SILAC-Based Quantification of TGFBR2-Regulated Protein Expression in Extracellular Vesicles of Microsatellite Unstable Colorectal Cancers

 , ,
, ,
Abstract
1. Introduction
2. Results
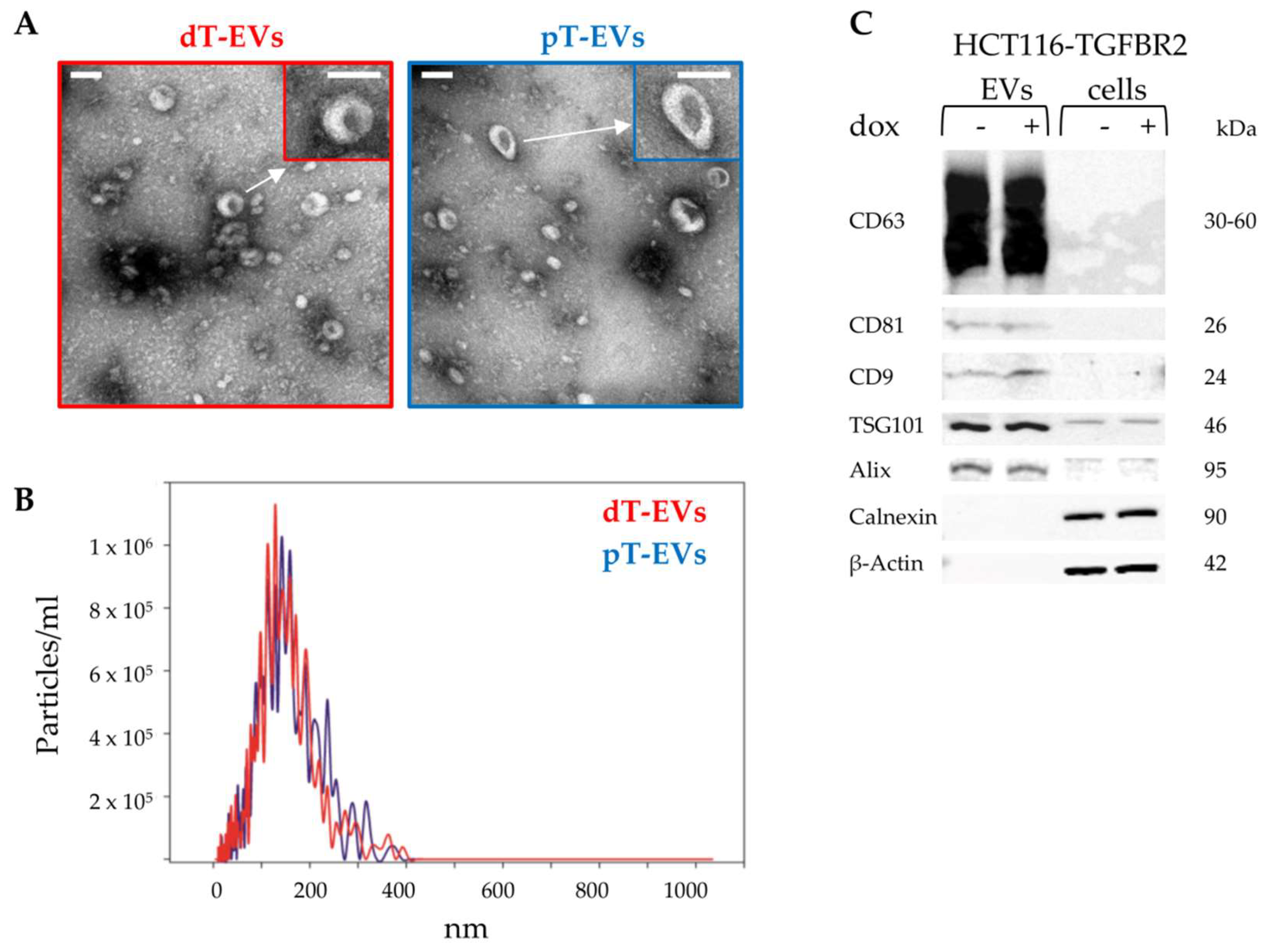
2.1. Isolation and Identification of EVs Secreted by TGFBR2-Deficient and -Proficient MSI Tumor Cells
2.2. SILAC-Based Quantification of EV Proteins Regulated in a TGFBR2-Dependent Manner
2.3. Validation of TGFBR2-Regulated Protein Expression in EVs
2.4. Protein-Protein Network Analysis and Functional Enrichment Analysis of EV Proteins Upregulated by TGFBR2 Deficiency
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)
4.3. Doxycycline Hyclate Treatment
4.4. EV Preparation
4.5. Transmission Electron Microscopy (TEM)
4.6. Nanoparticle Tracking Analysis (NTA)
4.7. Protein Extraction and Determination of Protein Concentration
4.8. In-Solution Tryptic Digestion
4.9. Nano-LC-ESI-MS/MS
4.10. Protein Identification and Quantification
4.11. Data Analysis
4.12. SDS Page and Western Blot
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| Dox | Doxycycline |
| dT | Transforming Growth Factor Beta Receptor Type 2-deficient |
| ECM | Extracellular matrix |
| EVs | Extracellular vesicles |
| FTMS | Fourier transform mass spectrometry |
| MISEV | Minimal information for studies of extracellular vesicles |
| MSI | Microsatellite instability |
| PPI | Protein-protein interaction |
| pT | Transforming Growth Factor Beta Receptor Type 2-proficient |
| SILAC | Stable isotope labeling with amino acids in cell culture |
| TGFBR2 | Transforming Growth Factor Beta Receptor Type 2 |
References
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Staffa, L.; Ahadova, A.; von Knebel Doeberitz, M. Clinical significance of microsatellite instability in colorectal cancer. Langenbecks Arch. Surg. 2014, 399, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Li, S.K.H.; Martin, A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol. Med. 2016, 22, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Buckowitz, A.; Knaebel, H.P.; Benner, A.; Blaker, H.; Gebert, J.; Kienle, P.; von Knebel Doeberitz, M.; Kloor, M. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br. J. Cancer 2005, 92, 1746–1753. [Google Scholar] [CrossRef] [PubMed]
- Malesci, A.; Laghi, L.; Bianchi, P.; Delconte, G.; Randolph, A.; Torri, V.; Carnaghi, C.; Doci, R.; Rosati, R.; Montorsi, M.; et al. Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clin. Cancer Res. 2007, 13, 3831–3839. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Devaud, N.; Gallinger, S. Chemotherapy of MMR-deficient colorectal cancer. Fam. Cancer 2013, 12, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Woerner, S.M.; Benner, A.; Sutter, C.; Schiller, M.; Yuan, Y.P.; Keller, G.; Bork, P.; von Knebel Doeberitz, M.; Gebert, J. Pathogenesis of DNA repair-deficient cancers: A statistical meta-analysis of putative Real Common Target genes. Oncogene 2003, 22, 2226–2235. [Google Scholar] [CrossRef]
