Transcriptome Sequencing and Metabolome Analysis Reveal Genes Involved in Pigmentation of Green-Colored Cotton Fibers


Abstract
:1. Introduction
2. Results
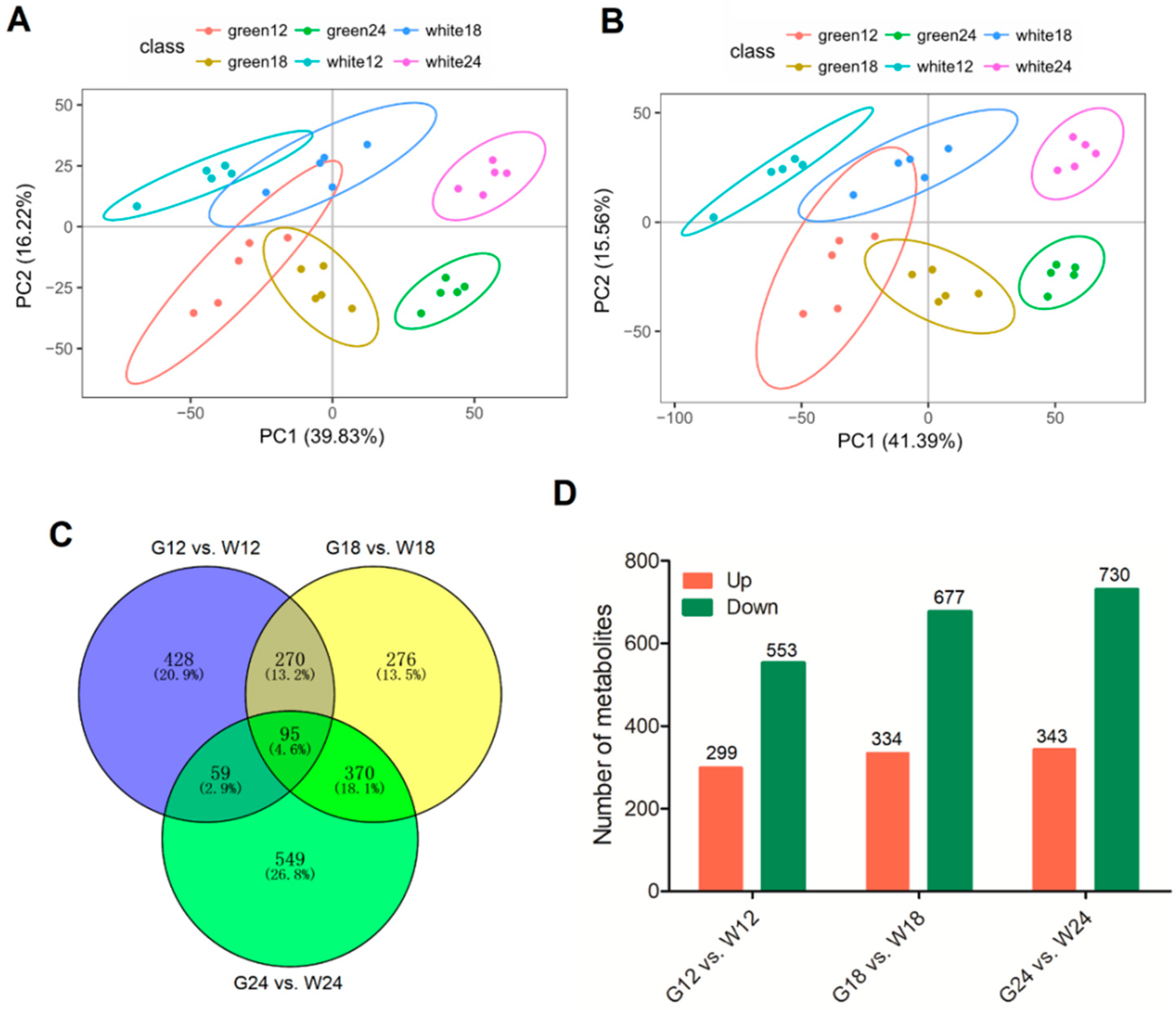
2.1. Overview of the Metabolomic Profiling
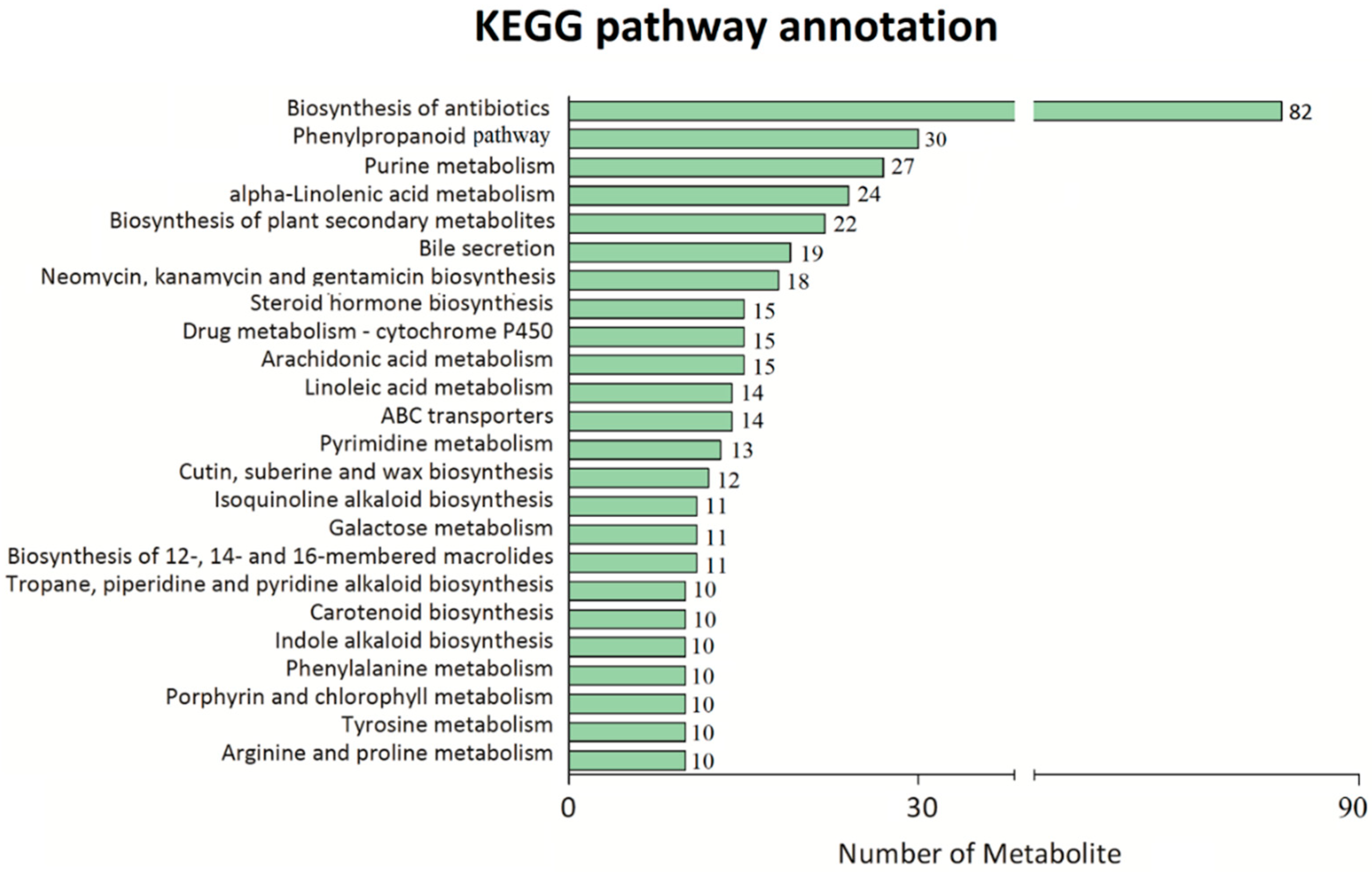
2.2. Untargeted Metabolomic Analysis of Phenylpropanoid Metabolite Content
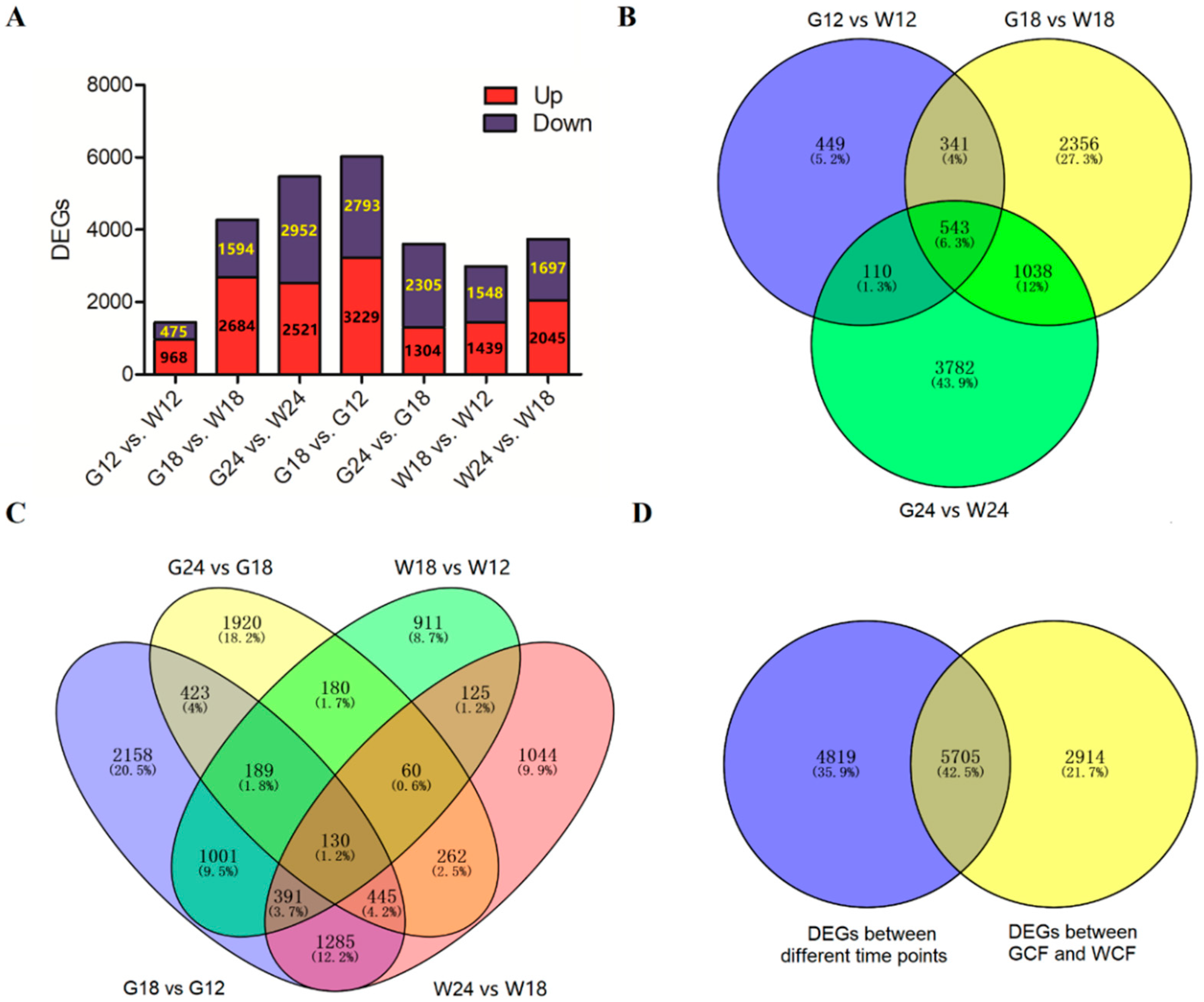
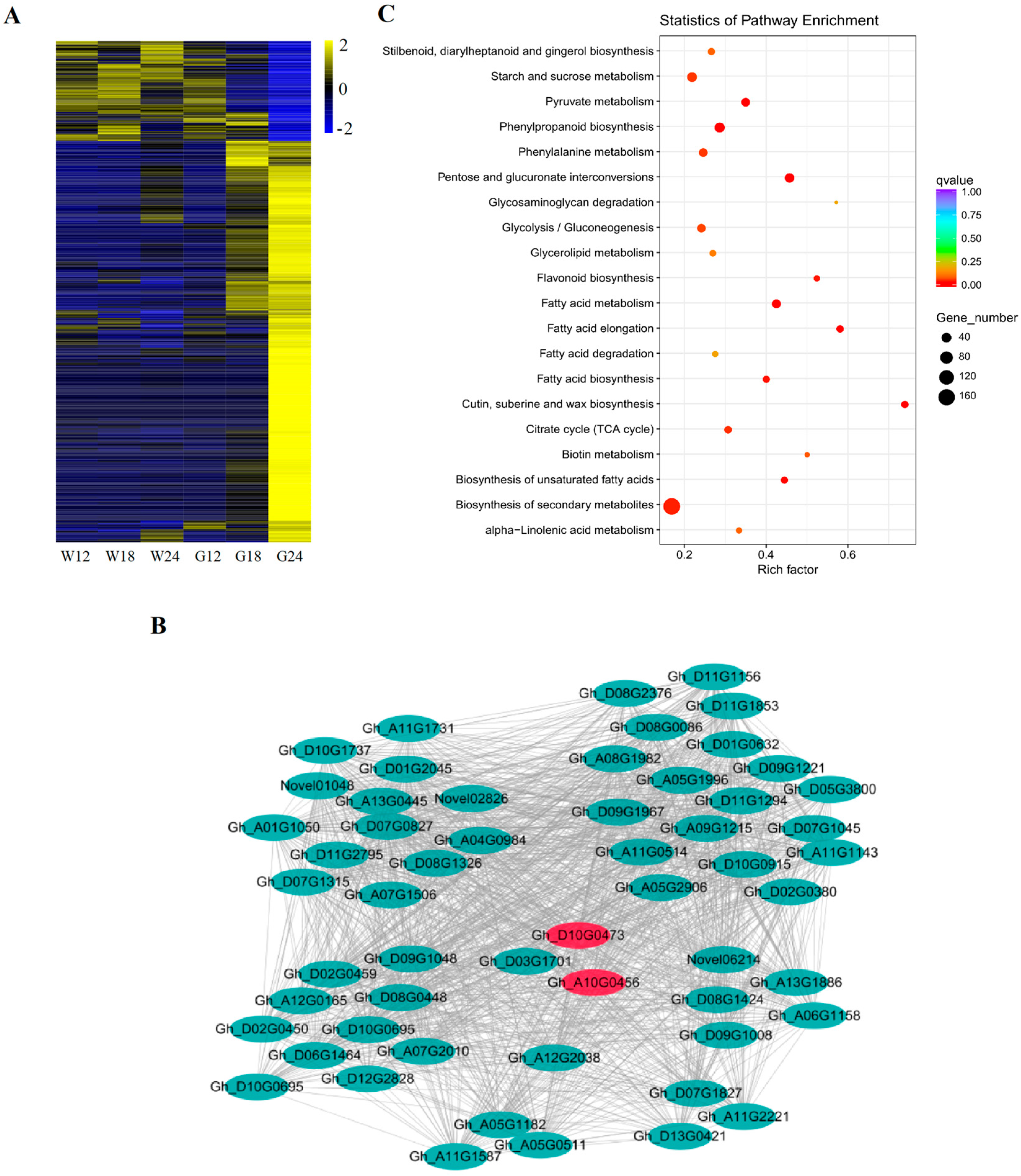
2.3. Global Transcriptome Changes During the Process of Fiber Pigmentation
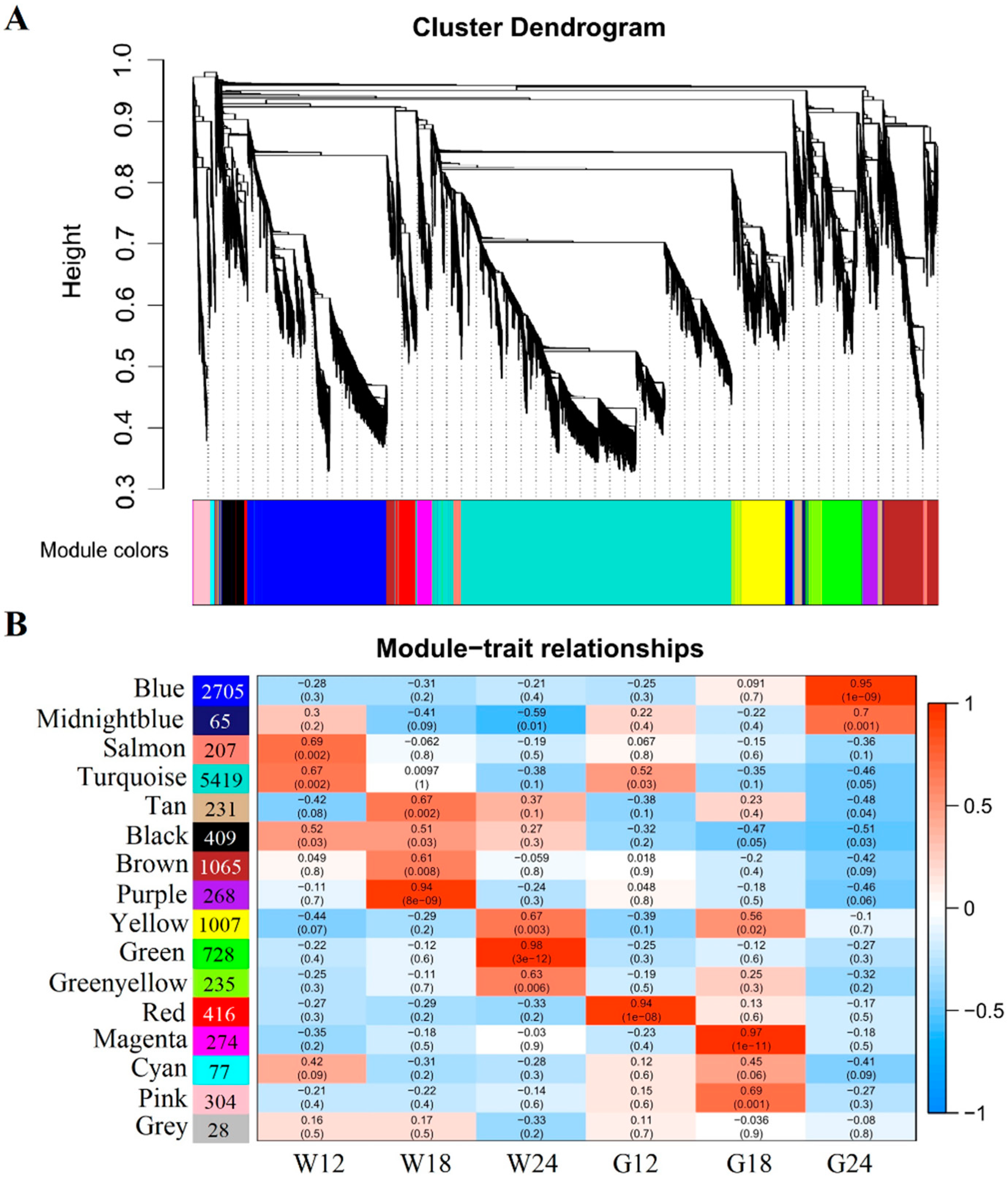
2.4. Co-Expression Network Analysis Identified Pigmentation-Related Differentially Expressed Genes (DEGs)
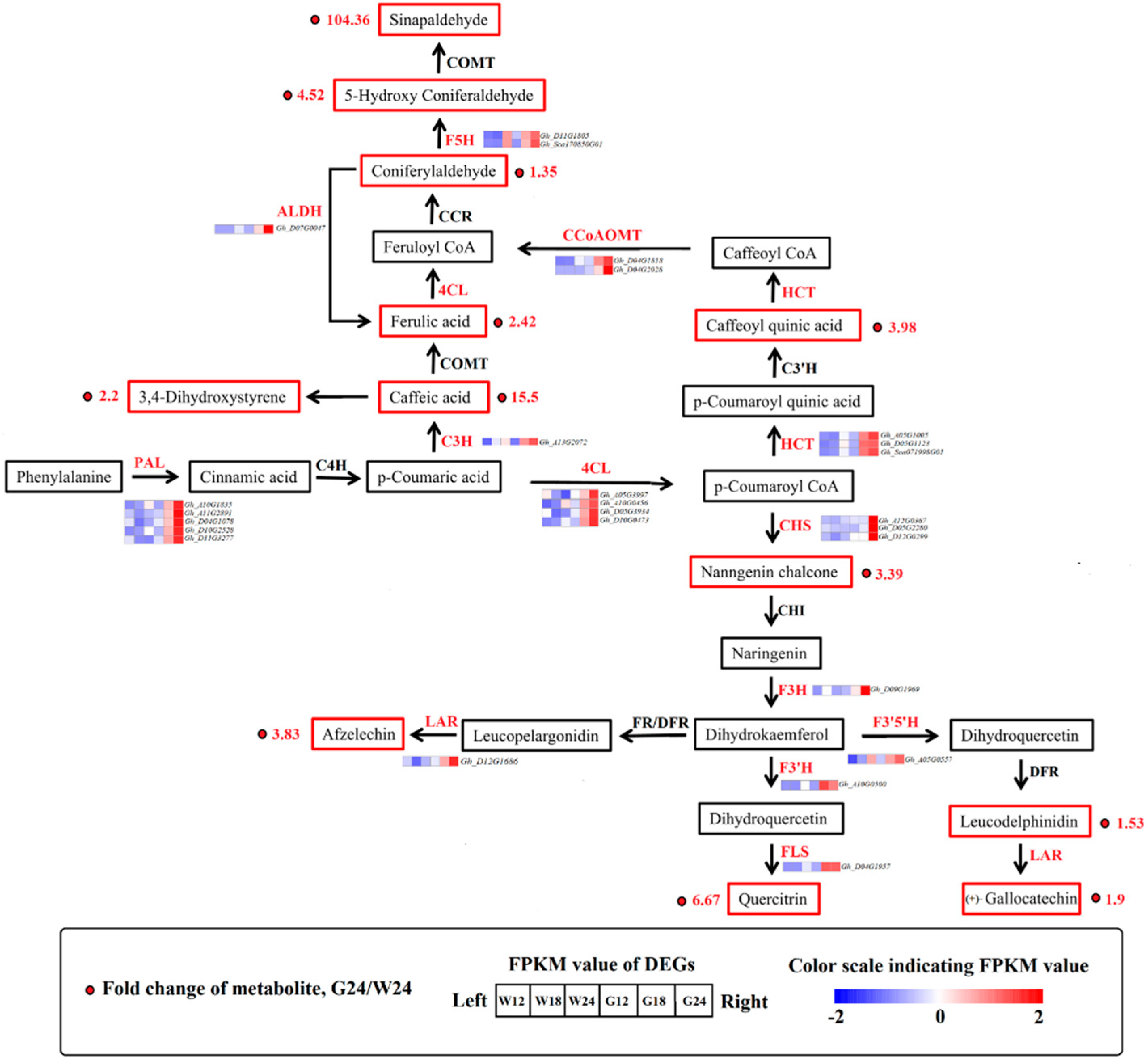
2.5. Phenylpropanoid Pathway
2.6. Lipid Metabolism Pathway
3. Discussion
3.1. Identifying Pigmentation-Related Metabolites in Green-Colored Fiber (GCF)
3.2. Identifying Gene-Expression Modules Associated with Pigmentation in GCF
4. Materials and Methods
4.1. Plant Materials and Treatments
4.2. Metabolite Extraction and Profiling
4.3. Metabolite Identification and Quantification
4.4. RNA Isolation and Illumina Sequencing
4.5. RNA Sequencing Data Analysis
4.6. Assignment of MapMan Bins and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways
4.7. Gene Network Construction and Visualization
4.8. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ma, M.; Hussain, M.; Memon, H.; Zhou, W. Structure of pigment compositions and radical scavenging activity of naturally green-colored cotton fiber. Cellulose 2016, 23, 955–963. [Google Scholar] [CrossRef]
- Vreeland, J.M. The revival of colored cotton. Sci. Am. 1999, 280, 112–118. [Google Scholar] [CrossRef]
- Dutt, Y.; Wang, X.D.; Zhu, Y.G.; Li, Y.Y. Breeding for high yield and fibre quality in colored cotton. Plant. Breed. 2004, 123, 145–151. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, X.Y.; Wang, F.X.; Yang, C.L.; Liu, F.; Xia, G.X.; Sun, J. A comparative proteomic analysis provides insights into pigment biosynthesis in brown color fiber. J. Proteom. 2013, 78, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Murthy, M.S.S. Never say dye: The story of coloured cotton. Resonance 2001, 6, 29–35. [Google Scholar] [CrossRef]
- Yuan, S.; Hua, S.; Malik, W.; Bibi, N.; Wang, X. Physiological and biochemical dissection of fiber development in colored cotton. Euphytica 2012, 187, 215–226. [Google Scholar] [CrossRef]
- Kohel, R.J. Genetic Analysis of Fiber Color Variants in Cotton 1. Crop. Sci. 1985, 25, 793–797. [Google Scholar] [CrossRef]
