apoptosis to necrosis via cyclooxygenase-2 upregulation in HeLa cell induced by hydrogen peroxide. Cancer Letters 2006, 232(2), 179–188. [Google Scholar]
- When peeled thin zinc particles were used, the reaction times were from 10 to 48 h. Usage of zinc powder significantly increases reaction times – up to 8 days.
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Dilber, S.P.; Žižak, Ž.S.; Stanojković, T.P.; Juranić, Z.D.; Drakulić, B.J.; Juranić, I.O. Antiproliferative Activity of β-Hydroxy-β-Arylalkanoic Acids. Int. J. Mol. Sci. 2007, 8, 214-228. https://doi.org/10.3390/i8030214
Dilber SP, Žižak ŽS, Stanojković TP, Juranić ZD, Drakulić BJ, Juranić IO. Antiproliferative Activity of β-Hydroxy-β-Arylalkanoic Acids. International Journal of Molecular Sciences. 2007; 8(3):214-228. https://doi.org/10.3390/i8030214
Chicago/Turabian StyleDilber, Sanda P., Željko S. Žižak, Tatjana P. Stanojković, Zorica D. Juranić, Branko J. Drakulić, and Ivan O. Juranić. 2007. "Antiproliferative Activity of β-Hydroxy-β-Arylalkanoic Acids" International Journal of Molecular Sciences 8, no. 3: 214-228. https://doi.org/10.3390/i8030214
APA StyleDilber, S. P., Žižak, Ž. S., Stanojković, T. P., Juranić, Z. D., Drakulić, B. J., & Juranić, I. O. (2007). Antiproliferative Activity of β-Hydroxy-β-Arylalkanoic Acids. International Journal of Molecular Sciences, 8(3), 214-228. https://doi.org/10.3390/i8030214