
Inhibitory Effects of Nobiletin on Voltage-Gated Na+ Channel in Rat Ventricular Myocytes Based on Electrophysiological Analysis and Molecular Docking Method

Abstract
:1. Introduction
2. Results
2.1. NOB Alleviated Fatal Ventricular Arrhythmia In Vivo
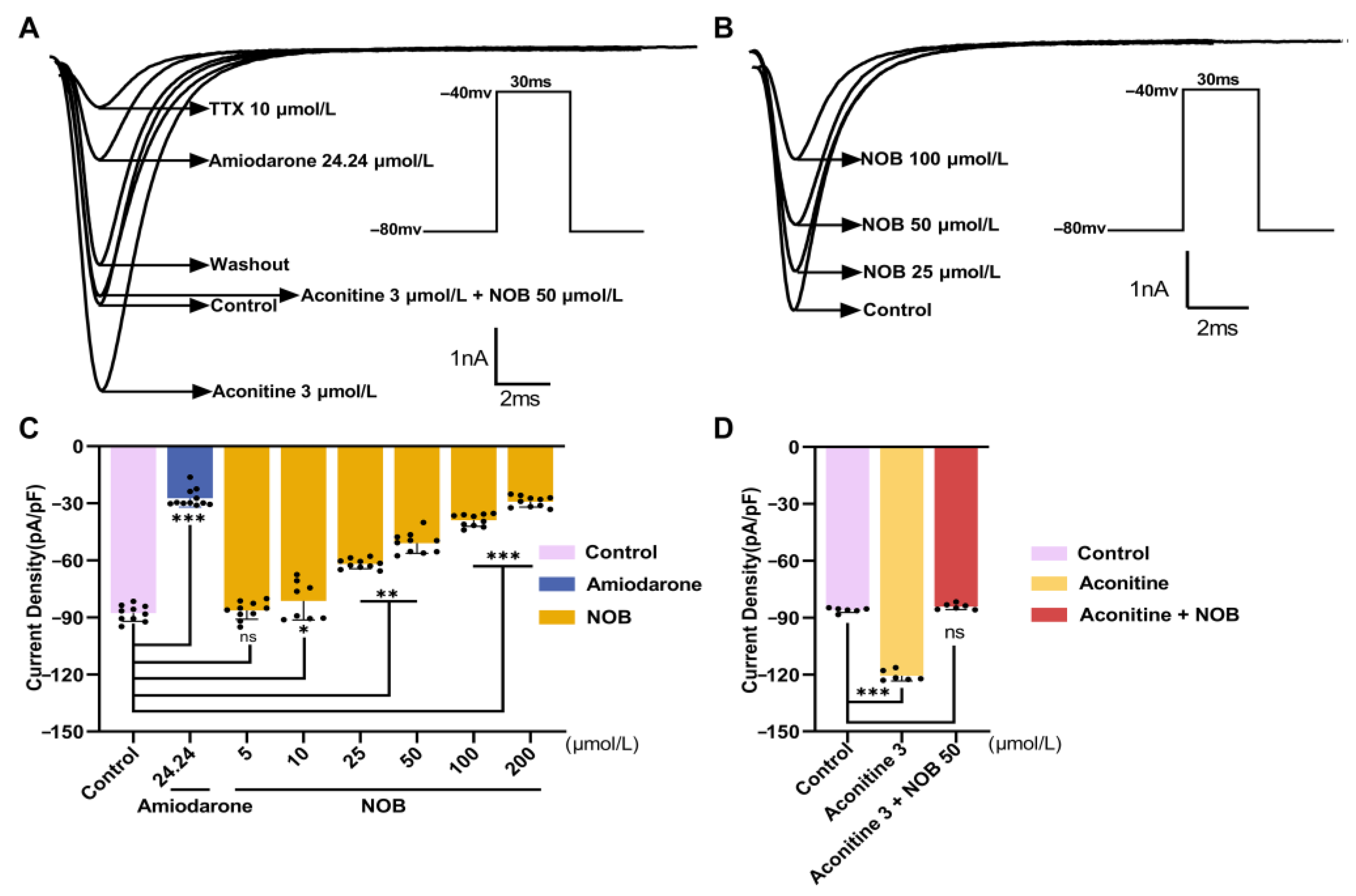
2.2. NOB Inhibited INa in a Concentration-Dependent Manner
2.3. Effects of NOB on the Current–Voltage Curve (I–V Curve) of INa
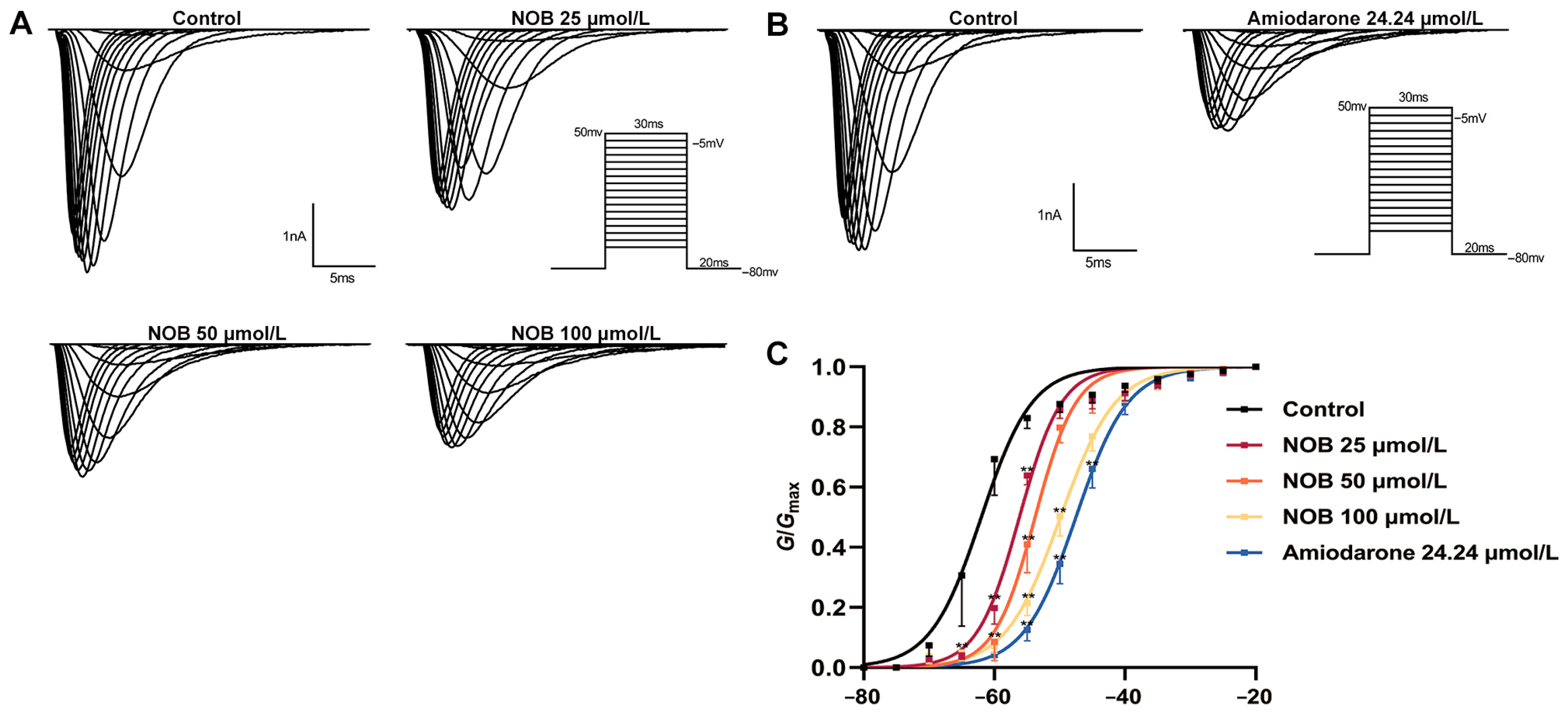
2.4. Effects of NOB on the Steady-State INa Activation Curve
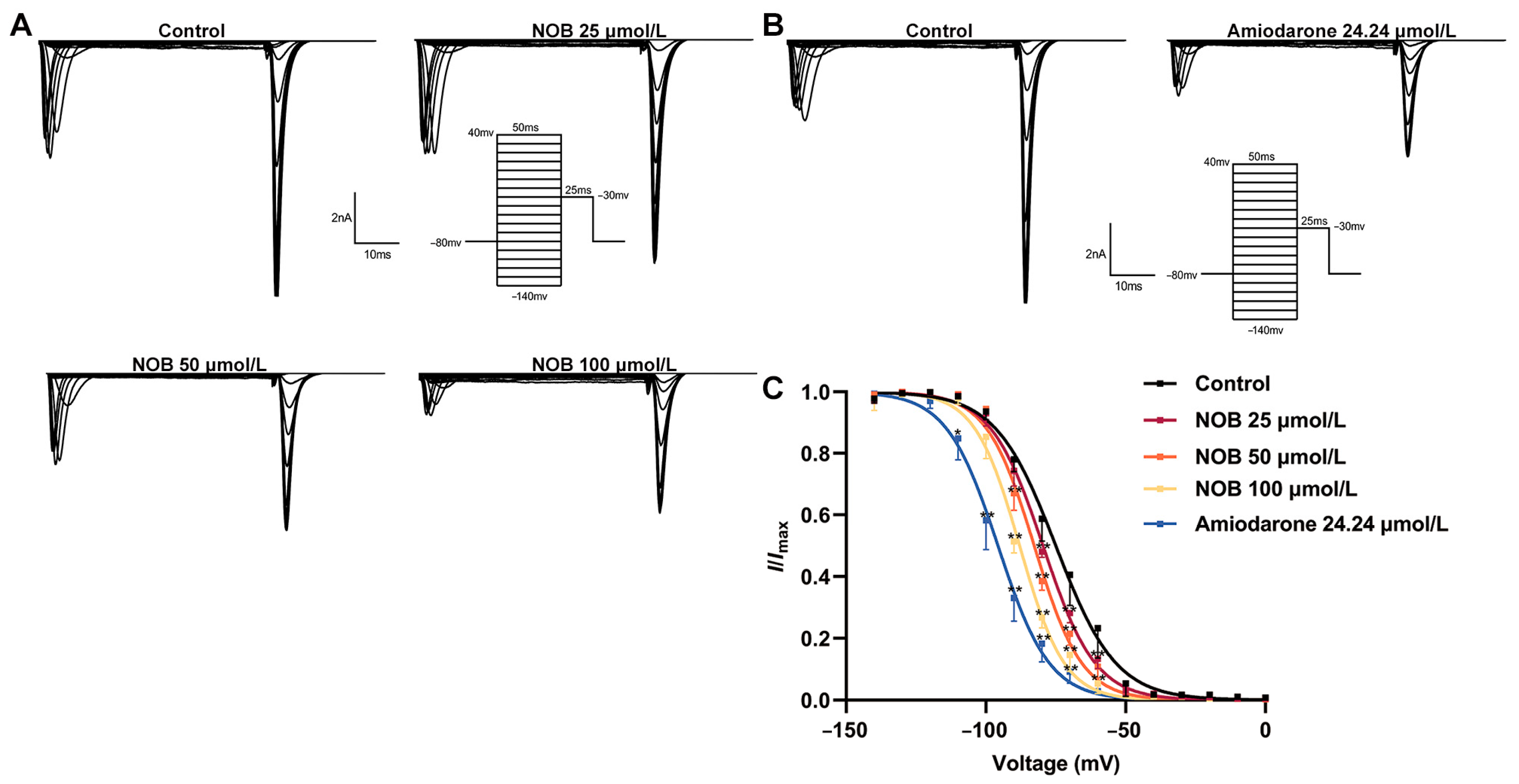
2.5. Effects of NOB on the INa Inactivation Curve
2.6. Effects of NOB on Recovery Curves after Inactivation of INa
2.7. Molecular Docking Simulation
3. Discussion
4. Materials and Methods
4.1. Animals and Ethics Statement
4.2. Drugs and Reagents
4.3. Solutions
4.4. Establishment of Ventricular Arrhythmia In Vivo
4.5. Acute Single-Cell Isolation
4.6. Stimulus Protocols
4.7. Whole-Cell Patch-Clamp Recording
4.8. Molecular Modeling and Computational Methods
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamilton, S.; Veress, R.; Belevych, A.; Terentyev, D. The role of calcium homeostasis remodeling in inherited cardiac arrhythmia syndromes. Pflugers Arch. 2021, 473, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Gumz, M.L.; Rabinowitz, L. Role of circadian rhythms in potassium homeostasis. Semin. Nephrol. 2013, 33, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Nolan, J.P.; Maconochie, I.; Soar, J.; Olasveengen, T.M.; Greif, R.; Wyckoff, M.H.; Singletary, E.M.; Aickin, R.; Berg, K.M.; Mancini, M.E.; et al. Executive Summary 2020 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science with Treatment Recommendations. Resuscitation 2020, 156, A1–A22. [Google Scholar] [CrossRef] [PubMed]
