EGFR-Specific Tyrosine Kinase Inhibitor Modifies NK Cell-Mediated Antitumoral Activity against Ovarian Cancer Cells



Abstract
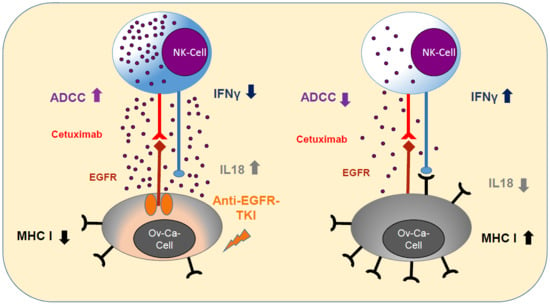
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
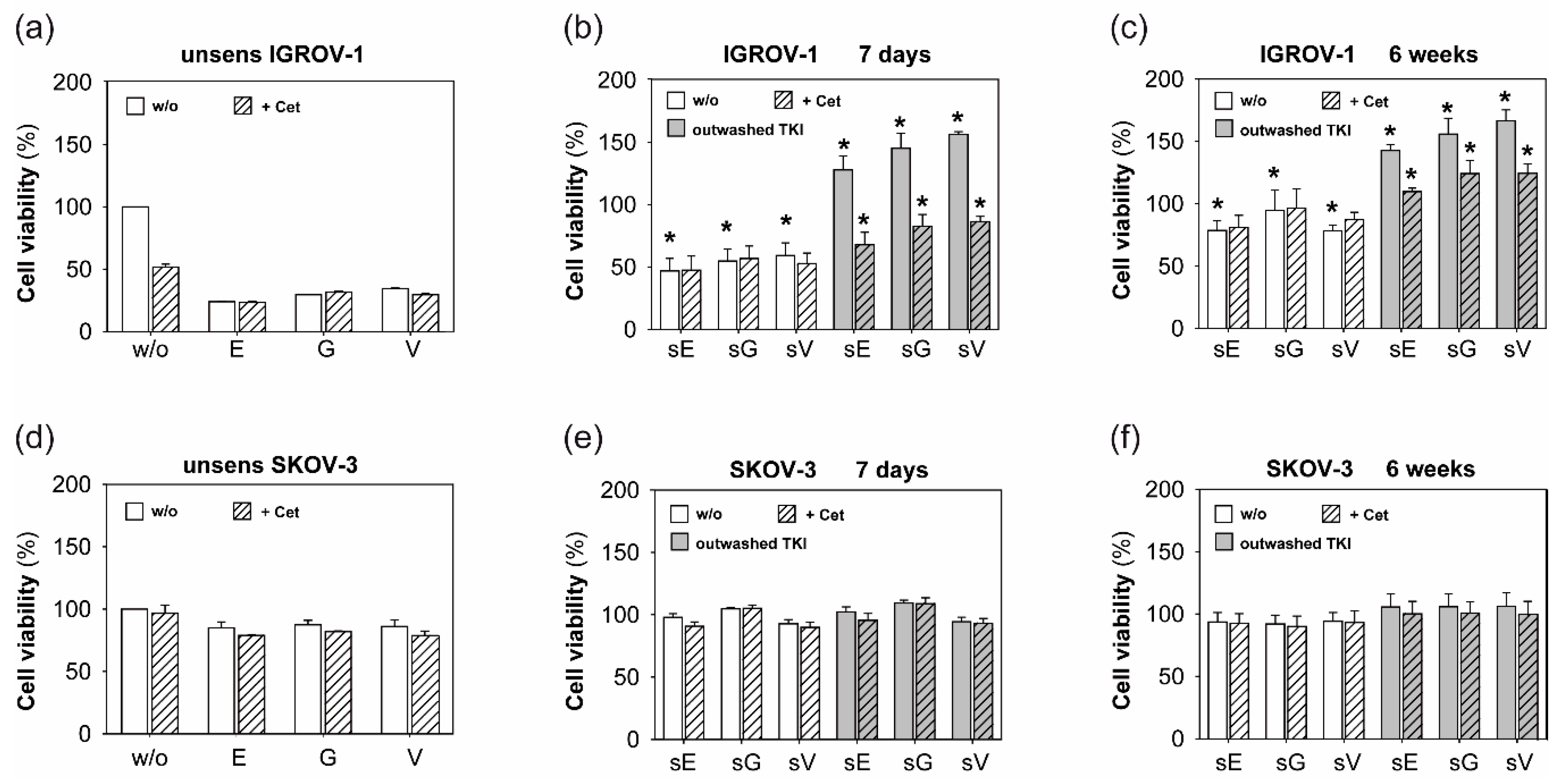
2.1. Anti-EGFR Sensitization of Ovarian Cancer Cells Enhanced Tumor Cell Viability and Increased Resistance to Cetuximab