- Woerner, S.M.; Gebert, J.; Yuan, Y.P.; Sutter, C.; Ridder, R.; Bork, P.; von Knebel Doeberitz, M. Systematic identification of genes with coding microsatellites mutated in DNA mismatch repair-deficient cancer cells. Int. J. Cancer 2001, 93, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Jonchere, V.; Marisa, L.; Greene, M.; Virouleau, A.; Buhard, O.; Bertrand, R.; Svrcek, M.; Cervera, P.; Goloudina, A.; Guillerm, E.; et al. Identification of Positively and Negatively Selected Driver Gene Mutations Associated With Colorectal Cancer With Microsatellite Instability. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Trobridge, P.; Romero-Gallo, J.; Billheimer, D.; Myeroff, L.L.; Willson, J.K.; Markowitz, S.D.; Grady, W.M. Mutational inactivation of TGFBR2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor beta resistant cells. Genes Chromosomes Cancer 2008, 47, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chytil, A.; Washington, K.; Romero-Gallo, J.; Gorska, A.E.; Wirth, P.S.; Gautam, S.; Moses, H.L.; Grady, W.M. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004, 64, 4687–4692. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Myeroff, L.L.; Swinler, S.E.; Rajput, A.; Thiagalingam, S.; Lutterbaugh, J.D.; Neumann, A.; Brattain, M.G.; Chang, J.; Kinzler, K.W.; et al. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999, 59, 320–324. [Google Scholar] [PubMed]
- Meng, X.; Vander Ark, A.; Lee, P.; Hostetter, G.; Bhowmick, N.A.; Matrisian, L.M.; Williams, B.O.; Miranti, C.K.; Li, X. Myeloid-specific TGF-beta signaling in bone promotes basic-FGF and breast cancer bone metastasis. Oncogene 2016, 35, 2370–2378. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Placencio, V.; Iturregui, J.M.; Uwamariya, C.; Sharif-Afshar, A.R.; Koyama, T.; Hayward, S.W.; Bhowmick, N.A. Prostate tumor progression is mediated by a paracrine TGF-beta/Wnt3a signaling axis. Oncogene 2008, 27, 7118–7130. [Google Scholar] [CrossRef]
- Copland, J.A.; Luxon, B.A.; Ajani, L.; Maity, T.; Campagnaro, E.; Guo, H.; LeGrand, S.N.; Tamboli, P.; Wood, C.G. Genomic profiling identifies alterations in TGFbeta signaling through loss of TGFbeta receptor expression in human renal cell carcinogenesis and progression. Oncogene 2003, 22, 8053–8062. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.D.; Raposo, G. Exosomes and extracellular vesicles: The path forward. Essays Biochem. 2018, 62, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Anand, S.; Samuel, M.; Kumar, S.; Mathivanan, S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta Proteins Proteom 2019. [Google Scholar] [CrossRef]
- Wu, A.Y.; Ueda, K.; Lai, C.P. Proteomic Analysis of Extracellular Vesicles for Cancer Diagnostics. Proteomics 2019, 19, e1800162. [Google Scholar] [CrossRef]
