
Direct Cardiac Epigenetic Reprogramming through Codelivery of 5′Azacytidine and miR-133a Nanoformulation

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
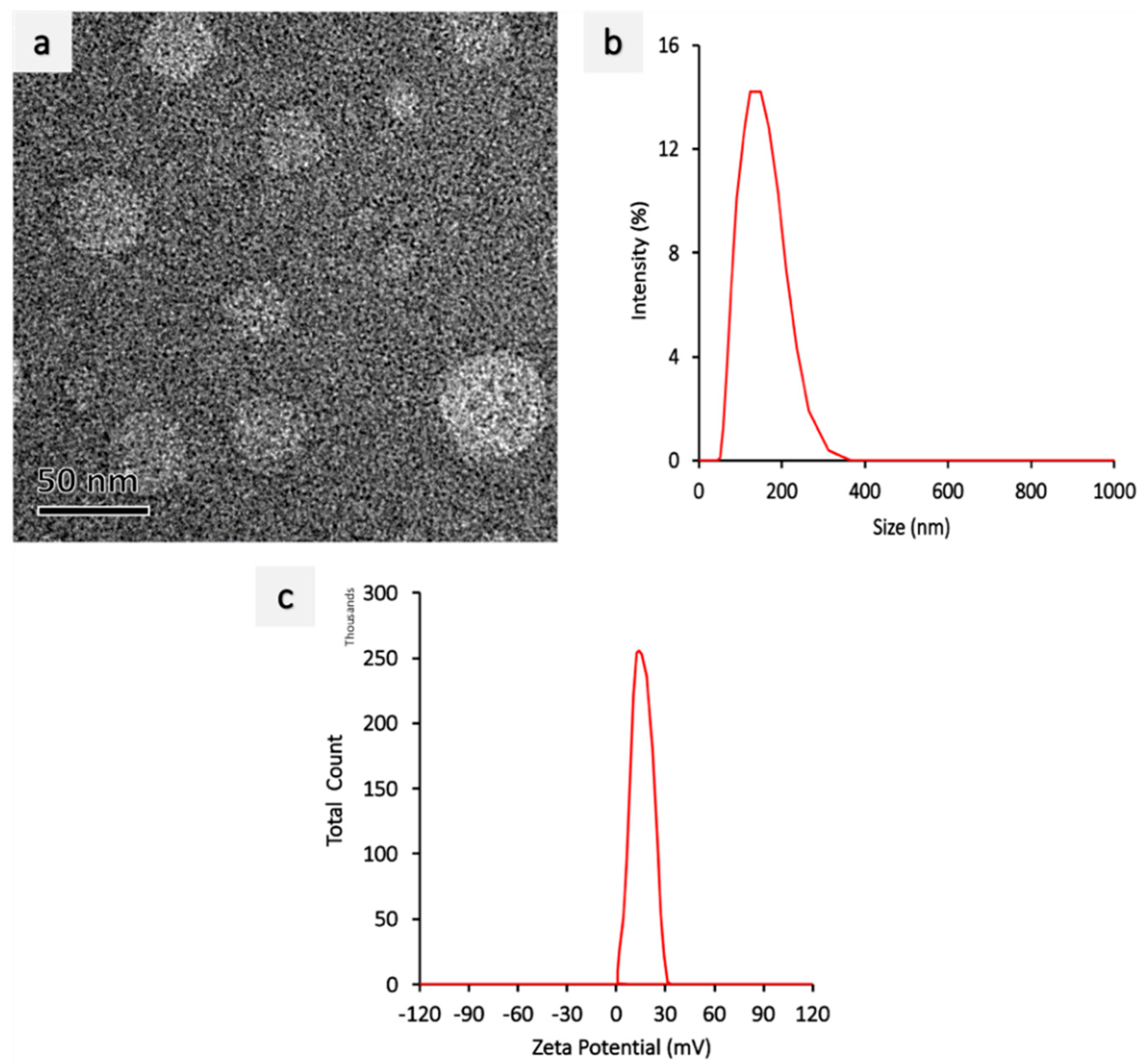
2.1. Synthesis and Characterization of PEI-miR-133a, PLGA, PLGA-AZA, PLGA-PEI-miR-133a, PEI-miR-133a-AZA PLGA Nanoparticles
2.2. Encapsulation and Stability of PEI-miR-133a Polyplexes
2.3. Invitro Release Studies
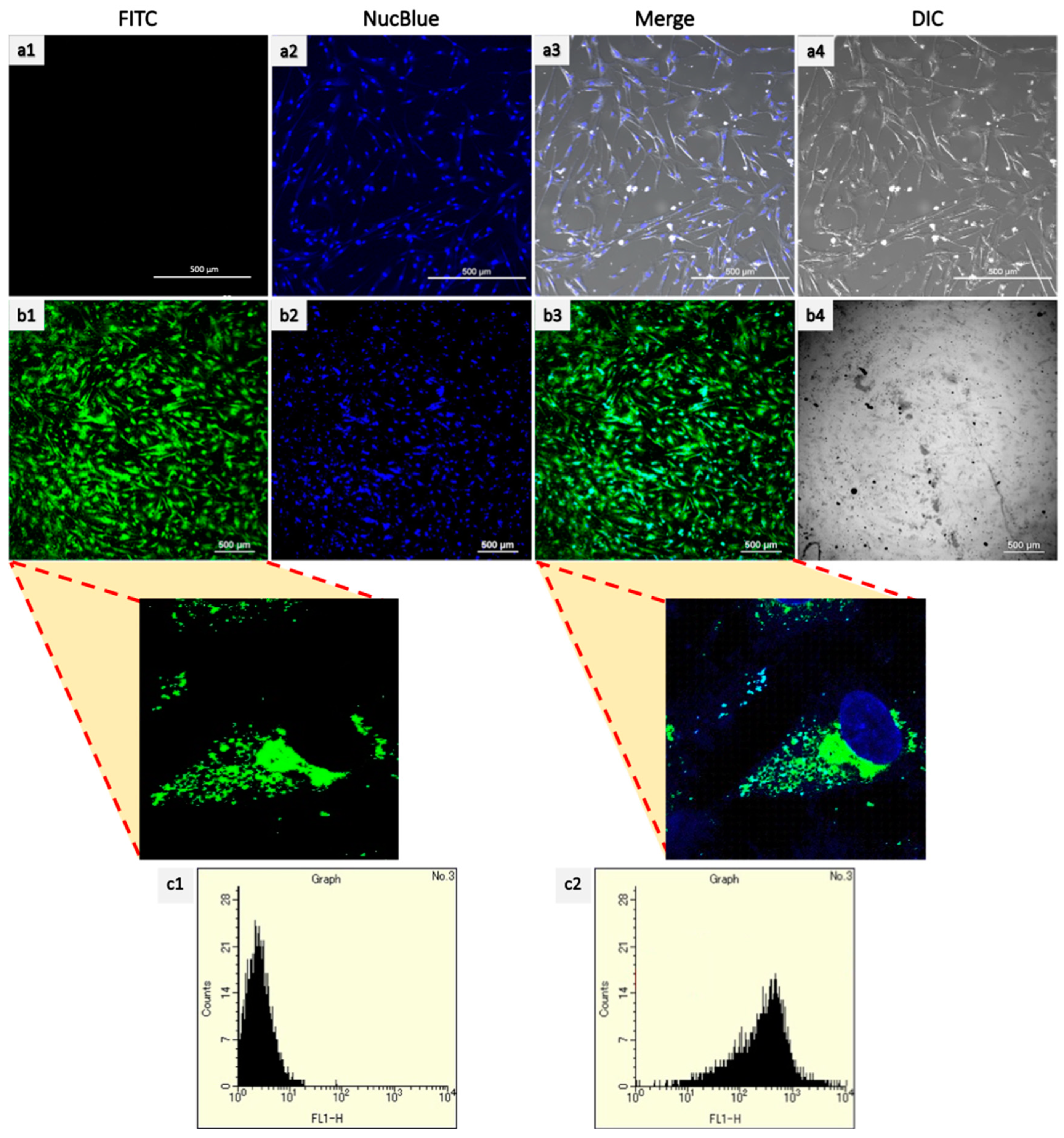
2.4. Cellular Uptake
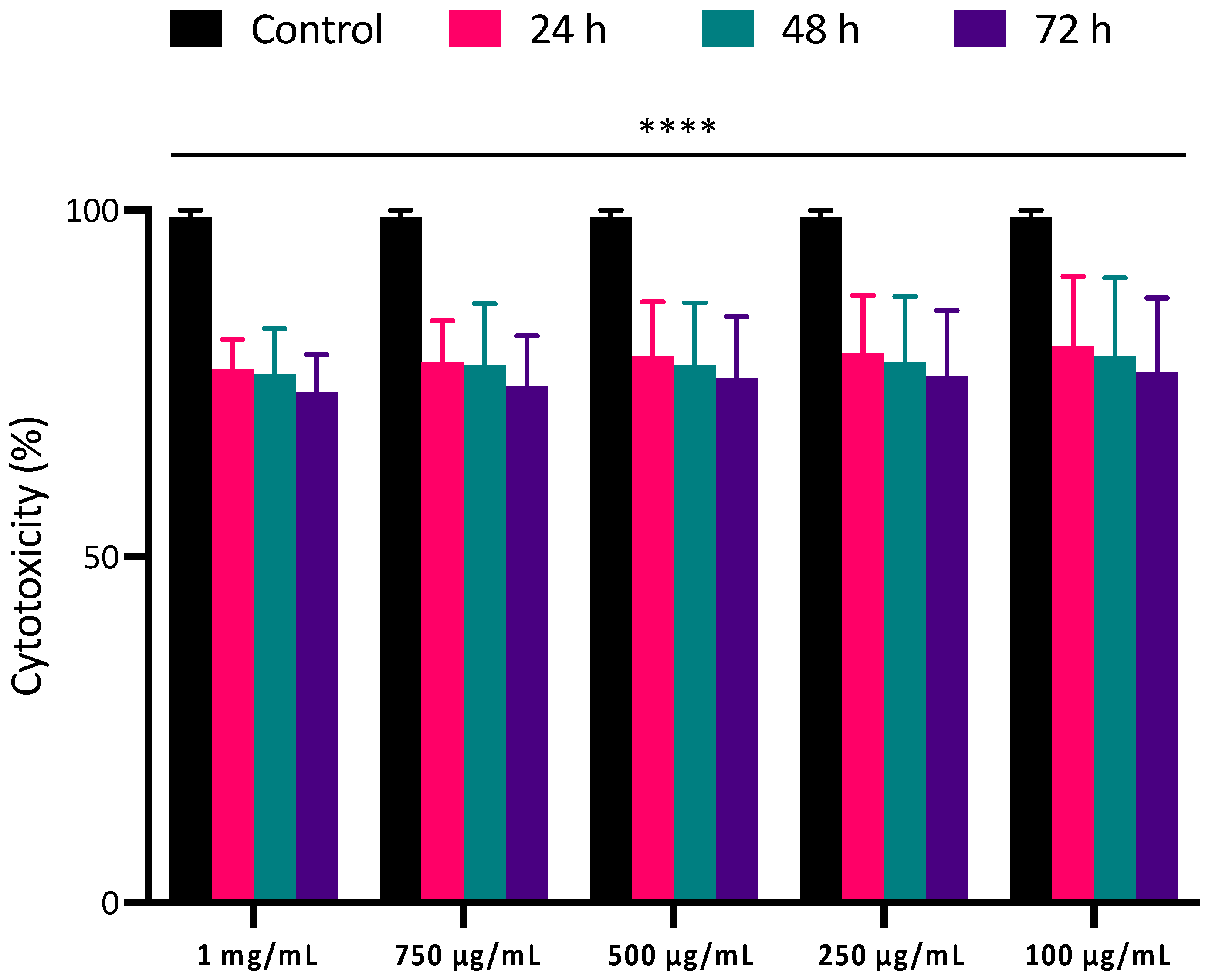
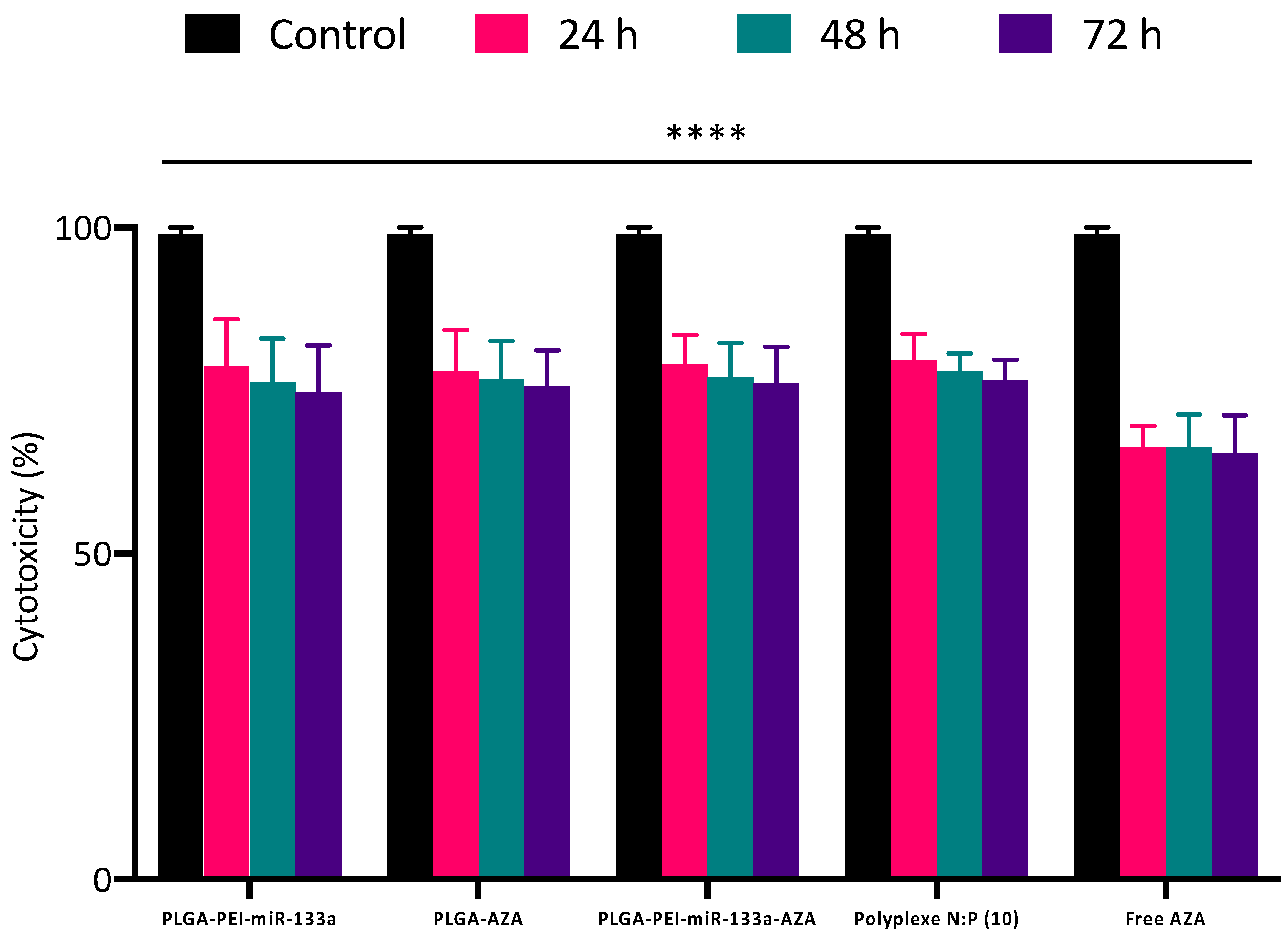
2.5. Cytocompatibility and Cell Viability of PLGA NPs
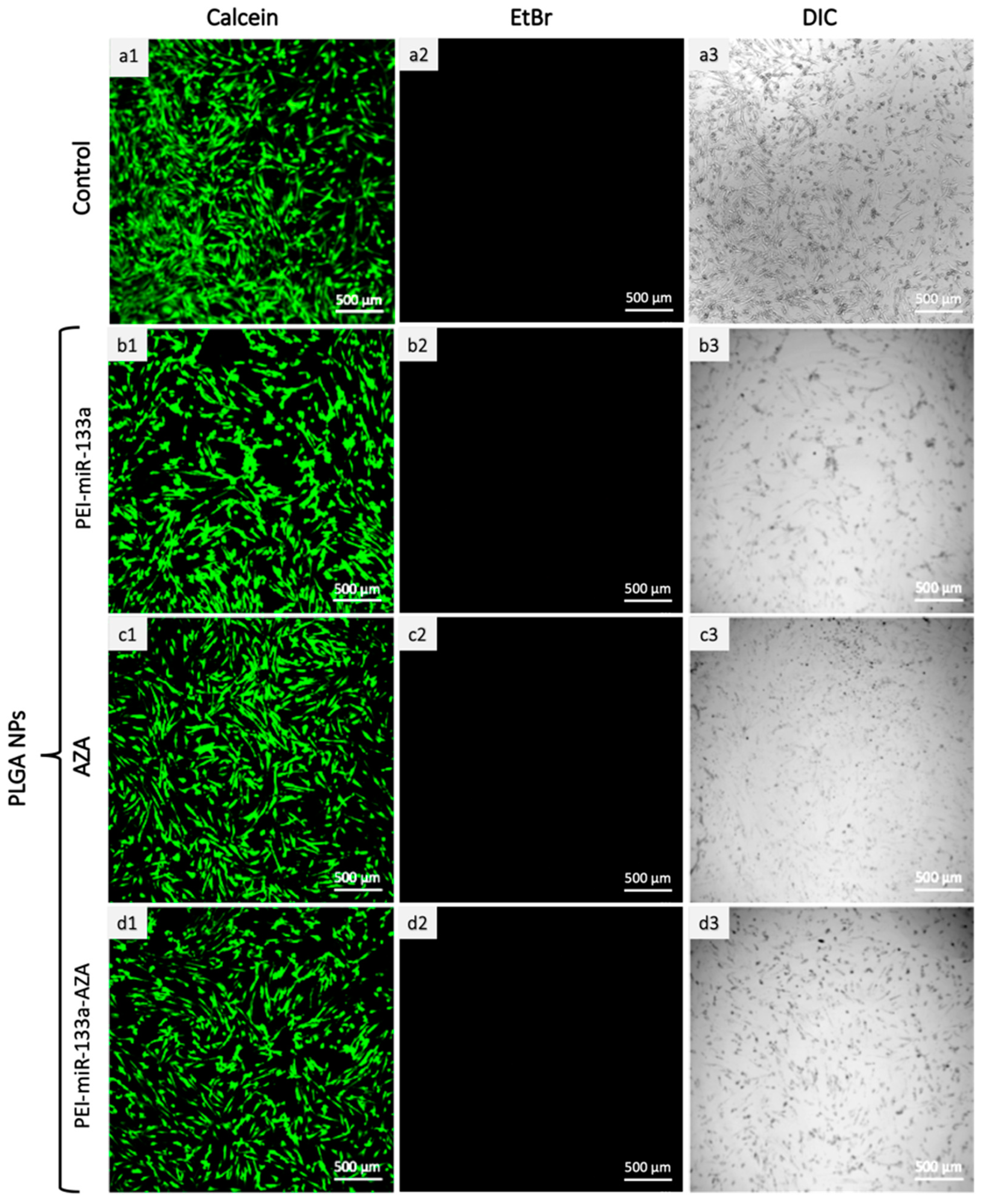
2.6. Live/Dead Cytotoxicity Analysis
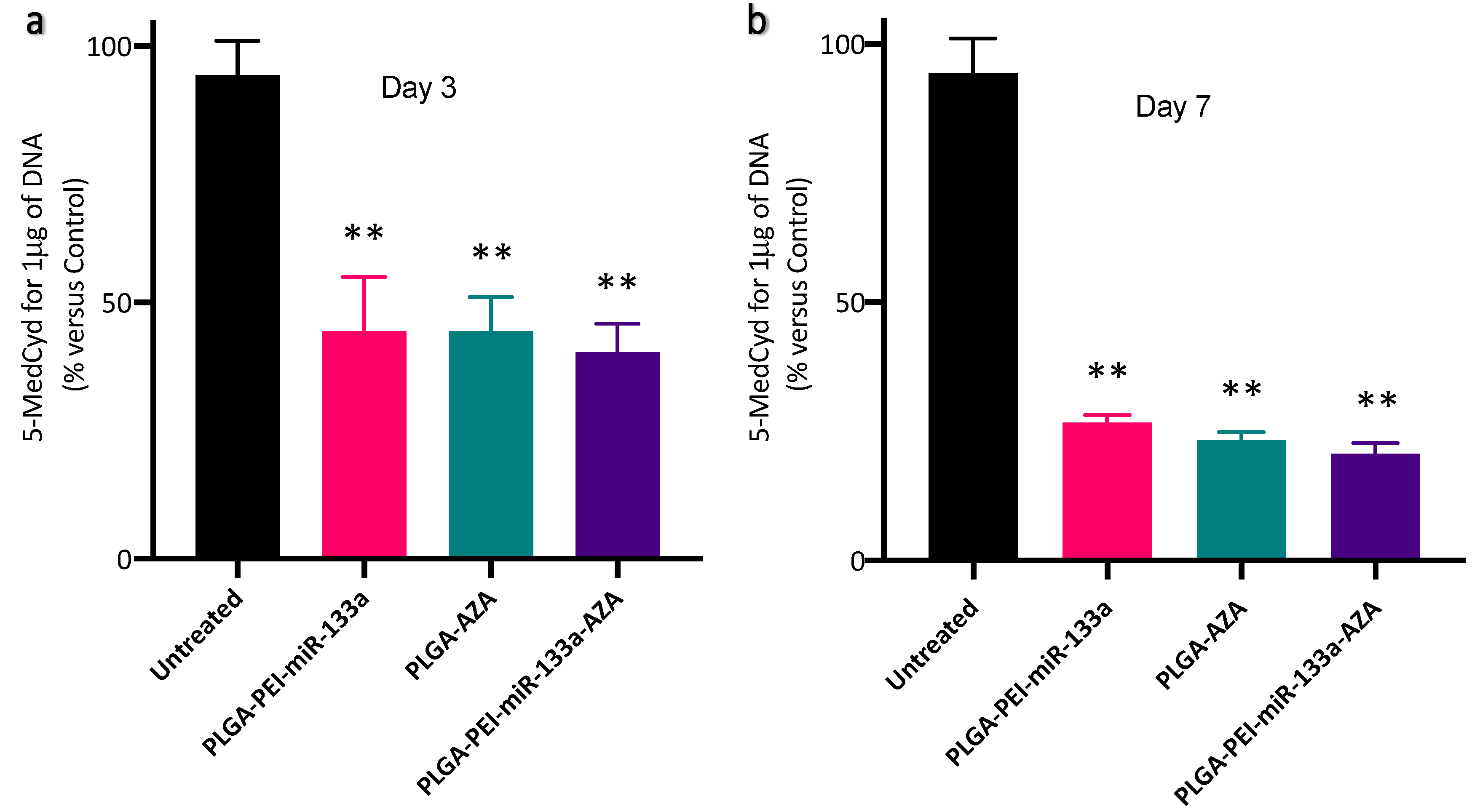
2.7. cTnT Expression and Global Methylation Analysis
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. miRNA Polyplexes Preparation and Gel Retardation Assay
3.2.2. Synthesis and Characterization of PEI-miR-133a, PLGA AZA, and PLGA-PEI-miRNA-AZA
3.2.3. Physico-Chemical Characterization
3.2.4. Percentage Yield and Encapsulation Efficiency
3.2.5. Gel Electrophoresis Assay to Confirm Encapsulation and Stability of miRNA Polyplexes to PLGA Nanoparticles
3.2.6. Invitro miRNA/Drug Release
3.2.7. Cell Culture
3.2.8. PLGA-PEI-miR133a Transfection
3.2.9. PrestoBlue Cell Viability Assay
3.2.10. Live/Dead Cell Cytotoxicity Assay
3.2.11. Cellular Uptake
3.2.12. cTnT Cardiac Late Marker Expression
3.2.13. RNA Isolation Quantitative Real-Time PCR (qRT-PCR)
3.2.14. Global Methylation Analysis
3.2.15. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Cardiovascular Diseases (CVDs); WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strzyz, P. Regeneration: Mending broken hearts. Nat. Rev. Mol. Cell Biol. 2018, 19, 277. [Google Scholar] [CrossRef]