- Bui, A.H.; Waks, J.W. Risk Stratification of Sudden Cardiac Death After Acute Myocardial Infarction. J. Innov. Card Rhythm Manag. 2018, 9, 3035–3049. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Liu, Y.; Yu, H.; Xu, N.; Huang, D.; Xue, Y.; Li, S.; Chen, H.; Liu, J.; et al. Fibroblast Growth Factor 21 Ameliorates NaV1.5 and Kir2.1 Channel Dysregulation in Human AC16 Cardiomyocytes. Front. Pharmacol. 2021, 12, 715466. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; He, Y.; Wang, X.; Ge, J.; Li, H. Ventricular voltage-gated ion channels: Detection, characteristics, mechanisms, and drug safety evaluation. Clin. Transl. Med. 2021, 11, e530. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.A. Sodium leak through K2P potassium channels and cardiac arrhythmia, an emerging theme. EMBO Mol. Med. 2017, 9, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Pieroni, M.; Fatah, M.; Charpentier, F.; Cunningham, K.S.; Spears, D.A.; Chatterjee, D.; Suna, G.; Bos, J.M.; Ackerman, M.J.; et al. An autoantibody profile detects Brugada syndrome and identifies abnormally expressed myocardial proteins. Eur. Heart J. 2020, 41, 2878–2890. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jin, X.; Wu, T.; Zhao, X.; Wang, W.; Lei, J.; Pan, X.; Yan, N. Structure of human Nav1.5 reveals the fast inactivation-related segments as a mutational hotspot for the long QT syndrome. Proc. Natl. Acad. Sci. USA 2021, 118, e21000691. [Google Scholar]
- Shimazu, R.; Anada, M.; Miyaguchi, A.; Nomi, Y.; Matsumoto, H. Evaluation of Blood-Brain Barrier Permeability of Polyphenols, Anthocyanins, and Their Metabolites. J. Agric. Food Chem. 2021, 69, 11676–11686. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.H.; Yang, S.F.; Cheng, C.C.; Hsu, K.C.; Chen, Y.S.; Chen, Y.Y.; Wang, C.W.; Guan, S.S.; Wu, C.T. Nobiletin Alleviates Ferroptosis-Associated Renal Injury, Inflammation, and Fibrosis in a Unilateral Ureteral Obstruction Mouse Model. Biomedicines 2022, 10, 595. [Google Scholar] [CrossRef] [PubMed]
- Iampanichakul, M.; Poasakate, A.; Potue, P.; Rattanakanokchai, S.; Maneesai, P.; Prachaney, P.; Settheetham-Ishida, W.; Pakdeechote, P. Nobiletin resolves left ventricular and renal changes in 2K-1C hypertensive rats. Sci. Rep. 2022, 12, 9289. [Google Scholar] [CrossRef] [PubMed]
