Sphingosine 1-Phosphate Receptor Blockade Affects Pro-Inflammatory Bone Marrow-Derived Macrophages and Relieves Mouse Fatty Liver Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
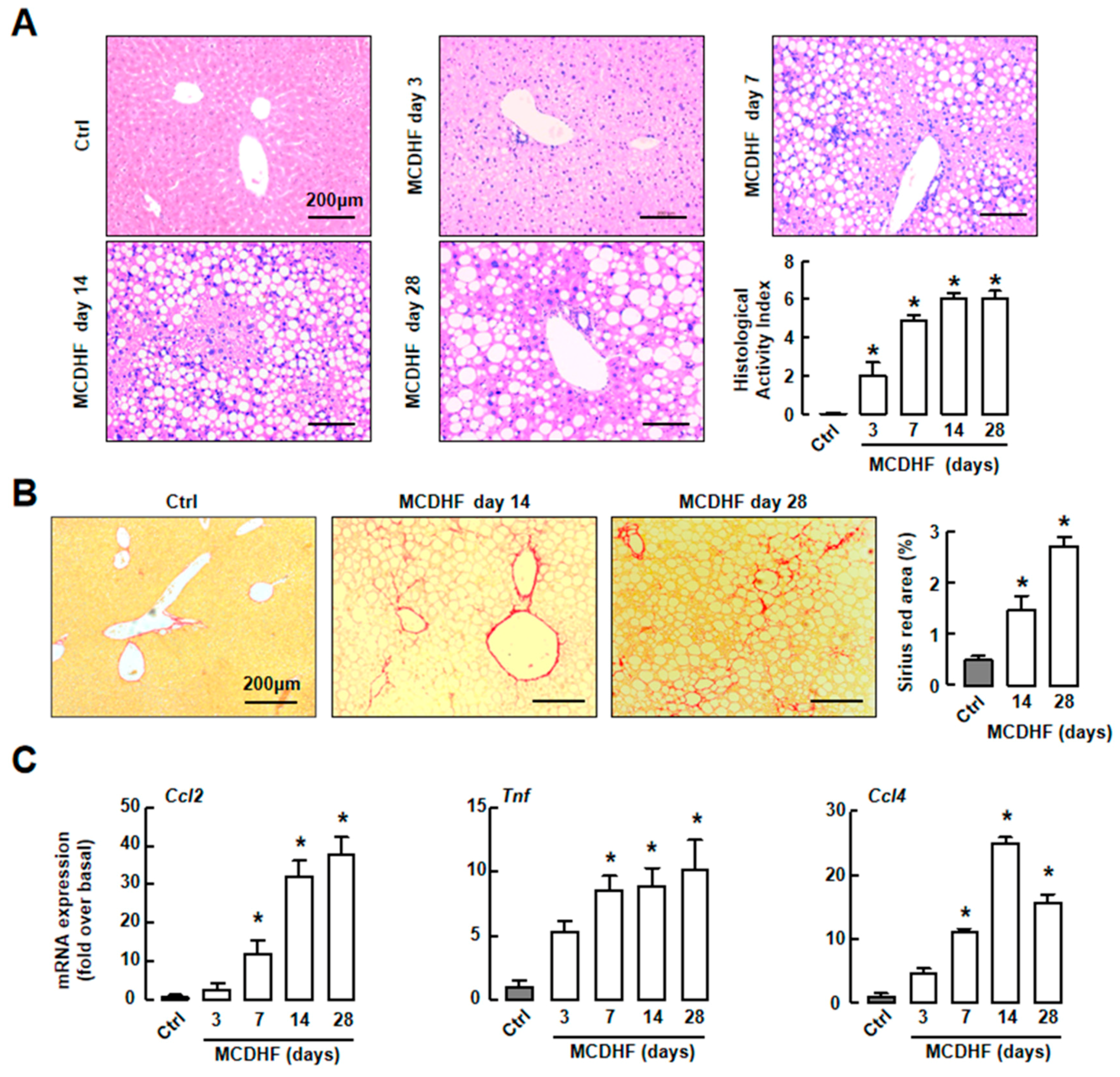
2.1. Mouse Fatty Liver Model Induced by MCDHF Diet Shows Progressive Hepatic Steatosis, Inflammation, and/then Fibrosis
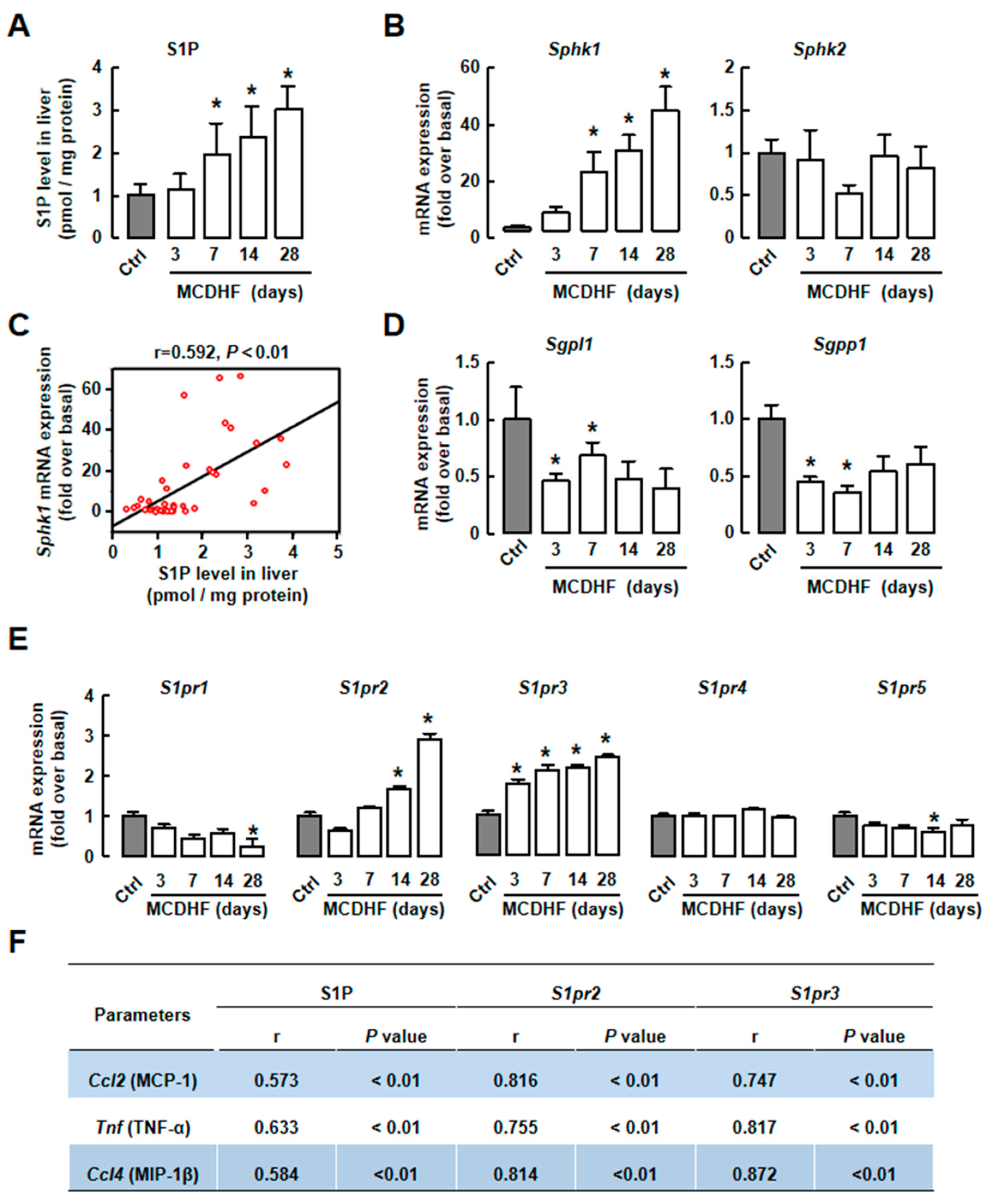
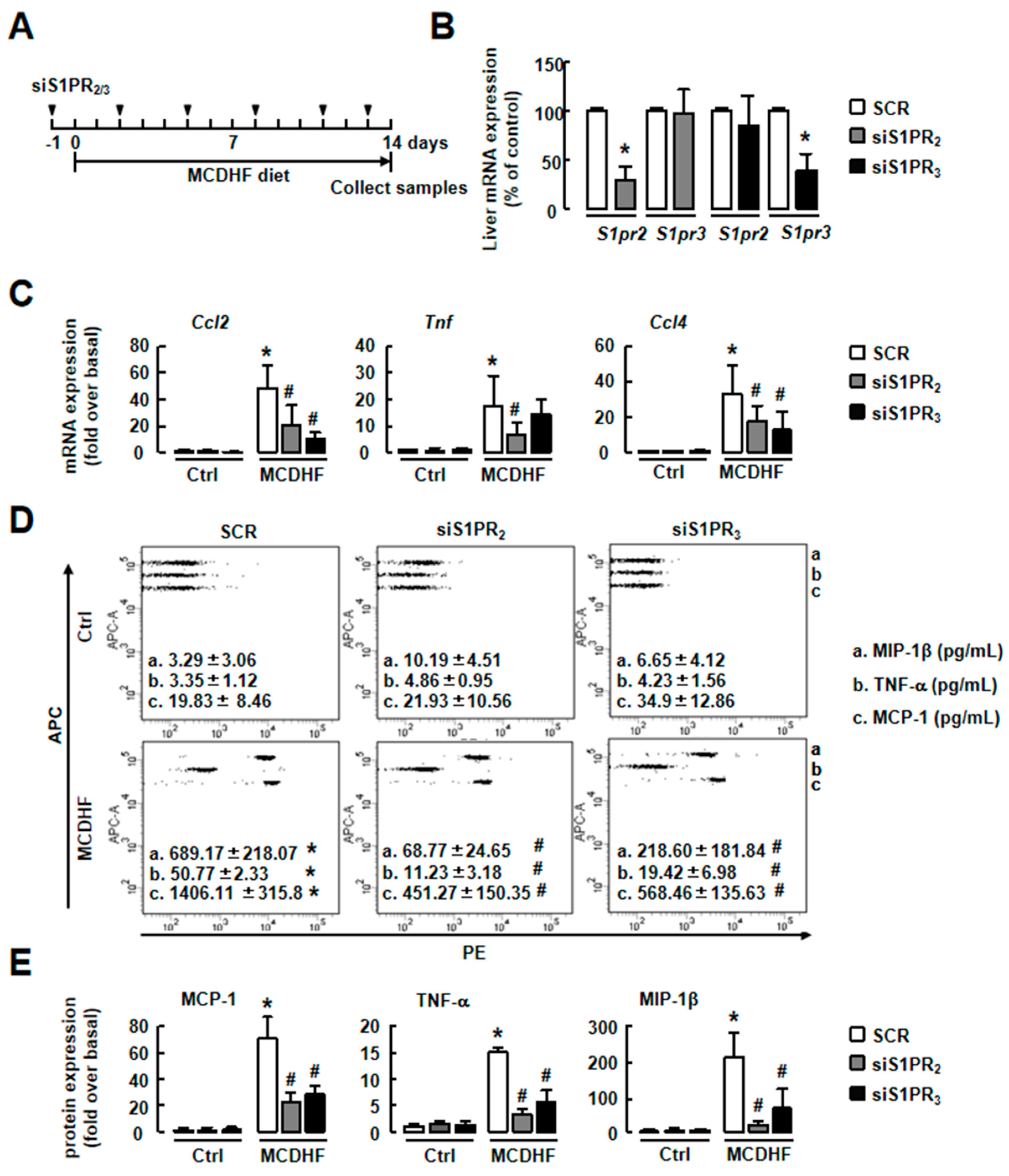
2.2. S1P/S1PR2/3 Axis was Associated with Pro-Inflammatory Factor Production in MCDHF Diet-Treated Livers
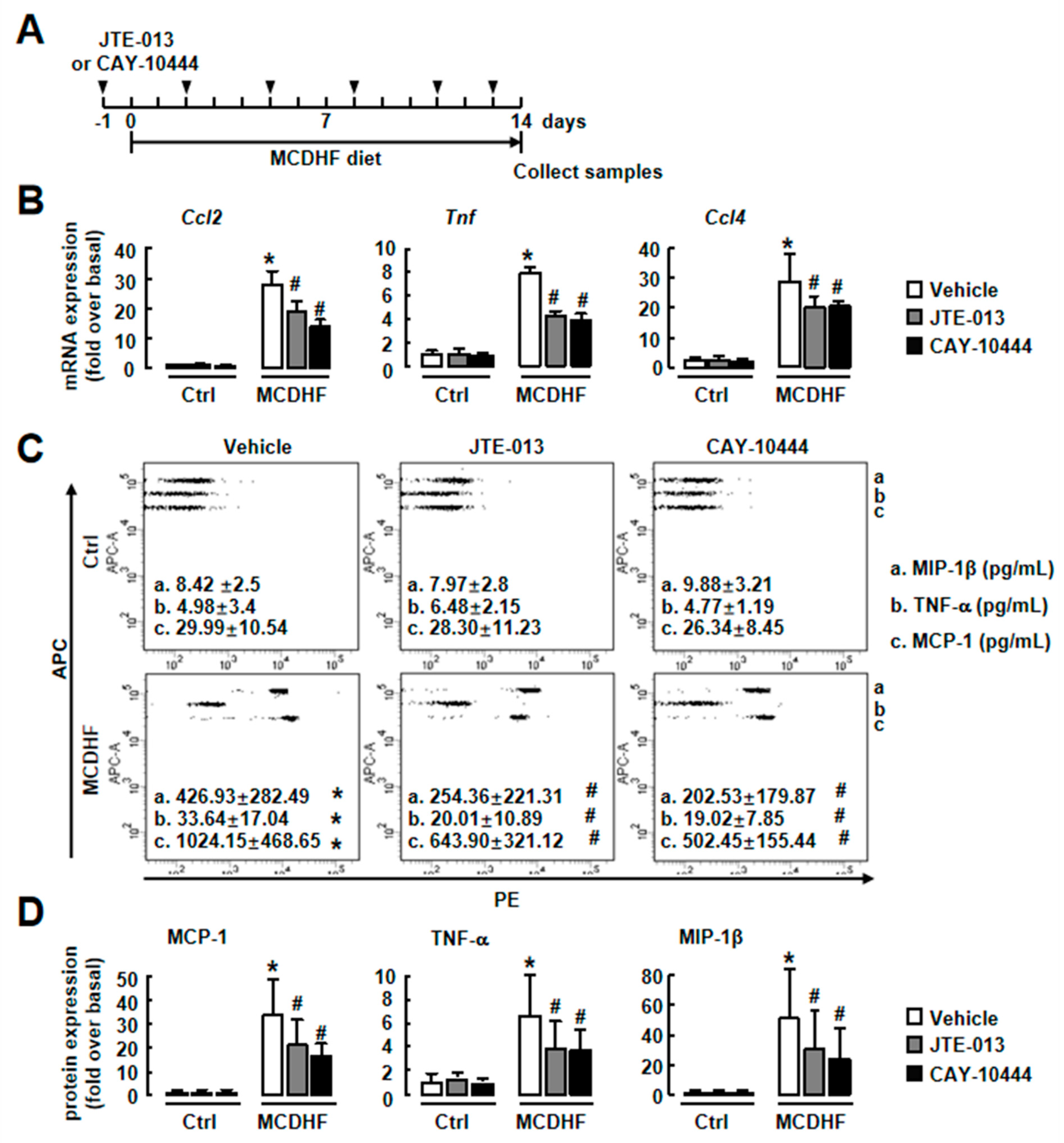
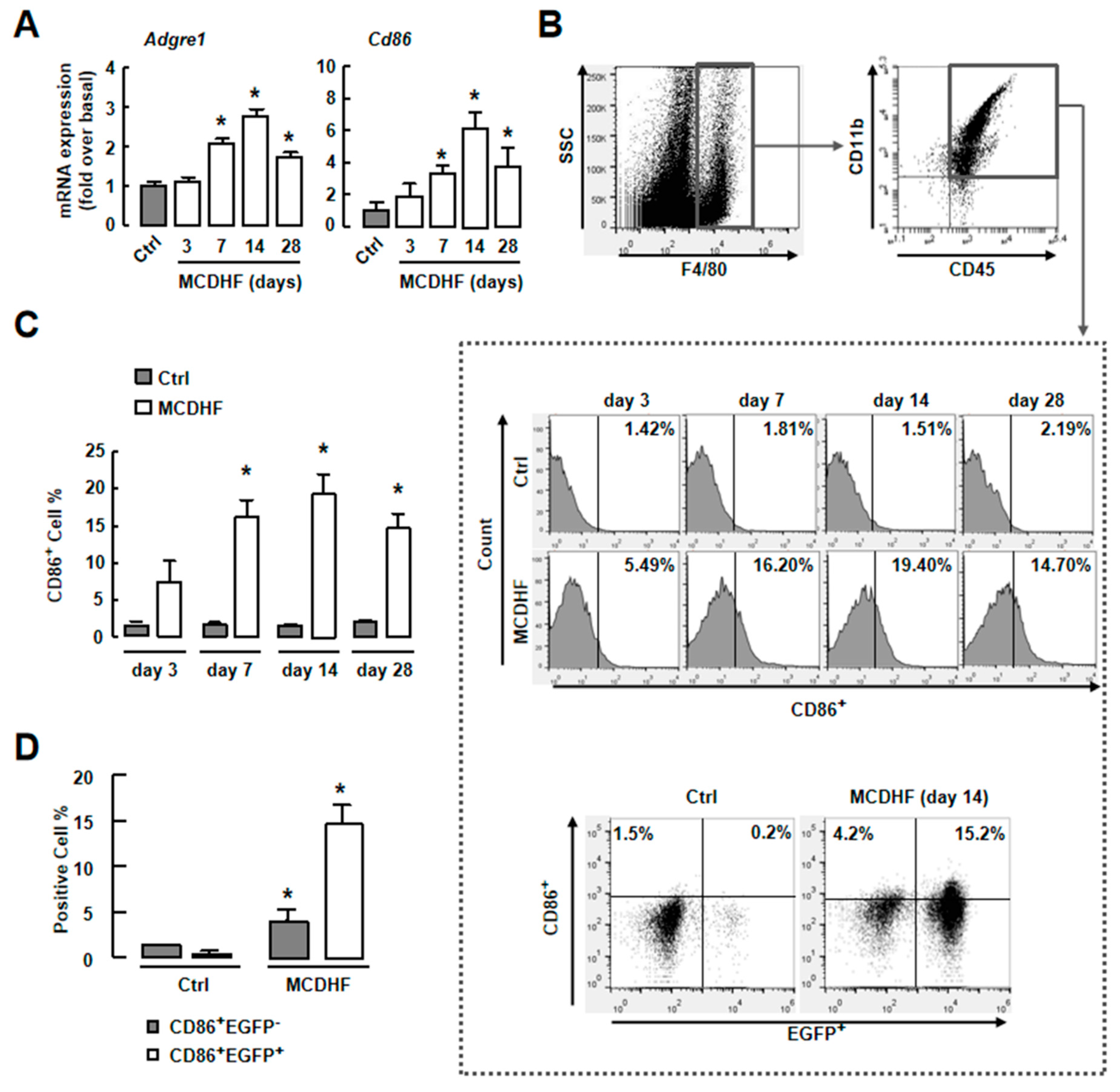
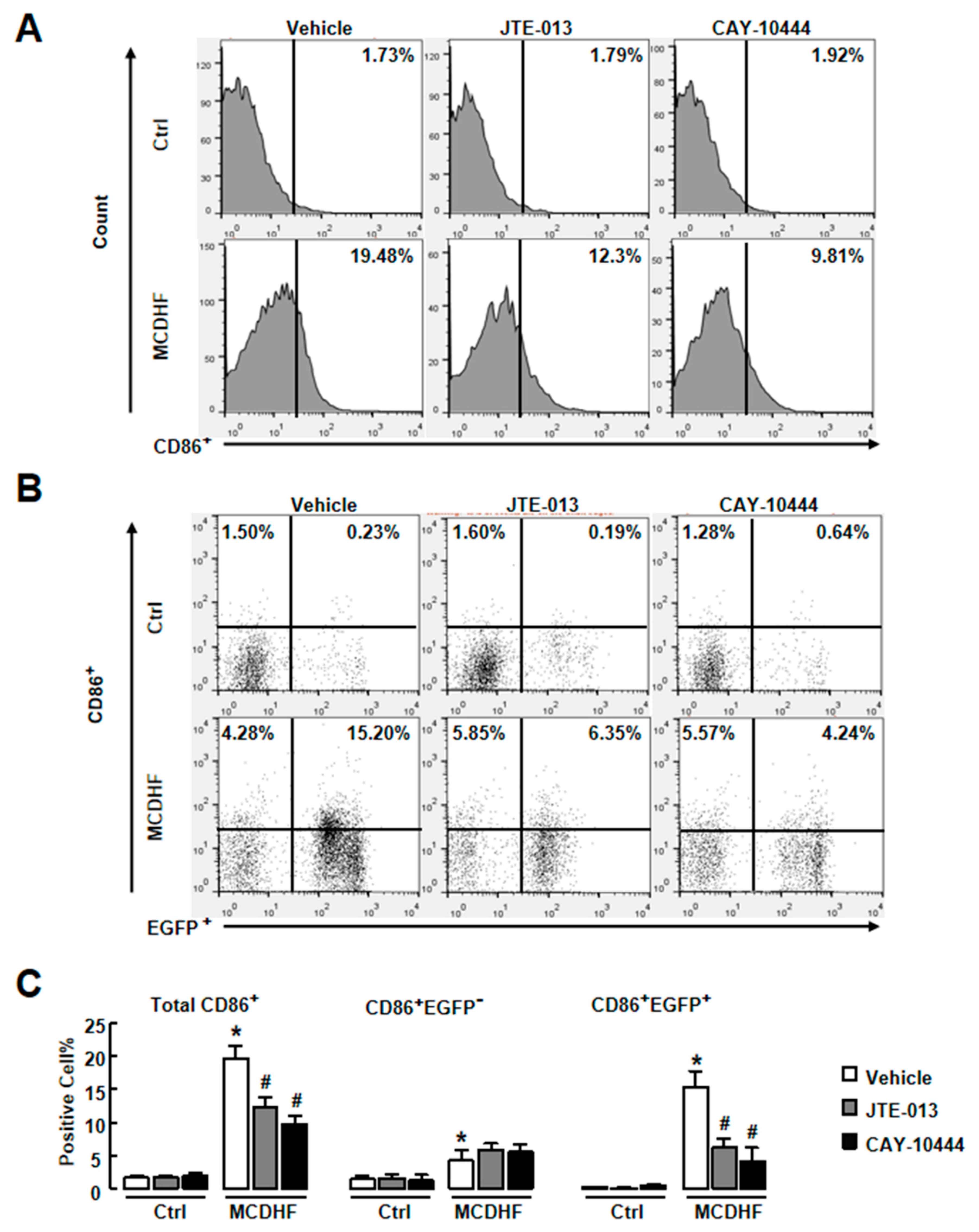
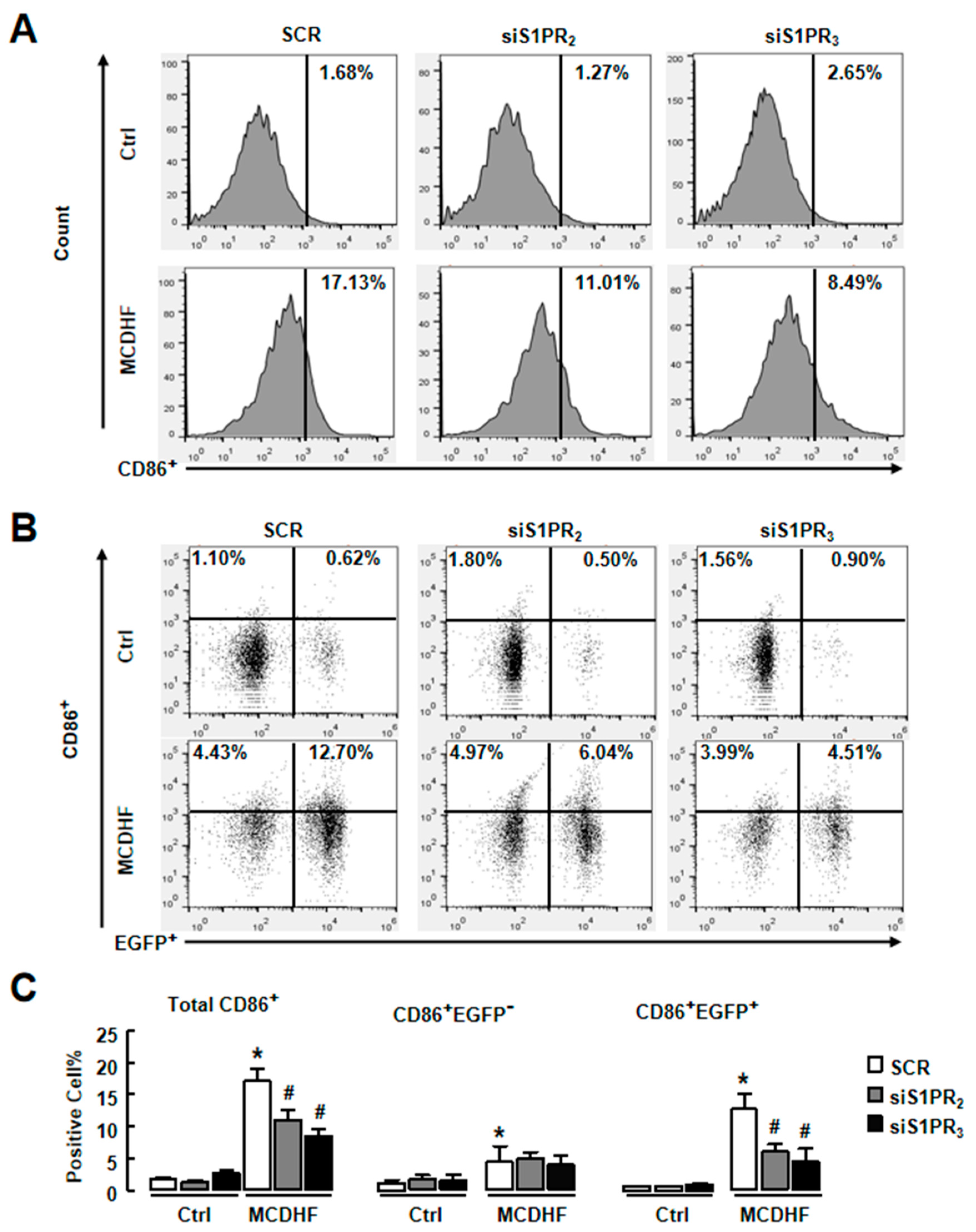
2.3. Blockade of S1PR2/3 Alleviates Hepatic Inflammation by Decreasing the Number of Pro-Inflammatory BMMs and Inflammatory Factor Expression in Fatty Liver Injury
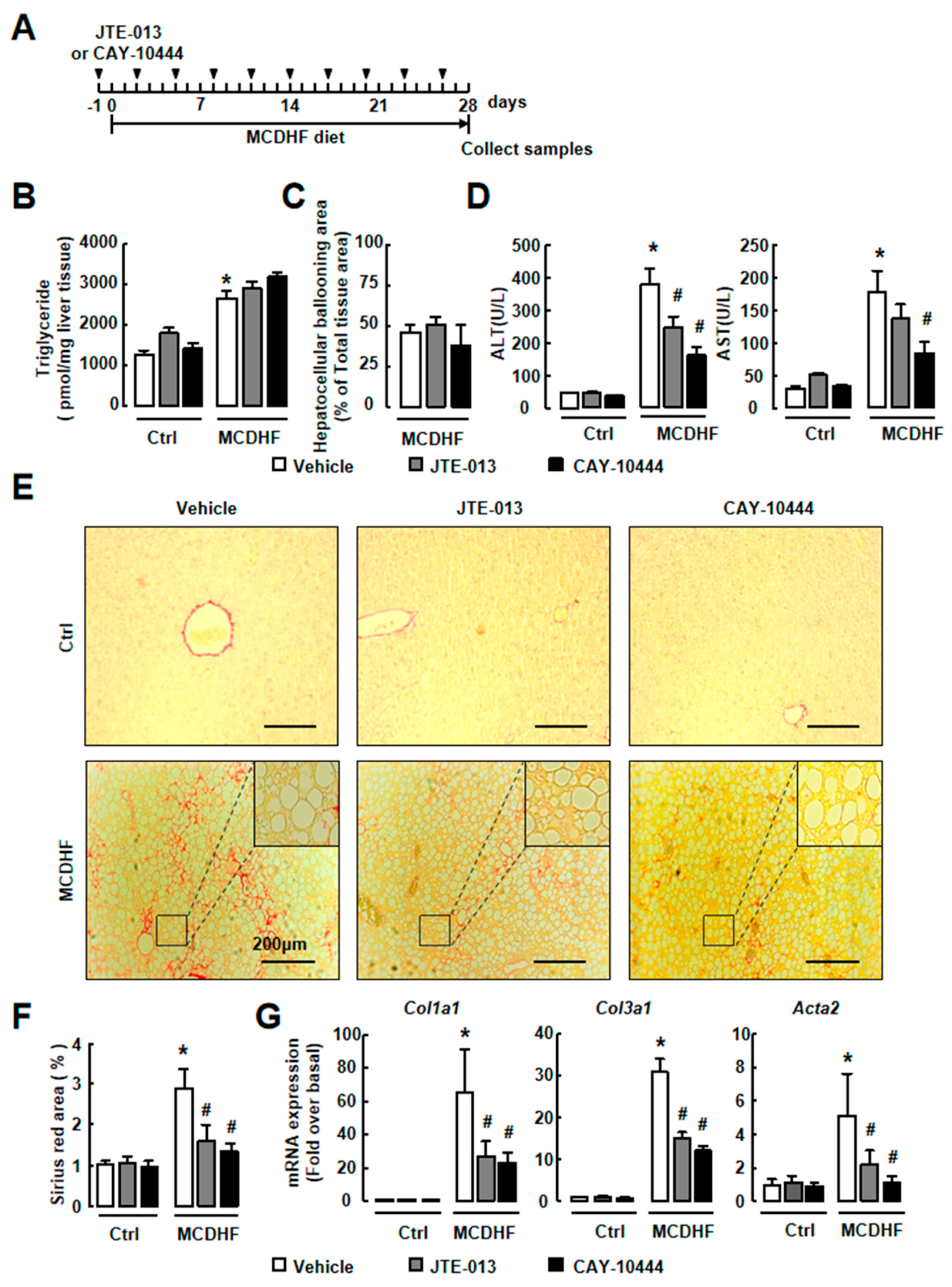
2.4. Antagonism of S1PR2/3 Attenuates Hepatic Injury and Fibrosis, Not Fat Accumulation In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Model
4.3. BM Transplantation
4.4. RNA Interference (RNAi) In Vivo
4.5. Fluorescence-Activated Cell Sorting (FACS)
4.6. S1P Quantitation
4.7. RT-qPCR
4.8. Histology Analysis
4.9. Measurement of Cytokine and Chemokine by CBA
4.10. Biochemical Assays