- Baumann, K. Mending broken hearts with the help of epigenetic remodellers. Nat. Rev. Mol. Cell Biol. 2021, 22, 371. [Google Scholar] [CrossRef] [PubMed]
- Muniyandi, P.; Maekawa, T.; Hanajiri, T.; Palaninathan, V. Direct Cardiac Reprogramming with Engineered miRNA Scaffolds. Curr. Pharm. Des. 2020, 26, 4285–4303. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Ma, W.; Solov’yov, I.A.; Chipot, C.; Schulten, K. Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res. 2012, 40, 2747–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wang, L.; Vaseghi, H.R.; Liu, Z.; Lu, R.; Alimohamadi, S.; Yin, C.; Fu, J.D.; Wang, G.G.; Liu, J.; et al. Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 2016, 18, 382–395. [Google Scholar] [CrossRef] [Green Version]
- Testa, G.; Russo, M.; Di Benedetto, G.; Barbato, M.; Parisi, S.; Pirozzi, F.; Tocchetti, C.G.; Abete, P.; Bonaduce, D.; Russo, T.; et al. Bmi1 inhibitor PTC-209 promotes Chemically-induced Direct Cardiac Reprogramming of cardiac fibroblasts into cardiomyocytes. Sci. Rep. 2020, 10, 7129. [Google Scholar] [CrossRef]
- Singh, V.P.; Pinnamaneni, J.P.; Pugazenthi, A.; Sanagasetti, D.; Mathison, M.; Wang, K.; Yang, J.; Rosengart, T.K. Enhanced Generation of Induced Cardiomyocytes Using a Small-Molecule Cocktail to Overcome Barriers to Cardiac Cellular Reprogramming. J. Am. Heart Assoc. 2020, 9, e015686. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Kikyo, N. Inhibitors of suppressive histone modification promote direct reprogramming of fibroblasts to cardiomyocyte-like cells. Cardiovasc. Res. 2014, 102, 188–190. [Google Scholar] [CrossRef]