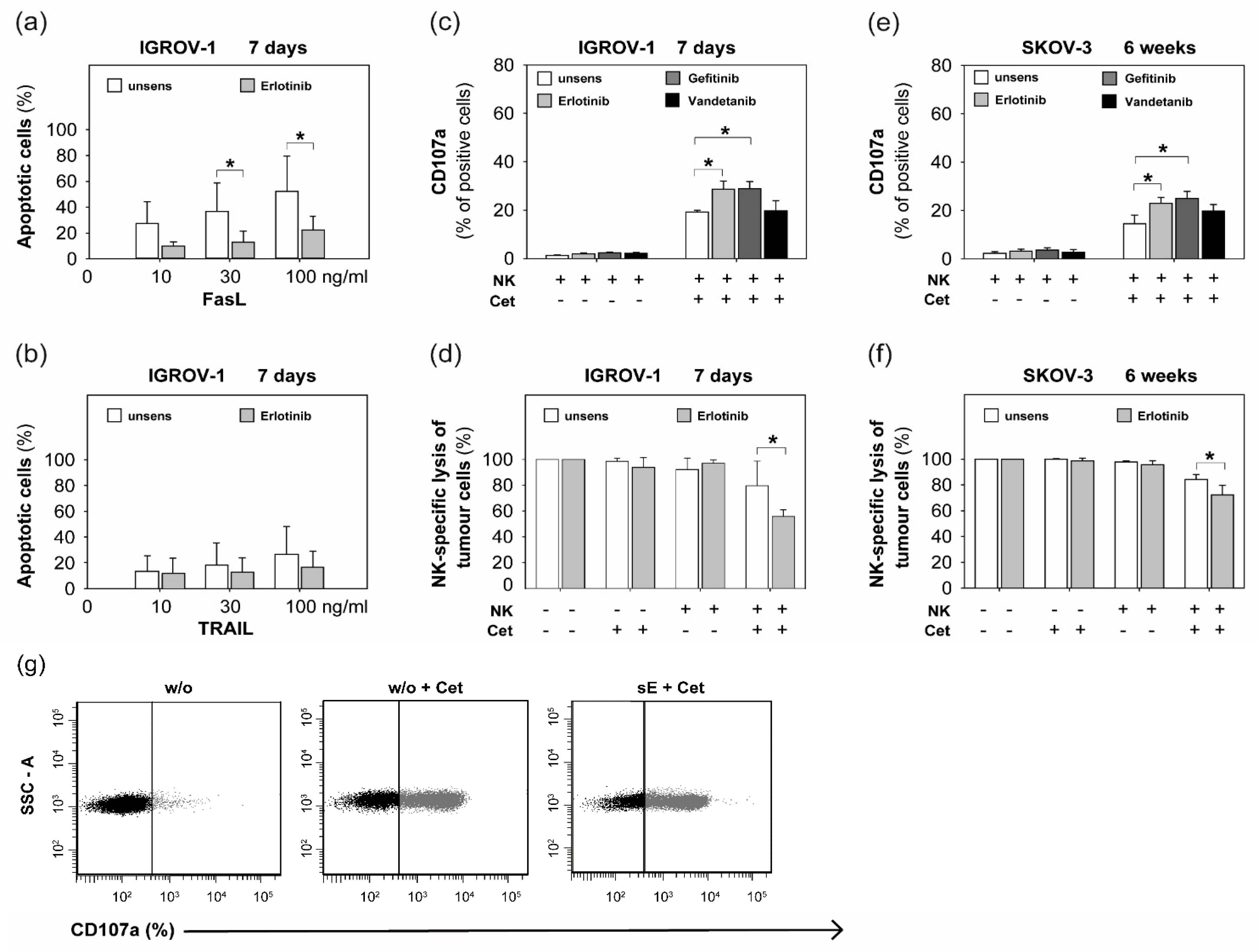
2.2. Sensitization with Anti-EGFR TKI Decreased Sensitivity to FasLigand but Enhanced Ovarian Cancer Cells for NK Cell-Mediated Cytotoxic Degranulation
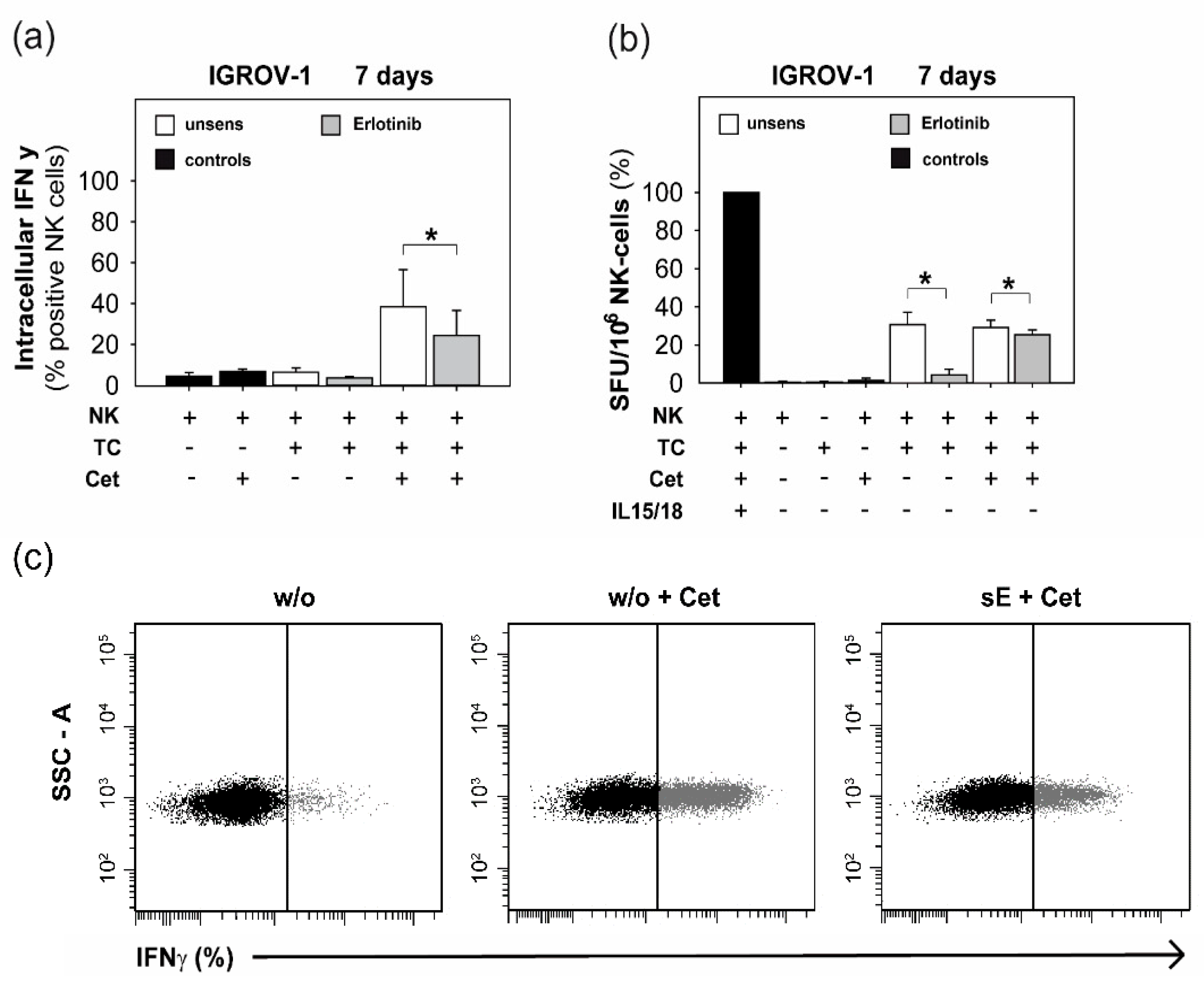
2.3. Anti-EGFR TKI Sensitized Ovarian Cancer Cells Led to Reduced Cytokine Release of Secretory NK Cells