- Parkar, N.A.; Bhatt, L.K.; Addepalli, V. Efficacy of nobiletin, a citrus flavonoid, in the treatment of the cardiovascular dysfunction of diabetes in rats. Food Funct. 2016, 7, 3121–3129. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, H.; Li, Y.; Lu, X. Nobiletin suppresses oxidative stress and apoptosis in H9c2 cardiomyocytes following hypoxia/reoxygenation injury. Eur. J. Pharmacol. 2019, 854, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gao, Z.; Zeng, L.; Liang, Z.; Zheng, D.; Wu, X. Nobiletin ameliorates cardiac impairment and alleviates cardiac remodeling after acute myocardial infarction in rats via JNK regulation. Pharmacol. Res. Perspect. 2021, 9, e00728. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A. Diabetes and Arrhythmias: Pathophysiology, Mechanisms and Therapeutic Outcomes. Front. Physiol. 2018, 9, 1669. [Google Scholar] [CrossRef]
- Jagu, B.; Charpentier, F.; Toumaniantz, G. Identifying potential functional impact of mutations and polymorphisms: Linking heart failure, increased risk of arrhythmias and sudden cardiac death. Front. Physiol. 2013, 4, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, H.F.; Zhang, H.; Wang, C.; Chen, Y.F.; Ma, R.; Xiang, J.Z.; Du, X.L.; Tang, Q. Inhibitory effects of hesperetin on Nav1.5 channels stably expressed in HEK 293 cells and on the voltage-gated cardiac sodium current in human atrial myocytes. Acta Pharmacol. Sin. 2016, 37, 1563–1573. [Google Scholar] [CrossRef] [Green Version]
- Li, M.T.; Du, Y.Y.; Zhong, F.; Wang, J.R.; Gu, Y.W.; Zhang, Y.; Huang, X.T.; Deng, Y.Z.; Xu, Z.X. Inhibitory effects of aloperine on voltage-gated Na(+) channels in rat ventricular myocytes. Naunyn-Schmiedeb. Arch. Pharmacol. 2021, 394, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ding, H.S.; Song, T.; Chen, Y.T.; Wang, T.; Tang, Y.H.; Barajas-Martinez, H.; Huang, C.X.; Hu, D. Abrogation of CC Chemokine Receptor 9 Ameliorates Ventricular Electrical Remodeling in Mice After Myocardial Infarction. Front. Cardiovasc. Med. 2021, 8, 716219. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.C.; Lee, S.H.; Cho, Y.K.; Park, H.S.; Kim, Y.N.; Lee, Y.S.; Shin, D.G. Role of the alternans of action potential duration and aconitine-induced arrhythmias in isolated rabbit hearts. J. Korean Med. Sci. 2011, 26, 1576–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppini, R.; Ferrantini, C.; Mazzoni, L.; Sartiani, L.; Olivotto, I.; Poggesi, C.; Cerbai, E.; Mugelli, A. Regulation of intracellular Na(+) in health and disease: Pathophysiological mechanisms and implications for treatment. Glob. Cardiol. Sci. Pract. 2013, 2013, 222–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.L.; Du, W.; Yu, Y.C.; Ju, W.Z.; Man, Y.L.; Li, X.R.; Chen, Y.; Wang, Z.D.; Gu, W.J.; et al. Hepatocyte growth factor modification enhances the anti-arrhythmic properties of human bone marrow-derived mesenchymal stem cells. PLoS ONE 2014, 9, e111246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, T.; Ma, A.; Zhou, X.; Gui, J.; Wan, H.; Shi, R.; Huang, C.; Grace, A.A.; Huang, C.L.; et al. Correlations between clinical and physiological consequences of the novel mutation R878C in a highly conserved pore residue in the cardiac Na+ channel. Acta Physiol. 2008, 194, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remme, C.A.; Wilde, A.A. Targeting sodium channels in cardiac arrhythmia. Curr. Opin. Pharmacol. 2014, 15, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Jin, X.; Zhao, Y.; Huang, G.; Huang, X.; Shen, Z.; Cao, Y.; Dong, M.; Lei, J.; et al. Comparative structural analysis of human Na(v)1.1 and Na(v)1.5 reveals mutational hotspots for sodium channelopathies. Proc. Natl. Acad. Sci. USA 2021, 118, e2100066118. [Google Scholar] [CrossRef]
- Jiang, D.; Zhang, J.; Xia, Z. Structural Advances in Voltage-Gated Sodium Channels. Front. Pharmacol. 2022, 13, 908867. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA 1996, 93, 9270–9275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazola, Y.; Márquez Montesinos, J.C.E.; Ramírez, D.; Zúñiga, L.; Decher, N.; Ravens, U.; Yarov-Yarovoy, V.; González, W. Common Structural Pattern for Flecainide Binding in Atrial-Selective K(v)1.5 and Na(v)1.5 Channels: A Computational Approach. Pharmaceutics 2022, 14, 1356. [Google Scholar] [CrossRef] [PubMed]