- Xiao, Y.H.; Yan, Q.; Ding, H.; Luo, M.; Hou, L.; Zhang, M.; Yao, D.; Liu, H.S.; Li, X.; Zhao, J.; et al. Transcriptome and biochemical analyses revealed a detailed proanthocyanidin biosynthesis pathway in brown cotton fiber. PLoS ONE 2014, 9, e86344. [Google Scholar] [CrossRef]
- Feng, H.; Li, Y.; Wang, S.; Zhang, L.; Liu, Y.; Xue, F.; Sun, Y.; Wang, Y.; Sun, J. Molecular analysis of proanthocyanidins related to pigmentation in brown cotton fibre (gossypium hirsutum L.). J. Exp. Bot. 2014, 65, 5759–5769. [Google Scholar] [CrossRef]
- Yan, Q.; Wang, Y.; Li, Q.; Zhang, Z.; Ding, H.; Zhang, Y.; Liu, H.; Luo, M.; Liu, D.; Song, W.; et al. Up-regulation of GhTT2-3A in cotton fibres during secondary wall thickening results in brown fibres with improved quality. Plant. Biotechnol. J. 2018, 16, 1735–1747. [Google Scholar] [CrossRef]
- Ryser, U.; Meier, H.; Holloway, P.J. Identification and locailzation of suberin in the cell wall of green (Gossypium hirsutum L. Var. green lint). Protoplasma 1983, 117, 196–205. [Google Scholar] [CrossRef]
- Yatsu, L.Y.; Espelie, K.E.; Kolattukudy, P.E. Ultrastructural and chemical evidence that the cell wall of green cotton fber is suberized. Plant. Physiol. 1983, 73, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.M. The high wax content of green lint cotton. Science 1941, 94, 113. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, A.; Jenny, T.; Amrhein, N.; Ryser, U. Caffeic acid and glycerol are constituents of the suberin layers in green cotton fibres. Planta 1993, 189, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, A.; Jenny, T.; Ryser, U. A caffeoyl-fatty acid-glycerol ester from wax associated with green cotton fibre suberin. Phytochemistry 1994, 36, 1343–1346. [Google Scholar] [CrossRef]
- Feng, H.J.; Yang, Y.L.; Sun, S.C.; Li, Y.; Zhang, L.; Tian, J.; Zhu, Q.; Feng, Z.; Zhu, H.; Sun, J. Molecular analysis of caffeoyl residues related to pigmentation in green cotton fibers. J. Exp. Bot. 2017, 68, 4559–4569. [Google Scholar] [CrossRef]
- Schmutz, A.; Buchala, A.J.; Ryser, U. Changing the dimensions of suberin lamellae of green cotton fibers with a specific inhibitor of the endoplasmic reticulum-associated fatty acid elongases. Plant. Physiol. 1996, 110, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Q.; Wang, X.D. Composition analysis of pigment in colored cotton fiber. Acta Agron. Sin. 2005, 31, 456–462. [Google Scholar]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (gossypium hirsutum l. acc. tm-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantifcation by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Li, Y.J.; Wang, F.X.; Wang, Y.Q.; Liu, Y.C.; Zhang, X.Y.; Sun, Y.Q.; Sun, J. Identification