- Dal-Pra, S.; Hodgkinson, C.P.; Dzau, V.J. Induced cardiomyocyte maturation: Cardiac transcription factors are necessary but not sufficient. PLoS ONE 2019, 14, e0223842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal-Pra, S.; Hodgkinson, C.P.; Mirotsou, M.; Kirste, I.; Dzau, V.J. Demethylation of H3K27 Is Essential for the Induction of Direct Cardiac Reprogramming by miR Combo. Circ Res. 2017, 120, 1403–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šorm, F.; Pískala, A.; Čihák, A.; Veselý, J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia 1964, 20, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Fukuda, K.; Miyoshi, S.; Konishi, F.; Kodama, H.; Pan, J.; Sano, M.; Takahashi, T.; Hori, S.; Abe, H.; et al. Cardiomyocytes can be generated from marrow stromal cells in vitro. J. Clin. Investig. 1999, 103, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakuno, D.; Fukuda, K.; Makino, S.; Konishi, F.; Tomita, Y.; Manabe, T.; Suzuki, Y.; Umezawa, A.; Ogawa, S. Bone marrow-derived regenerated cardiomyocytes (CMG Cells) express functional adrenergic and muscarinic receptors. Circulation 2002, 105, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Rangappa, S.; Fen, C.; Lee, E.H.; Bongso, A.; Sim, E.K. Transformation of adult mesenchymal stem cells isolated from the fatty tissue into cardiomyocytes. Ann. Thorac. Surg. 2003, 75, 775–779. [Google Scholar] [CrossRef]
- Palacios, D.; Puri, P.L. The epigenetic network regulating muscle development and regeneration. J. Cell. Physiol. 2006, 207, 1–11. [Google Scholar] [CrossRef]