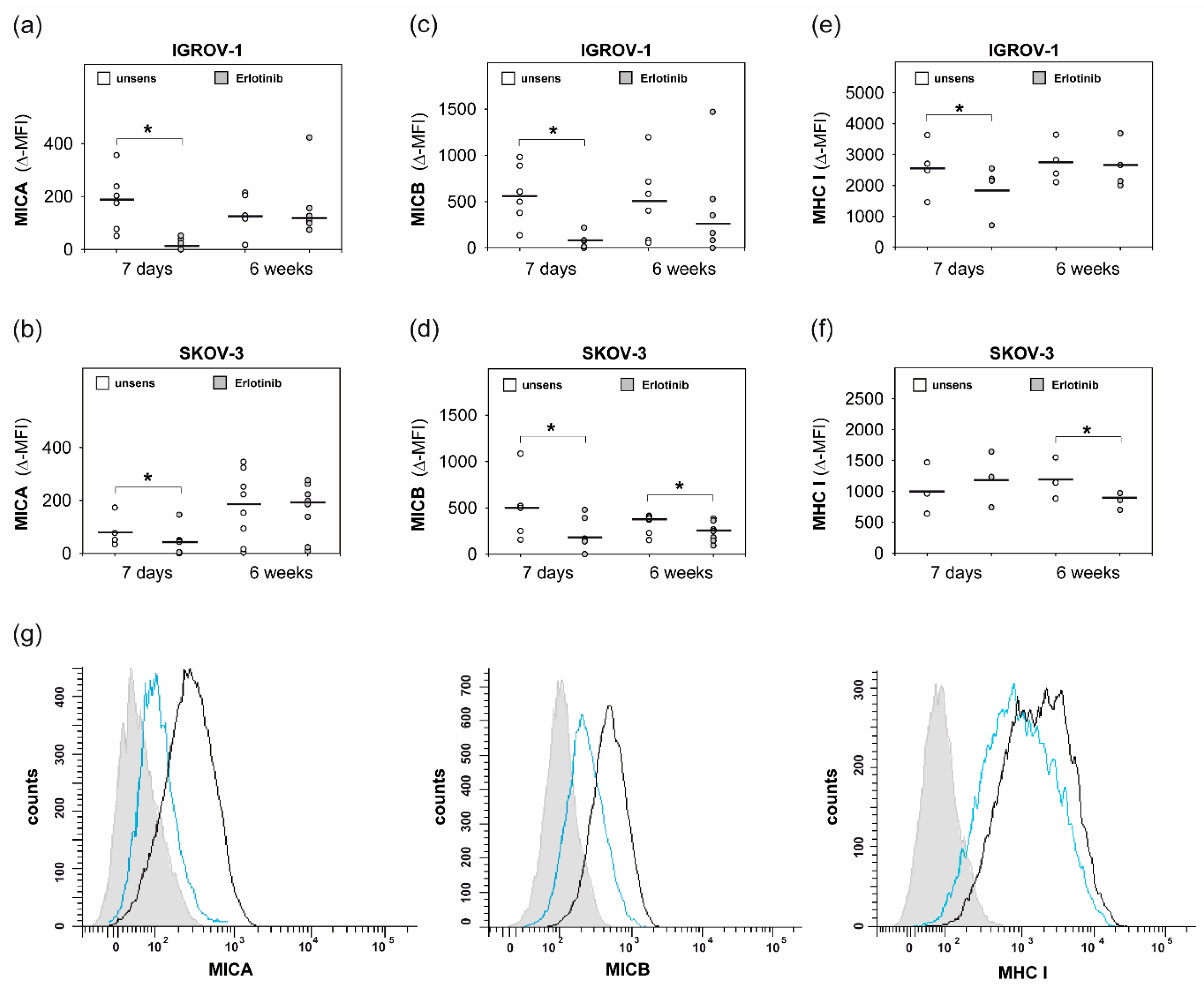
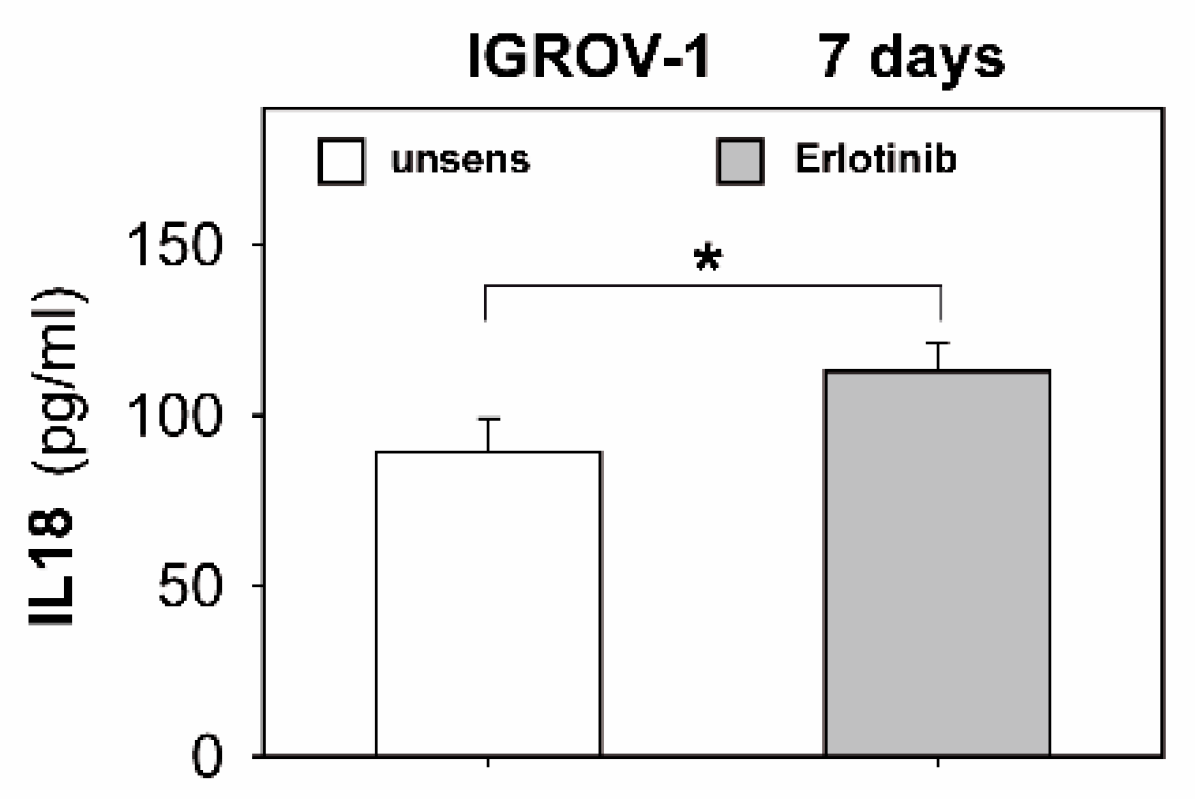
2.4. Anti-EGFR Sensitized Ovarian Cancer Cells Show Altered Expression of Stress-Induced Ligands and MHC I and Enhanced Cytokine Release
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Sensitization of Ovarian Cancer Cells with Anti-EGFR TKIs
4.3. MTT Proliferation Assay
4.4. Isolation of NK Cells of Healthy Donors
4.5. ELISA for Human IL18, IL12, and TNFα
4.6. Antibodies Used for Flow Cytometric Analysis (FACS)
4.7. Extracellular and Intracellular FACS Staining
4.8. CD107a Degranulation Assay
4.9. 7-AAD/Annexin Staining
4.10. ELISpot for IFNγ Detection
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howlader, N.N.A.; Krapcho, M.; Neyman, N.; Aminou, R.; Altekruse, S.F.; Kosary, C.L.; Ruhl, J.; Tatalovich, Z.; Cho, H.; Mariotto, A.; et al. (Eds.) SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). Available online: http://seer.cancer.gov/csr/1975_2009_pops09 (accessed on 20 August 2012).