- Gulsevin, A.; Glazer, A.M.; Shields, T.; Kroncke, B.M.; Roden, D.M.; Meiler, J. Veratridine Can Bind to a Site at the Mouth of the Channel Pore at Human Cardiac Sodium Channel NaV1.5. Int. J. Mol. Sci. 2022, 23, 2225. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jin, X.; Wu, T.; Huang, G.; Wu, K.; Lei, J.; Pan, X.; Yan, N. Structural Basis for Pore Blockade of the Human Cardiac Sodium Channel Nav 1.5 by the Antiarrhythmic Drug Quinidine*. Angew. Chem. Int. Ed. Engl. 2021, 60, 11474–11480. [Google Scholar] [CrossRef] [PubMed]
- Ruzieh, M.; Moroi, M.K.; Aboujamous, N.M.; Ghahramani, M.; Naccarelli, G.V.; Mandrola, J.; Foy, A.J. Meta-Analysis Comparing the Relative Risk of Adverse Events for Amiodarone Versus Placebo. Am. J. Cardiol. 2019, 124, 1889–1893. [Google Scholar] [CrossRef] [PubMed]
- Hageman, J.R. Long COVID-19 or Post-Acute Sequelae of SARS-CoV-2 Infection in Children, Adolescents, and Young Adults. Pediatr. Ann. 2021, 50, e232–e233. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Groff, D.; Sun, A.; Ssentongo, A.E.; Ba, D.M.; Parsons, N.; Poudel, G.R.; Lekoubou, A.; Oh, J.S.; Ericson, J.E.; Ssentongo, P.; et al. Short-term and Long-term Rates of Postacute Sequelae of SARS-CoV-2 Infection: A Systematic Review. JAMA Netw. Open 2021, 4, e2128568. [Google Scholar] [CrossRef] [PubMed]
- Giustino, G.; Pinney, S.P.; Lala, A.; Reddy, V.Y.; Johnston-Cox, H.A.; Mechanick, J.I.; Halperin, J.L.; Fuster, V. Coronavirus and Cardiovascular Disease, Myocardial Injury, and Arrhythmia: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 76, 2011–2023. [Google Scholar] [CrossRef] [PubMed]
- Mitrani, R.D.; Dabas, N.; Goldberger, J.J. COVID-19 cardiac injury: Implications for long-term surveillance and outcomes in survivors. Heart Rhythm 2020, 17, 1984–1990. [Google Scholar] [CrossRef]
- Clark, A.P.; Wei, S.; Kalola, D.; Krogh-Madsen, T.; Christini, D.J. An in silico-in vitro pipeline for drug cardiotoxicity screening identifies ionic pro-arrhythmia mechanisms. Br. J. Pharmacol. 2022, 179, 4829–4843. [Google Scholar] [CrossRef] [PubMed]