- Leptidis, S.; El Azzouzi, H.; Lok, S.I.; de Weger, R.; Olieslagers, S.; Kisters, N.; Silva, G.J.; Heymans, S.; Cuppen, E.; Berezikov, E.; et al. A deep sequencing approach to uncover the miRNOME in the human heart. PLoS ONE 2013, 8, e57800. [Google Scholar] [CrossRef]
- McCarthy, J.J.; Esser, K.A. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J. Appl. Physiol. 2007, 102, 306–313. [Google Scholar] [CrossRef]
- Melman, Y.F.; Shah, R.; Das, S. MicroRNAs in heart failure: Is the picture becoming less miRky? Circ. Heart Fail. 2014, 7, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Care, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Chavali, V.; Tyagi, S.C.; Mishra, P.K. MicroRNA-133a regulates DNA methylation in diabetic cardiomyocytes. Biochem. Biophys. Res. Commun. 2012, 425, 668–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Puthanveetil, P.; Feng, B.; Matkovich, S.J.; Dorn, G.W., 2nd; Chakrabarti, S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J. Cell. Mol. Med. 2014, 18, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaldi, A.; Zaglia, T.; Di Mauro, V.; Carullo, P.; Viggiani, G.; Borile, G.; Di Stefano, B.; Schiattarella, G.G.; Gualazzi, M.G.; Elia, L.; et al. MicroRNA-133 modulates the beta1-adrenergic receptor transduction cascade. Circ. Res. 2014, 115, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Ham, O.; Cha, M.J.; Song, B.W.; Choi, E.; Kim, I.K.; Chang, W.; Lim, S.; Lee, C.Y.; Park, J.H.; et al. The promotion of cardiogenic differentiation of hMSCs by targeting epidermal growth factor receptor using microRNA-133a. Biomaterials 2013, 34, 92–99. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef] [Green Version]