- Fricke, F.; Lee, J.; Michalak, M.; Warnken, U.; Hausser, I.; Suarez-Carmona, M.; Halama, N.; Schnölzer, M.; Kopitz, J.; Gebert, J. TGFBR2-dependent alterations of exosomal cargo and functions in DNA mismatch repair-deficient HCT116 colorectal cancer cells. Cell Commun. Signal. 2017, 15, 14. [Google Scholar] [CrossRef]
- Lee, J.; Ballikaya, S.; Schonig, K.; Ball, C.R.; Glimm, H.; Kopitz, J.; Gebert, J. Transforming growth factor beta receptor 2 (TGFBR2) changes sialylation in the microsatellite unstable (MSI) Colorectal cancer cell line HCT116. PLoS ONE 2013, 8, e57074. [Google Scholar] [CrossRef]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, 516–519. [Google Scholar] [CrossRef]
- Bosque, A.; Dietz, L.; Gallego-Lleyda, A.; Sanclemente, M.; Iturralde, M.; Naval, J.; Alava, M.A.; Martinez-Lostao, L.; Thierse, H.J.; Anel, A. Comparative proteomics of exosomes secreted by tumoral Jurkat T cells and normal human T cell blasts unravels a potential tumorigenic role for valosin-containing protein. Oncotarget 2016, 7, 29287–29305. [Google Scholar] [CrossRef]
- Cai, X.; Liu, C.; Zhang, T.N.; Zhu, Y.W.; Dong, X.; Xue, P. Down-regulation of FN1 inhibits colorectal carcinogenesis by suppressing proliferation, migration, and invasion. J. Cell Biochem. 2018, 119, 4717–4728. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y.; Xu, X.; Cao, H.; Sahengbieke, S.; Sheng, H.; Huang, Q.; Lai, M. Transcriptional activation of FN1 and IL11 by HMGA2 promotes the malignant behavior of colorectal cancer. Carcinogenesis 2016, 37, 511–521. [Google Scholar] [CrossRef]
- Ding, J.; Li, D.; Wang, X.; Wang, C.; Wu, T. Fibronectin promotes invasiveness and focal adhesion kinase tyrosine phosphorylation of human colon cancer cell. Hepatogastroenterology 2008, 55, 2072–2076. [Google Scholar]
- Ignotz, R.A.; Massague, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar]
- Lobert, V.H.; Brech, A.; Pedersen, N.M.; Wesche, J.; Oppelt, A.; Malerod, L.; Stenmark, H. Ubiquitination of alpha 5 beta 1 integrin controls fibroblast migration through lysosomal degradation of fibronectin-integrin complexes. Dev. Cell 2010, 19, 148–159. [Google Scholar] [CrossRef]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef]
- Atay, S.; Gercel-Taylor, C.; Taylor, D.D. Human trophoblast-derived exosomal fibronectin induces pro-inflammatory IL-1beta production by macrophages. Am. J. Reprod. Immunol. 2011, 66, 259–269. [Google Scholar] [CrossRef]
- Moon, P.G.; Lee, J.E.; Cho, Y.E.; Lee, S.J.; Chae, Y.S.; Jung, J.H.; Kim, I.S.; Park, H.Y.; Baek, M.C. Fibronectin on circulating extracellular vesicles as a liquid biopsy to detect breast cancer. Oncotarget 2016, 7, 40189–40199. [Google Scholar] [CrossRef]
- Purushothaman, A.; Bandari, S.K.; Liu, J.; Mobley, J.A.; Brown, E.E.; Sanderson, R.D. Fibronectin on the Surface of Myeloma Cell-derived Exosomes Mediates Exosome-Cell Interactions. J. Biol. Chem. 2016, 291, 1652–1663. [Google Scholar] [CrossRef]