4.11. Triglyceride Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nobili, V.; Alisi, A.; Newton, K.P.; Schwimmer, J.B. Comparison of the Phenotype and Approach to Pediatric vs Adult Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1798–1810. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Haukeland, J.W.; Damas, J.K.; Konopski, Z.; Loberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjoro, K.; Aukrust, P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Castillo, J.; Perugorria, M.J.; Latasa, M.U.; Prieto, J.; Avila, M.A. Inflammation and liver cancer: New molecular links. Ann. N. Y. Acad. Sci. 2009, 1155, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Klein, I.; Cornejo, J.C.; Polakos, N.K.; John, B.; Wuensch, S.A.; Topham, D.J.; Pierce, R.H.; Crispe, I.N. Kupffer cell heterogeneity: Functional properties of bone marrow derived and sessile hepatic macrophages. Blood 2007, 110, 4077–4085. [Google Scholar] [CrossRef]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J. Immunol. 2014, 193, 344–353. [Google Scholar] [CrossRef]
- Yang, L.; Han, Z.; Tian, L.; Mai, P.; Zhang, Y.; Wang, L.; Li, L. Sphingosine 1-Phosphate Receptor 2 and 3 Mediate Bone Marrow-Derived Monocyte/Macrophage Motility in Cholestatic Liver Injury in Mice. Sci. Rep. 2015, 5, 13423. [Google Scholar] [CrossRef]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Et Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chang, N.; Liu, X.; Han, Z.; Zhu, T.; Li, C.; Yang, L.; Li, L. Bone