of the proteins in green cotton fiber using a proteomics-based approach. Biotechnol. Lett. 2013, 35, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Ehlting, J.; Buttner, D.; Wang, Q.; Douglas, C.J.; Somssich, I.E.; Kombrink, E. Three 4-coumarate: Coenzyme A ligases in Arabidopsis thaliana represent two evolutionarily divergent classes in angiosperms. Plant. J. 1999, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gianoulis, T.A.; Griffin, M.A.; Spakowicz, D.J.; Dunican, B.F.; Alpha, C.J.; Sboner, A.; Sismour, A.M.; Kodira, C.; Egholm, M.; Church, G.M. Genomic Analysis of the Hydrocarbon-Producing, Cellulolytic, Endophytic Fungus Ascocoryne sarcoides. PLoS Genet. 2012, 8, e1002558. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.A.; Achnine, L.; Kota, P.; Liu, C.J.; Reddy, M.S.S.; Wang, L. The phenylpropanoid pathway and plant defence a genomics perspective. Mol. Plant. Pathol. 2002, 3, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Compagnon, V.; Diehl, P.; Benveniste, I.; Meyer, D.; Schaller, H.; Schreiber, L.; Franke, R.; Pinot, F. CYP86B1 is required for very long chain omega-hydroxyacid and alpha, omega-dicarboxylic acid synthesis in root and seed suberin polyester. Plant. Physiol. 2009, 150, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Rene, H.; Isabel, B.; Martina, B.; Franck, P.; Lukas, S.; Rochus, F. The arabidopsis cytochrome p450 cyp86a1 encodes a fatty acid ω-hydroxylase involved in suberin monomer biosynthesis. J. Exp. Bot. 2008, 59, 2347–2360. [Google Scholar]
- Lotfy, S.; Negrel, J.; Javelle, F. Formation of ω-feruloyloxypalmitic acid by an enzyme from wound-healing potato tuber discs. Phytochemistry 1994, 35, 1419–1424. [Google Scholar] [CrossRef]
- Hooker, T.S.; Millar, A.A.; Kunst, L. Significance of the expression of the CER6 condensing enzyme for cuticular wax production in Arabidopsis. Plant. Physiol. 2002, 129, 1568–1580. [Google Scholar] [CrossRef]
- Moire, L.; Schmutz, A.; Buchala, A.; Yan, B.; Stark, R.E.; Ryser, U. Glycerol is a suberin monomer: New experimental evidence for an old hypothesis. Plant. Physiol. 1999, 119, 1137–1146. [Google Scholar] [CrossRef]
- Huang, J.; Gu, M.; Lai, Z.; Fan, B.; Shi, K.; Zhou, Y.H.; Yu, J.Q.; Chen, Z.X. Functional analysis of the arabidopsis pal gene family in plant growth, development, and response to environmental stress. Plant. Physiol. 2010, 153, 1526–1538. [Google Scholar] [CrossRef]
- Blankenship, S.M.; Unrath, C.R. PAL and ethylene content during maturation of Red and Golden Delicious apples. Photochemistry 1988, 27, 1001–1003. [Google Scholar] [CrossRef]