- Vitali Serdoz, L.; Rittger, H.; Furlanello, F.; Bastian, D. Quinidine—A legacy within the modern era of antiarrhythmic therapy. Pharmacol. Res. 2019, 144, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kociol, R.D.; Cooper, L.T.; Fang, J.C.; Moslehi, J.J.; Pang, P.S.; Sabe, M.A.; Shah, R.V.; Sims, D.B.; Thiene, G.; Vardeny, O. Recognition and Initial Management of Fulminant Myocarditis. Circulation 2020, 141, e69–e92. [Google Scholar] [CrossRef] [PubMed]
- Harb, I.; Ashour, H.; Rashed, L.A.; Mostafa, A.; Samir, M.; Aboulhoda, B.E.; El-Hanbuli, H.; Rashwan, E.; Mahmoud, H. Nicorandil mitigates amiodarone-induced pulmonary toxicity and fibrosis in association with the inhibition of lung TGF-beta1/PI3K/Akt1-p/mTOR axis in rats. Clin. Exp. Pharmacol. Physiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Caruso, L.; Nadur, N.F.; da Fonseca, M.B.; Peixoto Ferreira, L.A.; Lacerda, R.B.; Graebin, C.S.; Kummerle, A.E. The Design of Multi-target Drugs to Treat Cardiovascular Diseases: Two (or more) Birds on One Stone. Curr. Top Med. Chem. 2022, 22, 366–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.; Zhang, C.; Bai, X.; Zhang, J.; Zhao, X.; Chen, L.; Wang, L.; Zhu, C.; Cui, L.; et al. Nobiletin promotes antioxidant and anti-inflammatory responses and elicits protection against ischemic stroke in vivo. Brain Res. 2016, 1636, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zheng, D.; Qin, Y.; Liu, Z.; Zhang, G.; Zhu, X.; Zeng, L.; Liang, Z. Nobiletin attenuates adverse cardiac remodeling after acute myocardial infarction in rats via restoring autophagy flux. Biochem. Biophys. Res. Commun. 2017, 492, 262–268. [Google Scholar] [CrossRef]
- Liu, B.; Li, S.; Su, Y.; Xiong, M.; Xu, Y. Comparative study of the protective effects of terfenadine and amiodarone on barium chloride/aconitine-induced ventricular arrhythmias in rats: A potential role of terfenadine. Mol. Med. Rep. 2014, 10, 3217–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinamard, R.; Hof, T.; Salle, L. Current recordings at the single channel level in adult mammalian isolated cardiomyocytes. Methods Mol. Biol. 2014, 1183, 291–307. [Google Scholar] [PubMed]