- Deng, Z.; Cheng, Z.; Xiang, X.; Yan, J.; Zhuang, X.; Liu, C.; Jiang, H.; Ju, S.; Zhang, L.; Grizzle, W.; et al. Tumor cell cross talk with tumor-associated leukocytes leads to induction of tumor exosomal fibronectin and promotes tumor progression. Am. J. Pathol. 2012, 180, 390–398. [Google Scholar] [CrossRef]
- Marimpietri, D.; Petretto, A.; Raffaghello, L.; Pezzolo, A.; Gagliani, C.; Tacchetti, C.; Mauri, P.; Melioli, G.; Pistoia, V. Proteome profiling of neuroblastoma-derived exosomes reveal the expression of proteins potentially involved in tumor progression. PLoS ONE 2013, 8, e75054. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, Y.; Xu, L.; Zhan, S.; Xiao, Y.; Gao, Y.; Wu, B.; Ge, W. Protein content and functional characteristics of serum-purified exosomes from patients with colorectal cancer revealed by quantitative proteomics. Int. J. Cancer 2017, 140, 900–913. [Google Scholar] [CrossRef]
- Kida, H.; Takano, Y.; Yamamoto, K.; Mori, M.; Yanaga, K.; Tanaka, J.; Kudo, S.E.; Mimori, K. A single nucleotide polymorphism in fibronectin 1 determines tumor shape in colorectal cancer. Oncol. Rep. 2014, 32, 548–552. [Google Scholar] [CrossRef]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef]
- Mosher, D. Fibronectin, 1st ed.; Elsevier: San Diego, CA, 1989; pp. 1–492. [Google Scholar]
- Ochieng, J.; Nangami, G.; Sakwe, A.; Rana, T.; Ingram, S.; Goodwin, J.S.; Moye, C.; Lammers, P.; Adunyah, S.E. Extracellular histones are the ligands for the uptake of exosomes and hydroxyapatite-nanoparticles by tumor cells via syndecan-4. FEBS Lett. 2018, 592, 3274–3285. [Google Scholar] [CrossRef]
- Burke, M.C.; Oei, M.S.; Edwards, N.J.; Ostrand-Rosenberg, S.; Fenselau, C. Ubiquitinated proteins in exosomes secreted by myeloid-derived suppressor cells. J. Proteome Res. 2014, 13, 5965–5972. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef]
- Ueberham, E.; Arendt, E.; Starke, M.; Bittner, R.; Gebhardt, R. Reduction and expansion of the glutamine synthetase expressing zone in livers from tetracycline controlled TGF-beta1 transgenic mice and multiple starved mice. J. Hepatol. 2004, 41, 75–81. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Tsang, M.; Weatherbee, J.; Molitor, T.W.; Anderson, W.R.; Peterson, P.K. Effects of transforming growth factor-beta on murine astrocyte glutamine synthetase activity. Implications in neuronal injury. J. Clin. Invest. 1992, 90, 1786–1793. [Google Scholar] [CrossRef]
- James, L.; Lunn, P.; Middleton, S.; Elia, M. Distribution of glutaminase and glutamine synthetase activities in the human gastrointestinal tract. Clin. Sci. 1998, 94, 313–319. [Google Scholar] [CrossRef]