marrow-derived mesenchymal stem cells differentiate to hepatic myofibroblasts by transforming growth factor-beta1 via sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis. Am. J. Pathol. 2012, 181, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.M.; Del Poeta, M. Sphingosine-1-phosphate receptors and innate immunity. Cell Microbiol. 2018, 20, e12836. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, Y.; Ikeda, H.; Okamoto, Y.; Takuwa, N.; Yoshioka, K. Sphingosine-1-phosphate as a mediator involved in development of fibrotic diseases. Biochim. Biophys. Acta 2013, 1831, 185–192. [Google Scholar] [CrossRef]
- O’Sullivan, S.; Dev, K.K. Sphingosine-1-phosphate receptor therapies: Advances in clinical trials for CNS-related diseases. Neuropharmacology 2017, 113, 597–607. [Google Scholar] [CrossRef]
- Koscielny, V. Phase III SUNBEAM and RADIANCE PART B trials for Ozanimod in relapsing multiple sclerosis demonstrate superiority versus interferon-beta-1a (Avonex((R))) in reducing annualized relapse rates and MRI brain lesions. Neurodegener. Dis. Manag. 2018, 8, 141–142. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A.; et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Christopher, R.; Behan, D.; Lassen, C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun. Rev. 2017, 16, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, L.; Tian, L.; Ji, X.; Yang, L.; Li, L. Sphingosine 1-Phosphate (S1P)/S1P Receptor2/3 Axis Promotes Inflammatory M1 Polarization of Bone Marrow-Derived Monocyte/Macrophage via G(alpha)i/o/PI3K/JNK Pathway. Cell. Physiol. Biochem. 2018, 49, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kong, Y.; Wang, H.; Wang, S.; Yu, H.; Liu, X.; Yang, L.; Jiang, X.; Li, L. Homing of bone marrow mesenchymal stem cells mediated by sphingosine 1-phosphate contributes to liver fibrosis. J. Hepatol. 2009, 50, 1174–1183. [Google Scholar] [CrossRef]
- Pinto, A.T.; Pinto, M.L.; Cardoso, A.P.; Monteiro, C.; Pinto, M.T.; Maia, A.F.; Castro, P.; Figueira, R.; Monteiro, A.; Marques, M.; et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci. Rep. 2016, 6, 18765. [Google Scholar] [CrossRef]
- Tian, L.; Li, W.; Yang, L.; Chang, N.; Fan, X.; Ji, X.; Xie, J.; Yang, L.; Li, L. Cannabinoid Receptor 1 Participates in Liver Inflammation by Promoting M1 Macrophage Polarization via RhoA/NF-kappaB p65 and ERK1/2 Pathways, Respectively, in Mouse Liver Fibrogenesis. Front. Immunol. 2017, 8, 1214. [Google Scholar] [CrossRef]
- Tiper, I.V.; East, J.E.; Subrahmanyam, P.B.; Webb, T.J. Sphingosine 1-phosphate signaling impacts lymphocyte migration, inflammation and infection. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef]