- Muniyandi, P.; Palaninathan, V.; Mizuki, T.; Maekawa, T.; Hanajiri, T.; Mohamed, M.S. Poly(lactic-co-glycolic acid)/Polyethylenimine Nanocarriers for Direct Genetic Reprogramming of MicroRNA Targeting Cardiac Fibroblasts. ACS Appl. Nano Mater. 2020, 3, 2491–2505. [Google Scholar] [CrossRef]
- Muniyandi, P.; Palaninathan, V.; Mizuki, T.; Mohamed, M.S.; Hanajiri, T.; Maekawa, T. Scaffold Mediated Delivery of Dual miRNAs to Transdifferentiate Cardiac Fibroblasts. Mater. Eng. C 2021, 128, 112323. [Google Scholar] [CrossRef]
- Zhang, Y.; Chan, H.F.; Leong, K.W. Advanced materials and processing for drug delivery: The past and the future. Adv. Drug Deliv. Rev. 2013, 65, 104–120. [Google Scholar] [CrossRef]
- Lim, M.P.A.; Lee, W.L.; Widjaja, E.; Loo, S.C.J. One-step fabrication of core-shell structured alginate–PLGA/PLLA microparticles as a novel drug delivery system for water soluble drugs. Biomater. Sci. 2013, 1, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lin, L.; Wang, W.; Yan, X.; Chen, B.; Jiang, S.; Liu, S.; Ma, X.; Tian, H.; Yu, X. Aza-crown ether locked on polyethyleneimine: Solving the contradiction between transfection efficiency and safety during in vivo gene delivery. Chem. Commun. 2020, 56, 5552–5555. [Google Scholar] [CrossRef]
- Fu, X.; Ping, Q.; Gao, Y. Effects of formulation factors on encapsulation efficiency and release behaviour in vitro of huperzine A-PLGA microspheres. J. Microencapsul. 2005, 22, 705–714. [Google Scholar] [CrossRef]