- Duffy, M.J. Urokinase-type plasminogen activator: A potent marker of metastatic potential in human cancers. Biochem. Soc. Trans. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Bekes, E.M.; Deryugina, E.I.; Kupriyanova, T.A.; Zajac, E.; Botkjaer, K.A.; Andreasen, P.A.; Quigley, J.P. Activation of pro-uPA is critical for initial escape from the primary tumor and hematogenous dissemination of human carcinoma cells. Neoplasia 2011, 13, 806–821. [Google Scholar] [CrossRef]
- Quigley, J.P.; Gold, L.I.; Schwimmer, R.; Sullivan, L.M. Limited cleavage of cellular fibronectin by plasminogen activator purified from transformed cells. Proc. Natl. Acad. Sci. USA 1987, 84, 2776–2780. [Google Scholar] [CrossRef]
- Lyons, R.M.; Gentry, L.E.; Purchio, A.F.; Moses, H.L. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J. Cell Biol. 1990, 110, 1361–1367. [Google Scholar] [CrossRef]
- Planus, E.; Barlovatz-Meimon, G.; Rogers, R.A.; Bonavaud, S.; Ingber, D.E.; Wang, N. Binding of urokinase to plasminogen activator inhibitor type-1 mediates cell adhesion and spreading. J. Cell Sci. 1997, 110, 1091–1098. [Google Scholar]
- Stampfer, M.R.; Yaswen, P.; Alhadeff, M.; Hosoda, J. TGF beta induction of extracellular matrix associated proteins in normal and transformed human mammary epithelial cells in culture is independent of growth effects. J. Cell Physiol. 1993, 155, 210–221. [Google Scholar] [CrossRef]
- Lang, D.S.; Marwitz, S.; Heilenkotter, U.; Schumm, W.; Behrens, O.; Simon, R.; Reck, M.; Vollmer, E.; Goldmann, T. Transforming growth factor-beta signaling leads to uPA/PAI-1 activation and metastasis: A study on human breast cancer tissues. Pathol. Oncol. Res. 2014, 20, 727–732. [Google Scholar] [CrossRef]
- Santibanez, J.F. Transforming growth factor-Beta and urokinase-type plasminogen activator: Dangerous partners in tumorigenesis-implications in skin cancer. ISRN Dermatol. 2013, 2013, 597927. [Google Scholar] [CrossRef]
- Lau, L.F. Cell surface receptors for CCN proteins. J. Cell Commun. Signal. 2016, 10, 121–127. [Google Scholar] [CrossRef]
- Brunner, A.; Chinn, J.; Neubauer, M.; Purchio, A.F. Identification of a gene family regulated by transforming growth factor-beta. DNA Cell Biol. 1991, 10, 293–300. [Google Scholar] [CrossRef]
- Leask, A.; Parapuram, S.K.; Shi-Wen, X.; Abraham, D.J. Connective tissue growth factor (CTGF, CCN2) gene regulation: A potent clinical bio-marker of fibroproliferative disease? J. Cell Commun. Signal. 2009, 3, 89–94. [Google Scholar] [CrossRef]
- Mori, T.; Kawara, S.; Shinozaki, M.; Hayashi, N.; Kakinuma, T.; Igarashi, A.; Takigawa, M.; Nakanishi, T.; Takehara, K. Role and interaction of connective tissue growth factor with transforming growth factor-β in persistent fibrosis: A mouse fibrosis model. J. Cell Physiol. 1999, 181, 153–159. [Google Scholar] [CrossRef]
- Wahab, N.A.; Weston, B.S.; Mason, R.M. Modulation of the TGFbeta/Smad signaling pathway in mesangial cells by CTGF/CCN2. Exp. Cell Res. 2005, 307, 305–314. [Google Scholar] [CrossRef]