- Kataoka, I.; Kubo, Y.; Sugiura, A.; Tomana, T. Changes in L-phenylalanine ammonia-lyase activity and anthocyanin synthesis during berry ripening of three grape cultivars. J. Jpn. Soc. Hort. Sci. 1983, 52, 273–279. [Google Scholar] [CrossRef]
- Cheng, G.W.; Breen, P.J. Activity of Phenylalanine Ammonia-Lyase (PAL) and Concentrations of Anthocyanins and Phenolics in Developing Strawberry Fruit. J. Amer. Soc. Hort. Sci. 1991, 116, 865–869. [Google Scholar] [CrossRef]
- Debeaujon, I.; Leon-Kloosterziel, K.M.; Koornneef, M. Influence of the testa on seed dormancy, germination, and longevity in Arabidopsis. Plant. Physiol. 2000, 122, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.A.; Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant. Cell 1995, 7, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Landry, L.G.; Chapple, C.C.S.; Last, R.L. Arabidopsis mutants lacking phenolic sunscreens exhibit enhanced ultraviolet-B injury and oxidative damage. Plant. Physiol. 1995, 109, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Winkel-Shirley, B. Flavonoid biosynthesis: A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant. Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments:anthocyanins, betalains and carotenoids. Plant. J. 2008, 54, 733–749. [Google Scholar]
- Koes, R.E.; Spelt, C.E.; Mol, J.N.M. The chalcone synthase multigene family ofpetunia hybrida(v30): Differential, light-regulated expression during flower development and uv light induction. Plant. Mol. Biol. 1989, 12, 213–225. [Google Scholar] [CrossRef]
- Ohno, S.; Hosokawa, M.; Kojima, M.; Kitamura, Y.; Hoshino, A.; Tatsuzawa, F.; Doi, M.; Yazawa, S. Simultaneous post-transcriptional gene silencing of two different chalcone synthase genes resulting in pure white flowers in the octoploid dahlia. Planta 2011, 234, 945–958. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Hori, W.; Hosokawa, M.; Tatsuzawa, F.; Doi, M. Post-transcriptional silencing of chalcone synthase is involved in phenotypic lability in petals and leaves of bicolor dahlia (Dahlia variabilis) ‘Yuino’. Planta 2018, 247, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, Y.; Suzuki, K.; Fukuchi-Mizutani, M.; Fukui, Y.; Miyazaki, K.; Ohkawa, H.; Kusumi, T.; Tanaka, Y. Molecular and biochemical characterization of torenia flavonoid 3′-hydroxylase and flavone synthase II and modification of flower color by modulating the expression of these genes. Plant. Sci. 2002, 163, 253–263. [Google Scholar] [CrossRef]