- Aoki, M.; Aoki, H.; Ramanathan, R.; Hait, N.C.; Takabe, K. Sphingosine-1-Phosphate Signaling in Immune Cells and Inflammation: Roles and Therapeutic Potential. Mediat. Inflamm. 2016, 2016, 8606878. [Google Scholar] [CrossRef]
- Li, C.; Jiang, X.; Yang, L.; Liu, X.; Yue, S.; Li, L. Involvement of sphingosine 1-phosphate (SIP)/S1P3 signaling in cholestasis-induced liver fibrosis. Am. J. Pathol. 2009, 175, 1464–1472. [Google Scholar] [CrossRef]
- Li, C.; Zheng, S.; You, H.; Liu, X.; Lin, M.; Yang, L.; Li, L. Sphingosine 1-phosphate (S1P)/S1P receptors are involved in human liver fibrosis by action on hepatic myofibroblasts motility. J. Hepatol. 2011, 54, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Mauer, A.S.; Hirsova, P.; Maiers, J.L.; Shah, V.H.; Malhi, H. Inhibition of sphingosine 1-phosphate signaling ameliorates murine nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G300–G313. [Google Scholar] [CrossRef] [PubMed]
- Geng, T.; Sutter, A.; Harland, M.D.; Law, B.A.; Ross, J.S.; Lewin, D.; Palanisamy, A.; Russo, S.B.; Chavin, K.D.; Cowart, L.A. SphK1 mediates hepatic inflammation in a mouse model of NASH induced by high saturated fat feeding and initiates proinflammatory signaling in hepatocytes. J. Lipid Res. 2015, 56, 2359–2371. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Gieseler, R.K.; Gores, G.J.; Gerken, G. The relationship between apoptosis and non-alcoholic fatty liver disease: An evolutionary cornerstone turned pathogenic. Z. Fur. Gastroenterol. 2005, 43, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Orci, L. Lipoapoptosis: Its mechanism and its diseases. Biochim. Biophys. Acta 2002, 1585, 202–212. [Google Scholar] [CrossRef]
- Liaskou, E.; Wilson, D.V.; Oo, Y.H. Innate immune cells in liver inflammation. Mediat. Inflamm. 2012, 2012, 949157. [Google Scholar] [CrossRef]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef]
- Bhargava, P.; Lee, C.H. Role and function of macrophages in the metabolic syndrome. Biochem. J. 2012, 442, 253–262. [Google Scholar] [CrossRef]
- Tacke, F. Functional role of intrahepatic monocyte subsets for the progression of liver inflammation and liver fibrosis in vivo. Fibrogenesis. Tissue Repair. 2012, 5, S27. [Google Scholar] [CrossRef]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Mai, P.; Yang, L.; Tian, L.; Wang, L.; Jia, S.; Zhang, Y.; Liu, X.; Li, L. Endocannabinoid System Contributes to Liver Injury and Inflammation by Activation of Bone Marrow-Derived Monocytes/Macrophages in a CB1-Dependent Manner. J. Immunol. 2015, 195, 3390–3401. [Google Scholar] [CrossRef] [PubMed]