- Qi, W.; Chen, X.; Twigg, S.; Zhang, Y.; Gilbert, R.E.; Kelly, D.J.; Pollock, C.A. The differential regulation of Smad7 in kidney tubule cells by connective tissue growth factor and transforming growth factor-beta1. Nephrology 2007, 12, 267–274. [Google Scholar] [CrossRef]
- van Rooyen, B.A.; Schafer, G.; Leaner, V.D.; Parker, M.I. Tumour cells down-regulate CCN2 gene expression in co-cultured fibroblasts in a Smad7- and ERK-dependent manner. Cell Commun. Signal. 2013, 11, 75. [Google Scholar] [CrossRef]
- Lin, B.R.; Chang, C.C.; Che, T.F.; Chen, S.T.; Chen, R.J.; Yang, C.Y.; Jeng, Y.M.; Liang, J.T.; Lee, P.H.; Chang, K.J.; et al. Connective tissue growth factor inhibits metastasis and acts as an independent prognostic marker in colorectal cancer. Gastroenterology 2005, 128, 9–23. [Google Scholar] [CrossRef]
- Jung, S.H.; Kim, S.H.; Kim, J.H. Prognostic Impact of Microsatellite Instability in Colorectal Cancer Presenting With Mucinous, Signet-Ring, and Poorly Differentiated Cells. Ann. Coloproctol. 2016, 32, 58–65. [Google Scholar] [CrossRef]
- Charrier, A.; Chen, R.; Chen, L.; Kemper, S.; Hattori, T.; Takigawa, M.; Brigstock, D.R. Exosomes mediate intercellular transfer of pro-fibrogenic connective tissue growth factor (CCN2) between hepatic stellate cells, the principal fibrotic cells in the liver. Surgery 2014, 156, 548–555. [Google Scholar] [CrossRef]
- Liu, J.H.; Wei, S.; Burnette, P.K.; Gamero, A.M.; Hutton, M.; Djeu, J.Y. Functional association of TGF-beta receptor II with cyclin B. Oncogene 1999, 18, 269–275. [Google Scholar] [CrossRef]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Ji, H.; Mathivanan, S.; Scott, A.M.; Simpson, R.J. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods 2012, 56, 293–304. [Google Scholar] [CrossRef]
- Miller, D.S.J.; Bloxham, R.D.; Jiang, M.; Gori, I.; Saunders, R.E.; Das, D.; Chakravarty, P.; Howell, M.; Hill, C.S. The Dynamics of TGF-beta Signaling Are Dictated by Receptor Trafficking via the ESCRT Machinery. Cell Rep. 2018, 25, 1841–1855. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Lossner, C.; Warnken, U.; Pscherer, A.; Schnölzer, M. Preventing arginine-to-proline conversion in a cell-line-independent manner during cell cultivation under stable isotope labeling by amino acids in cell culture (SILAC) conditions. Anal. Biochem. 2011, 412, 123–125. [Google Scholar] [CrossRef]
- Letzel, T. Protein and Peptide Analysis by LC-MS: Experimental Strategies. Anal. Bioanal. Chem. 2012, 402, 2481–2482. [Google Scholar]
- Ngoka, L.C. Sample prep for proteomics of breast cancer: Proteomics and gene ontology reveal dramatic differences in protein solubilization preferences of radioimmunoprecipitation assay and urea lysis buffers. Proteome Sci. 2008, 6, 30. [Google Scholar] [CrossRef]