- Devulapally, R.; Foygel, K.; Sekar, T.V.; Willmann, J.K.; Paulmurugan, R. Gemcitabine and Antisense-microRNA Co-encapsulated PLGA-PEG Polymer Nanoparticles for Hepatocellular Carcinoma Therapy. ACS Appl. Mater. Interfaces 2016, 8, 33412–33422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.; Dai, D.; Huang, B. Fourier Transform Infrared Spectroscopy for Natural Fibres. Fourier Transform.-Mater. Anal. 2012, 3, 45–68. [Google Scholar]
- Portaccio, M.; Menale, C.; Diano, N.; Mita, D.G.; Lepore, M. FT-IR microspectroscopy of cisplatin loaded PLGA nanoparticles. In Proceedings of the 2014 Fotonica AEIT Italian Conference on Photonics Technologies, Naples, Italy, 12–14 May 2014; pp. 1–3. [Google Scholar]
- Busatto, C.; Pesoa, J.; Helbling, I.; Luna, J.; Estenoz, D. Effect of particle size, polydispersity and polymer degradation on progesterone release from PLGA microparticles: Experimental and mathematical modeling. Int. J. Pharm. 2018, 536, 360–369. [Google Scholar] [CrossRef]
- Mahboob, T.; Nawaz, M.; de Lourdes Pereira, M.; Tian-Chye, T.; Samudi, C.; Sekaran, S.D.; Wiart, C.; Nissapatorn, V. PLGA nanoparticles loaded with Gallic acid—A constituent of Leea indica against Acanthamoeba triangularis. Sci. Rep. 2020, 10, 8954. [Google Scholar] [CrossRef]
- Ferreira, M.P.A.; Talman, V.; Torrieri, G.; Liu, D.; Marques, G.; Moslova, K.; Liu, Z.; Pinto, J.; Hirvonen, J.; Ruskoaho, H.; et al. Dual-Drug Delivery Using Dextran-Functionalized Nanoparticles Targeting Cardiac Fibroblasts for Cellular Reprogramming. Adv. Funct. Mater. 2018, 28, 1705134. [Google Scholar] [CrossRef]
- Cartiera, M.S.; Johnson, K.M.; Rajendran, V.; Caplan, M.J.; Saltzman, W.M. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials 2009, 30, 2790–2798. [Google Scholar] [CrossRef] [Green Version]