- Han, Y.; Vimolmangkang, S.; Soria-Guerra, R.E.; Rosales-Mendoza, S.; Zheng, D.; Korban, S.S. Ectopic expression of apple F3′H genes contributes to anthocyanin accumulation in the arabidopsis tt7 mutant grown under nitrogen stress. Plant. Physiol. 2010, 153, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Brugliera, F. Flower colour and cytochromes P450. Phytochem. Rev. 2006, 5, 283–291. [Google Scholar] [CrossRef]
- Tanner, G.J.; Francki, K.T.; Abrahams, S.; Watson, J.M.; Larkin, P.J.; Ashton, A.R. Proanthocyanidin biosynthesis in plants. Purification of legume leucoanthocyanidin reductase and molecular cloning of its cDNA. J. Biol. Chem. 2003, 278, 3147–3156. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.M.; Schwinn, K.E.; Deroles, S.C.; Manson, D.G.; Lewis, D.H.; Bloor, S.J.; Bradley, J.M. Enhancing anthocyanin production by altering competition for substrate between flavonol synthase and dihydroflavonol reductase. Euphytica 2003, 131, 259–268. [Google Scholar] [CrossRef]
- Beuerle, T.; Pichersky, E. Enzymatic synthesis and purification of aromatic coenzyme a esters. Anal. Biochem. 2002, 302, 305–312. [Google Scholar] [CrossRef]
- Lee, S.B.; Jung, S.J.; Go, Y.S.; Kim, H.U.; Kim, J.K.; Cho, H.J.; Park, O.K.; Suh, M.C. Two Arabidopsis 3-ketoacyl CoA synthase genes, KCS20 and KCS2/DAISY, are functionally redundant in cuticular wax and root suberin biosynthesis, but differentially controlled by osmotic stress. Plant. J. 2009, 60, 462–475. [Google Scholar] [CrossRef]
- Beaudoin, F.; Wu, X.Z.; Li, F.; Haslam, R.P.; Markham, J.E.; Zheng, H. Functional characterization of the arabidopsis β-ketoacyl-coenzyme a reductase candidates of the fatty acid elongase. Plant. Physiol. 2009, 150, 1174–1191. [Google Scholar] [CrossRef]
- Doan, T.T.P.; Carlsson, A.S.; Hamberg, M.; Leif, B.; Stymne, S.; Olsson, P. Functional expression of five arabidopsis fatty acyl-coa reductase genes in escherichia coli. J. Plant. Physiol. 2009, 166, 787–796. [Google Scholar] [CrossRef]
- Yang, M.; Gu, L.Q.; Qian, H.S.; Zha, L.S. Comparative study on trace elements in natural colored cotton and white cotton. Spectrosc Spect Anal. 2008, 28, 203–205. [Google Scholar]
- Gu, Y.; Cai, X.J.; Li, M.S.; Zhou, W.L. An approach to the color properties of naturally colored cottons after metal salts treatment. J. Zhejiang Sci.-Tech. Univ. 2008, 25, 10–14. [Google Scholar]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. Mapman: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant. J. 2010, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for Visualization and Analysis of Biological Networks. Methods Mol. Biol. 2011, 696, 291–303. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Metabolite Name | Log2 (Fold Change) | VIP | Exact Mass (m/z) |