- Wessel, D.; Flugge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, 1100–1106. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2018, 47, 442–450. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. A Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, 607–613. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Protein Ratio 1 (dT/pT) | T Test p-Value (adj.) | Peptides (unique) | Coverage (%) | Identification Score |
|---|---|---|---|---|---|---|
| FN1 | Fibronectin | 4.73 | 0.006 | 31 (31) | 19.7 | 109.14 |
| HIST1H4A | Histone H4 | 2.48 | 7.79 × 10-5 | 8 (8) | 53.4 | 54.26 |
| HIST1H2BL | Histone H2B type 1-L | 2.13 | 2.33 × 10-4 | 6 (2) | 41.3 | 72.66 |
| HIST2H2BE | Histone H2B type 2-E | 2.03 | 3.25 × 10-4 | 6 (2) | 41.3 | 4.33 |
| H2AFV | Histone H2A.V | 1.98 | 0.006 | 5 (3) | 53.9 | 44.88 |
| GLUL | Glutamine synthetase | 1.77 | 0.010 | 6 (6) | 20.1 | 24.52 |
| C18orf8 | Uncharacterized protein C18orf8 | 1.74 | 0.018 | 2 (2) | 4.4 | 2.61 |
| ANXA6 | Annexin A6 | 1.68 | 0.002 | 12 (12) | 22.3 | 23.43 |
| FBLN1 | Fibulin-1 | 1.64 | 0.018 | 26 (13) | 51.9 | 323.21 |
| SUB1 | Activated RNA polymerase II transcriptional coactivator p15 | 1.64 | 0.001 | 6 (6) | 33.9 | 21.71 |
| RAP1B | Ras-related protein Rap-1b | 1.62 | 0.008 | 5 (5) | 35.9 | 17.40 |
| LTBP4 | Latent-transforming growth factor beta-binding protein 4 | 1.61 | 3.27 × 10-5 | 32 (32) | 30.8 | 302.21 |
| MDK | Midkine | 1.58 | 7.74 × 10-6 | 5 (5) | 32.9 | 138.01 |
| SSBP1 | Single-stranded DNA-binding protein, mitochondrial | 1.57 | 0.004 | 4 (4) | 29.1 | 8.99 |
| COL6A1 | Collagen alpha-1(VI) chain | 1.57 | 4.37 × 10-6 | 20 (20) | 26.5 | 323.31 |
| THBS1 | Thrombospondin-1 | 1.57 | 6.54 × 10-5 | 50 (14) | 56.8 | 323.31 |
| CHRD | Chordin | 1.57 | 0.002 | 7 (7) | 12.5 | 14.76 |
| SEPT6 | Septin-6 | 1.56 | 0.007 | 5 (2) | 20.0 | 5.35 |
| TNFRSF6B | Tumor necrosis factor receptor superfamily member 6B | 1.55 | 0.001 | 3 (3) | 16.0 | 17.80 |
| LGI3 | Leucine-rich repeat LGI family member 3 | 1.55 | 0.002 | 10 (10) | 25.0 | 39.49 |
| PURA | Transcriptional activator protein Pur-alpha | 1.54 | 0.003 | 3 (3) | 17.7 | 6.24 |
| COL6A2 | Collagen alpha-2(VI) chain | 1.54 | 0.001 | 12 (12) | 18.4 | 58.43 |
| GDF15 | Growth/differentiation factor 15 | 1.53 | 0.001 | 20 (20) | 65.3 | 127.44 |
| HMGN2 | Non-histone chromosomal protein HMG-17 | 1.53 | 3.42 × 10-4 | 3 (3) | 16.7 | 6.75 |
| H2AFY | Core histone macro-H2A.1 | 1.51 | 0.006 | 8 (8) | 34.4 | 26.84 |
| MATN2 | Matrilin-2 | 1.50 | 0.001 | 23 (23) | 31.2 | 190.93 |
| Gene Name | Protein Name | Protein Ratio 1 (dT/pT) | T Test p-Value (adj.) | Peptides (unique) | Coverage (%) | Identification Score |
|---|---|---|---|---|---|---|
| PLAU | Urokinase-type plasminogen activator | 0.46 | 1.73 × 10-4 | 16 (16) | 50.6 | 323.31 |
| CTGF | Connective tissue growth factor | 0.51 | 2.95 × 10-4 | 15 (15) | 55.3 | 66.74 |
| PSMB1 | Proteasome subunit beta type-1 | 0.55 | 0.007 | 5 (5) | 29 | 12.98 |
| PSMB5 | Proteasome subunit beta type-5 | 0.55 | 0.001 | 5 (5) | 25.5 | 20.07 |