- Bieghs, V.; Trautwein, C. The innate immune response during liver inflammation and metabolic disease. Trends. Immunol. 2013, 34, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Teixeira-Clerc, F.; Chobert, M.N.; Deveaux, V.; Pavoine, C.; Zimmer, A.; Pecker, F.; Mallat, A.; Lotersztajn, S. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 2011, 54, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Shao, M.; Kim, M.Y.; Baik, S.K.; Cho, M.Y.; Utsumi, T.; Satoh, A.; Ouyang, X.; Chung, C.; Iwakiri, Y. An endoplasmic reticulum protein, Nogo-B, facilitates alcoholic liver disease through regulation of kupffer cell polarization. Hepatology 2017, 65, 1720–1734. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Aleffi, S.; Galastri, S.; Provenzano, A. Mononuclear cells in liver fibrosis. Semin. Immunopathol. 2009, 31, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zhu, T.; Liu, X.; Li, C.; Yue, S.; Yang, L.; Li, L. 15-deoxy-Delta12,14 -prostaglandin J2 reduces recruitment of bone marrow-derived monocyte/macrophages in chronic liver injury in mice. Hepatology 2012, 56, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M. Grading and staging the histopathological lesions of chronic hepatitis: The Knodell histology activity index and beyond. Hepatology 2000, 31, 241–246. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Chang, N.; Yang, L.; Ji, X.; Zhou, X.; Tian, L.; Ma, Y.; Yang, Y.; Liu, Y.; Yang, L.; et al. Sphingosine 1-Phosphate Receptor Blockade Affects Pro-Inflammatory Bone Marrow-Derived Macrophages and Relieves Mouse Fatty Liver Injury. Int. J. Mol. Sci. 2019, 20, 4695. https://doi.org/10.3390/ijms20194695
Yang J, Chang N, Yang L, Ji X, Zhou X, Tian L, Ma Y, Yang Y, Liu Y, Yang L, et al. Sphingosine 1-Phosphate Receptor Blockade Affects Pro-Inflammatory Bone Marrow-Derived Macrophages and Relieves Mouse Fatty Liver Injury. International Journal of Molecular Sciences. 2019; 20(19):4695. https://doi.org/10.3390/ijms20194695
Chicago/Turabian StyleYang, Jingjing, Na Chang, Le Yang, Xiaofang Ji, Xuan Zhou, Lei Tian, Yuehan Ma, Yuanru Yang, Yuran Liu, Lin Yang, and et al. 2019. "Sphingosine 1-Phosphate Receptor Blockade Affects Pro-Inflammatory Bone Marrow-Derived Macrophages and Relieves Mouse Fatty Liver Injury" International Journal of Molecular Sciences 20, no. 19: 4695. https://doi.org/10.3390/ijms20194695
APA StyleYang, J., Chang, N., Yang, L., Ji, X., Zhou, X., Tian, L., Ma, Y., Yang, Y., Liu, Y., Yang, L., & Li, L. (2019). Sphingosine 1-Phosphate Receptor Blockade Affects Pro-Inflammatory Bone Marrow-Derived Macrophages and Relieves Mouse Fatty Liver Injury. International Journal of Molecular Sciences, 20(19), 4695. https://doi.org/10.3390/ijms20194695