|---|---|---|---|---|
| (G24/W24) | ||||
| Intermediates | Caffeic acid | 16.06 | 3.55 | 179.0336 |
| Ferulic acid | 4.52 | 2.86 | 193.049 | |
| Sinapaldehyde | 104.36 | 3.46 | 207.0648 | |
| 3,4-Dihydroxystyrene | 2.21 | 2.1 | 136.0529 | |
| Coniferyl aldehyde | 1.35 | 1.12 | 161.0601 | |
| 5-Hydroxyconiferaldehyde | 4.52 | 2.86 | 177.055 | |
| Caffeoyl quinic acid | 3.98 | 2.2 | 355.0999 | |
| Flavone | Naringenin chalcone | 3.39 | 1.44 | 271.0596 |
| Flavanol | (+)-Gallocatechin | 1.9 | 1.2 | 305.066 |
| Flavonol | Afzelin | 3.83 | 2.24 | 455.0991 |
| Quercitrin | 6.67 | 2.64 | 449.1087 | |
| Anthocyanin | Leucodelphinidin | 1.54 | 1 | 323.0751 |
| Metabolite Name | Log2 (Fold Change) | VIP | Exact Mass (m/z) |
|---|---|---|---|
| (G24/W24) | |||
| Docosanedioate | 11.85 | 2.5 | 369.2993 |
| 22-Oxo-docosanoate | 1.85 | 1.37 | 353.3067 |
| 22-Hydroxydocosanoate | 8.82 | 2.38 | 355.3204 |
| 9,10-Dihydroxystearate | 3.03 | 2 | 315.2526 |
| 9,10-Epoxy-18-hydroxystearate | 1.65 | 1.34 | 313.2365 |
| cis-9,10-Epoxystearic acid | 1.72 | 1.16 | 297.2418 |
| 18-Hydroxyoleate | 2.01 | 1.61 | 298.2513 |
| Hexadecanedioate | 2.56 | 1.95 | 285.2057 |
| 16-Hydroxypalmitic acid | 1.54 | 1.2 | 271.2261 |
| 16-Oxo-palmitate | 1.45 | 1.01 | 269.2104 |
| Gene ID | Description | KME Value |
| Lipid Metabolism | ||
| Gh_D11G1853 | Epoxide hydrolase 3, EPHX3 | 0.997 |
| Gh_A11G1143 | Non-specific lipid-transfer protein-like protein, At2g13820 | 0.997 |
| Gh_D02G0380 | Lipid binding protein | 0.997 |
| Gh_D09G1221 | Fatty acyl-CoA reductase, FAR | 0.997 |
| Gh_A05G1996 | Probable glucan endo-1,3-beta-glucosidase 1, BG1 | 0.997 |
| Gh_D08G0086 | Fatty acyl-CoA reductase, FAR | 0.995 |
| Gh_D11G1294 | Non-specific lipid-transfer protein-like protein, At2g13820 | 0.995 |
| Gh_A08G1982 | Sterol 3-beta-Glucosyltransferase, UGT80A2 | 0.995 |
| Gh_A11G0514 | Diacylglycerol O-acyltransferase 2, DGAT2 | 0.994 |
| Gh_D01G0632 | 1-acyl-sn-glycerol-3-phosphate acyltransferase, AGPAT | 0.993 |
| Gh_D01G2045 | Probable glycosyltransferase, At5g03795 | 0.993 |
| Gh_D05G3800 | Probable glucan endo-1,3-beta-glucosidase, BG1 | 0.992 |
| Gh_A13G0445 | Lipid binding protein | 0.992 |
| Gh_A09G1215 | Fatty acyl-CoA reductase, FAR | 0.992 |
| Gh_D10G0915 | GDSL glycine (G), aspartic acid (D), serine (S) and leucine (L) esterase/lipase, At2g23540 | 0.991 |
| Gh_D09G1967 | Xyloglucan Galactosyltransferase KATAMARI1 homolog, Os03g0144800 | 0.991 |
| Gh_D08G2376 | Sterol 3-beta-Glucosyltransferase, UGT80A2 | 0.991 |