| PSMA2 | Proteasome subunit alpha type-2 | 0.58 | 0.010 | 4 (4) | 21.4 | 20.44 |
| MCM7 | DNA replication licensing factor MCM7 | 0.59 | 1.51 × 10-5 | 12 (12) | 22.4 | 34.56 |
| SAFB | Scaffold attachment factor B1 | 0.60 | 0.023 | 6 (6) | 14.6 | 46.12 |
| PSMB2 | Proteasome subunit beta type-2 | 0.61 | 0.002 | 3 (3) | 17.9 | 27.04 |
| PSMB4 | Proteasome subunit beta type-4 | 0.61 | 0.007 | 3 (3) | 14.8 | 4.97 |
| CCAR2 | Cell cycle and apoptosis regulator protein 2 | 0.62 | 0.002 | 8 (8) | 13.2 | 22.57 |
| TARDBP | TAR DNA-binding protein 43 | 0.63 | 0.006 | 2 (2) | 7.2 | 5.87 |
| APRT | Adenine phosphoribosyltransferase | 0.64 | 0.004 | 5 (5) | 37.2 | 43.68 |
| PSMB3 | Proteasome subunit beta type-3 | 0.64 | 2.35 × 10-4 | 4 (4) | 30.7 | 14.65 |
| ALDH1A3 | Aldehyde dehydrogenase family 1 member A3 | 0.65 | 0.001 | 12 (12) | 28.5 | 57.51 |
| PSMA1 | Proteasome subunit alpha type-1 | 0.65 | 0.001 | 8 (8) | 36.9 | 14.21 |
| UHRF1 | E3 ubiquitin-protein ligase UHRF1 | 0.65 | 0.006 | 8 (8) | 13.7 | 9.60 |
| CDK1 | Cyclin-dependent kinase 1 | 0.65 | 0.006 | 9 (8) | 39.4 | 22.58 |
| PSMA3 | Proteasome subunit alpha type-3 | 0.65 | 0.002 | 8 (8) | 34.1 | 23.82 |
| MCM6 | DNA replication licensing factor MCM6 | 0.66 | 0.002 | 16 (16) | 26.4 | 43.74 |
| HSPD1 | 60 kDa heat shock protein, mitochondrial | 0.66 | 0.001 | 14 (14) | 34.0 | 108.37 |
| CTBP2 | C-terminal-binding protein 2 | 0.66 | 0.012 | 5 (4) | 18.2 | 16.52 |
| PSMA5 | Proteasome subunit alpha type-5 | 0.66 | 0.001 | 7 (7) | 40.2 | 34.82 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fricke, F.; Michalak, M.; Warnken, U.; Hausser, I.; Schnölzer, M.; Kopitz, J.; Gebert, J. SILAC-Based Quantification of TGFBR2-Regulated Protein Expression in Extracellular Vesicles of Microsatellite Unstable Colorectal Cancers. Int. J. Mol. Sci. 2019, 20, 4162. https://doi.org/10.3390/ijms20174162
Fricke F, Michalak M, Warnken U, Hausser I, Schnölzer M, Kopitz J, Gebert J. SILAC-Based Quantification of TGFBR2-Regulated Protein Expression in Extracellular Vesicles of Microsatellite Unstable Colorectal Cancers. International Journal of Molecular Sciences. 2019; 20(17):4162. https://doi.org/10.3390/ijms20174162
Chicago/Turabian StyleFricke, Fabia, Malwina Michalak, Uwe Warnken, Ingrid Hausser, Martina Schnölzer, Jürgen Kopitz, and Johannes Gebert. 2019. "SILAC-Based Quantification of TGFBR2-Regulated Protein Expression in Extracellular Vesicles of Microsatellite Unstable Colorectal Cancers" International Journal of Molecular Sciences 20, no. 17: 4162. https://doi.org/10.3390/ijms20174162
APA StyleFricke, F., Michalak, M., Warnken, U., Hausser, I., Schnölzer, M., Kopitz, J., & Gebert, J. (2019). SILAC-Based Quantification of TGFBR2-Regulated Protein Expression in Extracellular Vesicles of Microsatellite Unstable Colorectal Cancers. International Journal of Molecular Sciences, 20(17), 4162. https://doi.org/10.3390/ijms20174162