- Mora-Huertas, C.E.; Fessi, H.; Elaissari, A. Influence of process and formulation parameters on the formation of submicron particles by solvent displacement and emulsification-diffusion methods critical comparison. Adv. Colloid Interface Sci. 2011, 163, 90–122. [Google Scholar] [CrossRef]
- Ghavimi, M.A.; Bani Shahabadi, A.; Jarolmasjed, S.; Memar, M.Y.; Maleki Dizaj, S.; Sharifi, S. Nanofibrous asymmetric collagen/curcumin membrane containing aspirin-loaded PLGA nanoparticles for guided bone regeneration. Sci. Rep. 2020, 10, 18200. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sato, K.; Kitakami, E.; Kobayashi, S.; Hoshiba, T.; Fukushima, K. Design of biocompatible and biodegradable polymers based on intermediate water concept. Polym. J. 2015, 47, 114–121. [Google Scholar] [CrossRef]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.M.; Jones, P.A. Changes in phenotypic expression in embryonic and adult cells treated with 5-azacytidine. J. Cell Physiol. 1982, 111, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dal-Pra, S.; Mirotsou, M.; Jayawardena, T.M.; Hodgkinson, C.P.; Bursac, N.; Dzau, V.J. Tissue-engineered 3-dimensional (3D) microenvironment enhances the direct reprogramming of fibroblasts into cardiomyocytes by microRNAs. Sci. Rep. 2016, 6, 38815. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Li, H.; Zhu, Y.; Xu, P.; Zuo, Q.; Li, B.; Gu, X. 5-Azacytidine-Induced Cardiomyocyte Differentiation of Very Small Embryonic-Like Stem Cells. Stem Cells Int. 2020, 2020, 5162350. [Google Scholar] [CrossRef]