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.M.; Langdon, S.P.; Simpson, B.J.; Stewart, M.; Katsaros, D.; Sismondi, P.; Love, S.; Scott, W.N.; Williams, A.R.; Lessells, A.M.; et al. The prognostic value of epidermal growth factor receptor mRNA expression in primary ovarian cancer. Br. J. Cancer 1996, 73, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Colbrie, J.; Witt, A.; Heinzl, H.; Speiser, P.; Czerwenka, K.; Sevelda, P.; Zeillinger, R. EGFR and steroid receptors in ovarian carcinoma: Comparison with prognostic parameters and outcome of patients. Anticancer Res. 1997, 17, 613–619. [Google Scholar] [PubMed]
- Mendelsohn, J.; Baselga, J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 2003, 21, 2787–2799. [Google Scholar] [CrossRef]
- Phelps, S.L.B.; Schorge, J.O.; Peyton, M.J.; Shigematsu, H.; Xiang, L.L.; Miller, D.S.; Lea, J.S. Implications of EGFR inhibition in ovarian cancer cell proliferation. Gynecol. Oncol. 2008, 109, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Konner, J.; Schilder, R.J.; DeRosa, F.A.; Gerst, S.R.; Tew, W.P.; Sabbatini, P.J.; Hensley, M.L.; Spriggs, D.R.; Aghajanian, C.A. A phase II study of cetuximab/paclitaxel/carboplatin for the initial treatment of advanced-stage ovarian, primary peritoneal, or fallopian tube cancer. Gynecol. Oncol. 2008, 110, 140–145. [Google Scholar] [CrossRef]
- Secord, A.A.; Blessing, J.A.; Armstrong, D.K.; Rodgers, W.H.; Miner, Z.; Barnes, M.N.; Lewandowski, G.; Mannel, R.S.; Gynecologic Oncology, G. Phase II trial of cetuximab and carboplatin in relapsed platinum-sensitive ovarian cancer and evaluation of epidermal growth factor receptor expression: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 108, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Schilder, R.J.; Pathak, H.B.; Lokshin, A.E.; Holloway, R.W.; Alvarez, R.D.; Aghajanian, C.; Min, H.; Devarajan, K.; Ross, E.; Drescher, C.W.; et al. Phase II trial of single agent cetuximab in patients with persistent or recurrent epithelial ovarian or primary peritoneal carcinoma with the potential for dose escalation to rash. Gynecol. Oncol. 2009, 113, 21–27. [Google Scholar] [CrossRef]
- Dai, Q.; Ling, Y.H.; Lia, M.; Zou, Y.Y.; Kroog, G.; Iwata, K.K.; Perez-Soler, R. Enhanced sensitivity to the HER1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib hydrochloride in chemotherapy-resistant tumor cell lines. Clin. Cancer Res. 2005, 11, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Gaikwad, A.; Yu, J.; Wolf, J.K.; Brown, J.; Ramondetta, L.M.; Stewart, C.F. In vitro evaluation of the effects of gefitinib on the modulation of cytotoxic activity of selected anticancer agents in a panel of human ovarian cancer cell lines. Cancer Chemother. Pharmacol. 2008, 62, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.N.; Finkler, N.; Edwards, R.P.; Garcia, A.A.; Crozier, M.; Irwin, D.H.; Barrett, E. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: Results from a phase II multicenter study. Int. J. Gynecol. Cancer 2005, 15, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Blank, S.V.; Christos, P.; Curtin, J.P.; Goldman, N.; Runowicz, C.D.; Sparano, J.A.; Liebes, L.; Chen, H.X.; Muggia, F.M. Erlotinib added to carboplatin and paclitaxel as first-line treatment of ovarian cancer: A phase II study based on surgical reassessment. Gynecol. Oncol. 2010, 119, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.K.; Clouser, M.C.; Baker, A.F.; Roe, D.J.; Cui, H.; Brewer, M.A.; Hatch, K.D.; Gordon, M.S.; Janicek, M.F.; Isaacs, J.D.; et al. Overexpression of tumor vascular endothelial growth factor A may portend an increased likelihood of progression in a phase II trial of bevacizumab and erlotinib in resistant ovarian cancer. Clin. Cancer Res. 2010, 16, 5320–5328. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.B.; Jimeno, A.; Joly, F.; Katsaros, D.; Coens, C.; Despierre, E.; Marth, C.; Hall, M.; Steer, C.B.; Colombo, N.; et al. Randomized phase III study of erlotinib versus observation in patients with no evidence of disease progression after first-line platin-based chemotherapy for ovarian carcinoma: A European Organisation for Research and Treatment of Cancer-Gynaecological Cancer Group, and Gynecologic Cancer Intergroup study. J. Clin. Oncol. 2014, 32, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Posadas, E.M.; Liel, M.S.; Kwitkowski, V.; Minasian, L.; Godwin, A.K.; Hussain, M.M.; Espina, V.; Wood, B.J.; Steinberg, S.M.; Kohn, E.C. A phase II and pharmacodynamic study of gefitinib in patients with refractory or recurrent epithelial ovarian cancer. Cancer 2007, 109, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef]