| Gh_D11G1156 | Triacylglycerol lipase | 0.991 |
| Gh_D07G1045 | GDSL esterase/lipase, At5g22810 | 0.990 |
| Gh_A05G2906 | Lipid transfer-like protein, VAS | 0.990 |
| Phenylpropanoid Biosynthesis | ||
| Gh_A10G0456 | 4-coumarate-CoA ligase, 4CL | 0.996 |
| Gh_D10G0473 | 4-coumarate-CoA ligase, 4CL | 0.995 |
| Gh_D03G1701 | Caffeoylshikimate esterase, CSE | 0.991 |
| RNA | ||
| Gh_A13G1886 | Scarecrow-like protein, SCL3 | 0.995 |
| Novel06214 | NAC domain protein | 0.994 |
| Gh_D08G1424 | Probable WRKY transcription factor 43, WRKY43 | 0.994 |
| Gh_D09G1008 | LOB domain-containing protein 1, LBD1 | 0.993 |
| Gh_A06G1158 | Putative Myb family transcription factor, At1g14600 | 0.991 |
| Gene ID | Description | KME Value |
| Protein | ||
| Gh_A05G0511 | Aspartyl protease family protein 2, NEP2 | 0.998 |
| Gh_A05G1182 | RING-H2 finger protein, ATL3 | 0.991 |
| Gh_A11G1587 | Aspartic proteinase-like protein, At5g10080 | 0.990 |
| Signalling | ||
| Gh_A11G2221 | Cysteine-rich repeat secretory protein, CRRSP3 | 0.992 |
| Gh_D13G0421 | LRR receptor-like serine/threonine-protein kinase, LRR–RLK | 0.992 |
| Gh_D07G1827 | Receptor-like protein kinase, FER | 0.991 |
| Transport | ||
| Gh_A12G0165 | Nucleobase-ascorbate transporter, NAT | 0.997 |
| Gh_D08G0448 | Heavy metal-associated isoprenylated plant protein, HIPP | 0.996 |
| Gh_A07G2010 | Aquaporin, SIP | 0.994 |
| Gh_D02G0459 | Protein NRT1/ PTR family, NPF | 0.992 |
| Gh_D02G0450 | Phosphate transporter, PHO | 0.992 |
| Gh_D06G1464 | Aquaporin, SIP | 0.992 |
| Gh_D10G0695 | ADP,ATP carrier protein 1, chloroplastic, AATP | 0.991 |
| Gh_D12G2828 | Nucleobase-ascorbate transporter, NAT | 0.991 |
| Gh_D10G0695 | ADP,ATP carrier protein 1, chloroplastic, AATP | 0.991 |
| Gh_D09G1048 | ABC transporter G family member 23, ABCG23 | 0.990 |
| Hormone | ||
| Gh_A12G2038 | Probable aminotransferase 10, ACS10 | 0.990 |
| Others | ||
| Gh_A07G1506 | Pentatricopeptide repeat-containing protein, At3g22150 | 0.994 |
| Gh_D10G1737 | Uncharacterized protein | 0.994 |
| Gh_D08G1326 | Uncharacterized protein | 0.994 |
| Gh_A04G0984 | Periaxin, Prx | 0.993 |
| Novel01048 | Uncharacterized protein | 0.992 |
| Gh_A11G1731 | Uncharacterized protein | 0.992 |
| Gh_D07G0827 | Condensin complex subunit | 0.992 |
| Gh_D07G1315 | Uncharacterized protein | 0.992 |
| Novel02826 | Uncharacterized protein | 0.991 |
| Gh_A01G1050 | Uncharacterized protein | 0.991 |
| Gh_D11G2795 | Uncharacterized protein | 0.990 |
| Log2 (Fold Change G/W) | Annotation | Symbol | |||
|---|---|---|---|---|---|
| 12 DPA | 18 DPA | 24 DPA | |||