- Abbey, D.; Seshagiri, P.B. Aza-induced cardiomyocyte differentiation of P19 EC-cells by epigenetic co-regulation and ERK signaling. Gene 2013, 526, 364–373. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Finch, E.A.; Zhang, L.; Zhang, H.; Hodgkinson, C.P.; Pratt, R.E.; Rosenberg, P.B.; Mirotsou, M.; Dzau, V.J. MicroRNA induced cardiac reprogramming in vivo: Evidence for mature cardiac myocytes and improved cardiac function. Circ. Res. 2015, 116, 418–424. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Z-Average Diameter (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) | Yield (%) | Encapsulation Efficiency (%) |
|---|---|---|---|---|---|
| PLGA void | 186 ± 3.1 | 0.063 ± 0.06 | −15.6 ± 0.04 | 37.13% | - |
| PEI miRNA | 124 ± 0.6 | 0.301 ± 0.04 | 27.35 ± 0.03 | - | |
| PLGA AZA | 298 ± 2.0 | 0.034 ± 0.03 | −15.8 ± 0.86 | 37.35% | 96% |
| PLGA AZA-miRNA | 298 ± 2.4 | 0.031 ± 0.02 | −15.7 ± 0.65 | 38.15% | 97% |
| PLGA-miRNA | 298 ± 2.6 | 0.032 ± 0.02 | −15.6 ± 0.34 | 37.35% | 96% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muniyandi, P.; Palaninathan, V.; Hanajiri, T.; Maekawa, T. Direct Cardiac Epigenetic Reprogramming through Codelivery of 5′Azacytidine and miR-133a Nanoformulation. Int. J. Mol. Sci. 2022, 23, 15179. https://doi.org/10.3390/ijms232315179
Muniyandi P, Palaninathan V, Hanajiri T, Maekawa T. Direct Cardiac Epigenetic Reprogramming through Codelivery of 5′Azacytidine and miR-133a Nanoformulation. International Journal of Molecular Sciences. 2022; 23(23):15179. https://doi.org/10.3390/ijms232315179
Chicago/Turabian StyleMuniyandi, Priyadharshni, Vivekanandan Palaninathan, Tatsuro Hanajiri, and Toru Maekawa. 2022. "Direct Cardiac Epigenetic Reprogramming through Codelivery of 5′Azacytidine and miR-133a Nanoformulation" International Journal of Molecular Sciences 23, no. 23: 15179. https://doi.org/10.3390/ijms232315179