- Learn, C.A.; Hartzell, T.L.; Wikstrand, C.J.; Archer, G.E.; Rich, J.N.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin. Cancer Res. 2004, 10, 3216–3224. [Google Scholar] [CrossRef]
- Weickhardt, A.J.; Price, T.J.; Chong, G.; Gebski, V.; Pavlakis, N.; Johns, T.G.; Azad, A.; Skrinos, E.; Fluck, K.; Dobrovic, A.; et al. Dual Targeting of the Epidermal Growth Factor Receptor Using the Combination of Cetuximab and Erlotinib: Preclinical Evaluation and Results of the Phase II DUX Study in Chemotherapy-Refractory, Advanced Colorectal Cancer. J. Clin. Oncol. 2012, 30, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Azzoli, C.G.; Krug, L.M.; Pereira, L.K.; Rizvi, N.A.; Pietanza, M.C.; Kris, M.G.; Ginsberg, M.S.; Pao, W.; Miller, V.A.; et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin. Cancer Res. 2011, 17, 2521–2527. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, N.; Kimmig, R.; Lang, S.; Singh, M.; Brandau, S. Anti-Epidermal Growth Factor Receptor (EGFR) Antibodies Overcome Resistance of Ovarian Cancer Cells to Targeted Therapy and Natural Cytotoxicity. Int. J. Mol. Sci. 2012, 13, 12000–12016. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Screpanti, V.; Wallin, R.P.; Grandien, A.; Ljunggren, H.G. Impact of FASL-induced apoptosis in the elimination of tumor cells by NK cells. Mol. Immunol. 2005, 42, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Pietra, G.; Montaldo, E.; Vacca, P.; Pende, D.; Falco, M.; Del Zotto, G.; Locatelli, F.; Moretta, A.; Mingari, M.C. Human NK Cells: From Surface Receptors to the Therapy of Leukemias and Solid Tumors. Front. Immunol. 2014, 5, 87. [Google Scholar] [CrossRef]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Shah, M.H.; Turner, M.J.; VanDeusen, J.B.; Whitman, S.P.; Cooper, M.A.; Suzuki, K.; Wechser, M.; Goodsaid, F.; Caligiuri, M.A. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: Implications for the innate immune response. J. Immunol. 1999, 162, 4511–4520. [Google Scholar]
- Petri, R.M.; Hackel, A.; Hahnel, K.; Dumitru, C.A.; Bruderek, K.; Flohe, S.B.; Paschen, A.; Lang, S.; Brandau, S. Activated Tissue-Resident Mesenchymal Stromal Cells Regulate Natural Killer Cell Immune and Tissue-Regenerative Function. Stem Cell Rep. 2017, 9, 985–998. [Google Scholar] [CrossRef]
- Thomas, H.; Jager, M.; Mauel, K.; Brandau, S.; Lask, S.; Flohe, S.B. Interaction with mesenchymal stem cells provokes natural killer cells for enhanced IL-12/IL-18-induced interferon-gamma secretion. Mediat. Inflamm. 2014, 2014, 143463. [Google Scholar] [CrossRef]
- Gottschalk, N.; Lang, S.; Kimmig, R.; Singh, M.; Brandau, S. Monocytes and the 38kDa-antigen of mycobacterium tuberculosis modulate natural killer cell activity and their cytolysis directed against ovarian cancer cell lines. BMC Cancer 2012, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.M.; Li, H.; Yu, J.P.; Ren, X.B.; Wang, C.L. Combined Erlotinib and Cetuximab overcome the acquired resistance to epidermal growth factor receptors tyrosine kinase inhibitor in non-small-cell lung cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Armstrong, E.A.; Benavente, S.; Chinnaiyan, P.; Harari, P.M. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): Combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 2004, 64, 5355–5362. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Gettinger, S.; Camidge, D.R.; Smit, E.F.; Janjigian, Y.Y.; Miller, V.A.; Pao, W.; Freiwald, M.; Fan, J.; Wang, B.; et al. Continued use of afatinib with the addition of cetuximab after progression on afatinib in patients with EGFR mutation-positive non-small-cell lung cancer and acquired resistance to gefitinib or erlotinib. Lung Cancer 2017, 113, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wilken, J.A.; Webster, K.T.; Maihle, N.J. Trastuzumab Sensitizes Ovarian Cancer Cells to EGFR-targeted Therapeutics. J. Ovarian Res. 2010, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Ducano, N.; Lupo, B.; Vigna, E.; Avanzato, D.; Perera, T.; Trusolino, L.; Lanzetti, L.; Comoglio, P.M. Rebound Effects Caused by Withdrawal of MET Kinase Inhibitor Are Quenched by a MET Therapeutic Antibody. Cancer Res. 2016, 76, 5019–5029. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef]