| Gh_A10G1835 | 0.74938 | 3.1288 | 3.1633 | Phenylalanine ammonia lyase | PAL |
| Gh_A11G2891 | 0.27697 | 5.7791 | 6.3045 | PAL | |
| Gh_D04G1078 | 0.18112 | 4.0366 | 5.2906 | PAL | |
| Gh_D10G2528 | 0.76809 | 3.1311 | 3.7176 | PAL | |
| Gh_D11G3277 | −0.0074771 | 6.0276 | 9.165 | PAL | |
| Gh_A05G3997 | −0.49978 | 3.4355 | 8.1142 | 4-coumarate:CoA ligase | 4CL |
| Gh_A10G0456 | 3.8493 | 6.0506 | 4.0219 | 4CL | |
| Gh_D05G3934 | −0.23208 | 2.4448 | 3.4508 | 4CL | |
| Gh_D10G0473 | 1.5417 | 4.7604 | 5.0489 | 4CL | |
| Gh_A13G2072 | 0.14536 | 1.6208 | 2.1007 | Coumarate 3-hydroxylase | C3H |
| Gh_A05G1005 | 0.5165 | 3.8158 | 3.4893 | Shikimate/quinate hydroxycinnamoyl transferase | HCT |
| Gh_D05G1123 | 0.55687 | 3.5151 | 2.4623 | HCT | |
| Gh_Sca071998G01 | 0.7392 | 4.052 | 3.1351 | HCT | |
| Gh_D04G1818 | 4.3784 | 5.9612 | 3.5403 | Caffeoyl CoA O-methyltransfersae | CCoAOMT |
| Gh_D04G2028 | Inf | Inf | Inf | CCoAOMT | |
| Gh_D07G0047 | 0.90754 | 4.2483 | 3.475 | Aldehyde dehydrogenase | ALDH |
| Gh_D11G1805 | 2.5784 | 7.8403 | 1.8926 | Ferulate 5-hydroxylase | F5H |
| Gh_Sca170850G01 | 1.1641 | Inf | 1.5256 | F5H | |
| Gh_A12G0367 | 1.5724 | 2.819 | 4.867 | Chalcone and stilbene synthase family protein | CHS |
| Gh_D05G2280 | −1.449 | 1.9241 | 5.4836 | CHS | |
| Gh_D12G0299 | 1.2695 | 3.4081 | 4.445 | CHS | |
| Gh_D09G1969 | 0.69249 | 0.86727 | 4.5918 | Flavanone 3-hydroxylase | F3H |
| Gh_A10G0500 | 0.40181 | 4.5526 | 1.7541 | Flavonoid 3′-hydroxylase | F3′H |
| Gh_A05G0557 | 2.5078 | 3.5865 | 1.8235 | Flavonoid 3′,5′-hydroxylase | F3′5′H |
| Gh_D04G1975 | 0.26368 | −3.009 | 0.10962 | Flavonol synthase | FLS |
| Gh_D12G1686 | 0.30587 | 2.7314 | 3.453 | Leucoanthocyantin reducase | LAR |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, S.; Xiong, X.-p.; Zhu, Q.; Li, Y.-j.; Sun, J. Transcriptome Sequencing and Metabolome Analysis Reveal Genes Involved in Pigmentation of Green-Colored Cotton Fibers. Int. J. Mol. Sci. 2019, 20, 4838. https://doi.org/10.3390/ijms20194838
Sun S, Xiong X-p, Zhu Q, Li Y-j, Sun J. Transcriptome Sequencing and Metabolome Analysis Reveal Genes Involved in Pigmentation of Green-Colored Cotton Fibers. International Journal of Molecular Sciences. 2019; 20(19):4838. https://doi.org/10.3390/ijms20194838
Chicago/Turabian StyleSun, Shichao, Xian-peng Xiong, Qianhao Zhu, Yan-jun Li, and Jie Sun. 2019. "Transcriptome Sequencing and Metabolome Analysis Reveal Genes Involved in Pigmentation of Green-Colored Cotton Fibers" International Journal of Molecular Sciences 20, no. 19: 4838. https://doi.org/10.3390/ijms20194838