- Rigsby, R.E.; Parker, A.B. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem. Mol. Biol. Educ. 2016, 44, 433–437. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Saline | NOB | Amiodarone |
|---|---|---|
| 95% | 10% ## | 5% ## |
| Group (Concentration μmol/L) | Current Density (pA/pF) |
|---|---|
| Control | −87.79 ± 4.21 |
| NOB (5) | −86.36 ± 4.63 |
| NOB (10) | −81.30 ± 5.01 * |
| NOB (25) | −61.87 ± 2.54 ** |
| NOB (50) | −50.88 ± 5.41 ** |
| NOB (100) | −38.90 ± 3.21 *** |
| NOB (200) | −29.09 ± 2.85 *** |
| Amiodarone (24.24) | −27.34 ± 4.63 *** |
| Ion Channel Proteins | Binding Affinity (kcal/mol) | MM-GBSA Binding Energy (kcal/mol) | π-π Stacking | Hydrogen Bonding | Hydrophobic Interaction |
|---|---|---|---|---|---|
| rNav1.5 | −6.655 | −36.44 | Phe-1762 | Ser-1712 | Ile-1468, Leu-1464, Ser-1460, Phe-1420, Lys-1421 |
| rNav1.5/QQQ | −6.562 | −29.32 | Phe-1762 | Ser-1712 | Ile-1468, Leu-1464, Ser-1460, Phe-1420, Lys-1421 |
| hNav1.5 | −5.693 | −51.71 | Gln-371, Lys-1419, Ser-1458 | Phe-1418, Leu-1462, Phe-1760 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Y.; Wang, J.; Li, M.; Zhong, F.; Xiang, J.; Xu, Z. Inhibitory Effects of Nobiletin on Voltage-Gated Na+ Channel in Rat Ventricular Myocytes Based on Electrophysiological Analysis and Molecular Docking Method. Int. J. Mol. Sci. 2022, 23, 15175. https://doi.org/10.3390/ijms232315175
Gu Y, Wang J, Li M, Zhong F, Xiang J, Xu Z. Inhibitory Effects of Nobiletin on Voltage-Gated Na+ Channel in Rat Ventricular Myocytes Based on Electrophysiological Analysis and Molecular Docking Method. International Journal of Molecular Sciences. 2022; 23(23):15175. https://doi.org/10.3390/ijms232315175
Chicago/Turabian StyleGu, Youwei, Jieru Wang, Mengting Li, Fei Zhong, Jie Xiang, and Zhengxin Xu. 2022. "Inhibitory Effects of Nobiletin on Voltage-Gated Na+ Channel in Rat Ventricular Myocytes Based on Electrophysiological Analysis and Molecular Docking Method" International Journal of Molecular Sciences 23, no. 23: 15175. https://doi.org/10.3390/ijms232315175