- Bjorkelund, H.; Gedda, L.; Malmqvist, M.; Andersson, K. Resolving the EGF-EGFR interaction characteristics through a multiple-temperature, multiple-inhibitor, real-time interaction analysis approach. Mol. Clin. Oncol. 2013, 1, 343–352. [Google Scholar] [CrossRef]
- Cavazzoni, A.; Alfieri, R.R.; Cretella, D.; Saccani, F.; Ampollini, L.; Galetti, M.; Quaini, F.; Graiani, G.; Madeddu, D.; Mozzoni, P.; et al. Combined use of anti-ErbB monoclonal antibodies and erlotinib enhances antibody-dependent cellular cytotoxicity of wild-type erlotinib-sensitive NSCLC cell lines. Mol. Cancer 2012, 11, 91. [Google Scholar] [CrossRef]
- Bae, J.H.; Kim, S.J.; Kim, M.J.; Oh, S.O.; Chung, J.S.; Kim, S.H.; Kang, C.D. Susceptibility to natural killer cell-mediated lysis of colon cancer cells is enhanced by treatment with epidermal growth factor receptor inhibitors through UL16-binding protein-1 induction. Cancer Sci. 2012, 103, 7–16. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.H.; Kim, M.J.; Kim, S.J.; Park, S.J.; Chung, J.S.; Bae, J.H.; Kang, C.D. EGFR inhibitors enhanced the susceptibility to NK cell-mediated lysis of lung cancer cells. J. Immunother. 2011, 34, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Hayashi, H.; Haratani, K.; Shimizu, S.; Tanizaki, J.; Sakai, K.; Kawakami, H.; Yonesaka, K.; Tsurutani, J.; Togashi, Y.; et al. Mutational activation of the epidermal growth factor receptor down-regulates major histocompatibility complex class I expression via the extracellular signal-regulated kinase in non-small cell lung cancer. Cancer Sci. 2019, 110, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.Z.; Liu, G.J.; Zhang, X.J.; Zhao, J.Z.; Feng, R.T. Erlotinib enhances the CIK cell-killing sensitivity of lung adenocarcinoma A549 cells. Genet. Mol. Res. 2015, 14, 3082–3089. [Google Scholar] [CrossRef] [PubMed]
- Salih, J.; Hilpert, J.; Placke, T.; Grunebach, F.; Steinle, A.; Salih, H.R.; Krusch, M. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int. J. Cancer 2010, 127, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef]
- Geller, M.A.; Knorr, D.A.; Hermanson, D.A.; Pribyl, L.; Bendzick, L.; McCullar, V.; Miller, J.S.; Kaufman, D.S. Intraperitoneal delivery of human natural killer cells for treatment of ovarian cancer in a mouse xenograft model. Cytotherapy 2013, 15, 1297–1306. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Chen, R.; Yin, M.; Zheng, Q. Cetuximab intensifies the ADCC activity of adoptive NK cells in a nude mouse colorectal cancer xenograft model. Oncol. Lett. 2016, 12, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallmann-Gottschalk, N.; Sax, Y.; Kimmig, R.; Lang, S.; Brandau, S. EGFR-Specific Tyrosine Kinase Inhibitor Modifies NK Cell-Mediated Antitumoral Activity against Ovarian Cancer Cells. Int. J. Mol. Sci. 2019, 20, 4693. https://doi.org/10.3390/ijms20194693
Mallmann-Gottschalk N, Sax Y, Kimmig R, Lang S, Brandau S. EGFR-Specific Tyrosine Kinase Inhibitor Modifies NK Cell-Mediated Antitumoral Activity against Ovarian Cancer Cells. International Journal of Molecular Sciences. 2019; 20(19):4693. https://doi.org/10.3390/ijms20194693
Chicago/Turabian StyleMallmann-Gottschalk, Nina, Yvonne Sax, Rainer Kimmig, Stephan Lang, and Sven Brandau. 2019. "EGFR-Specific Tyrosine Kinase Inhibitor Modifies NK Cell-Mediated Antitumoral Activity against Ovarian Cancer Cells" International Journal of Molecular Sciences 20, no. 19: 4693. https://doi.org/10.3390/ijms20194693
APA StyleMallmann-Gottschalk, N., Sax, Y., Kimmig, R., Lang, S., & Brandau, S. (2019). EGFR-Specific Tyrosine Kinase Inhibitor Modifies NK Cell-Mediated Antitumoral Activity against Ovarian Cancer Cells. International Journal of Molecular Sciences, 20(19), 4693. https://doi.org/10.3390/ijms20194693