The Expression Profile and Prognostic Significance of Metallothionein Genes in Colorectal Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. mRNA Expression of Six Metallothionein (MT) Genes in Various Human Cancers
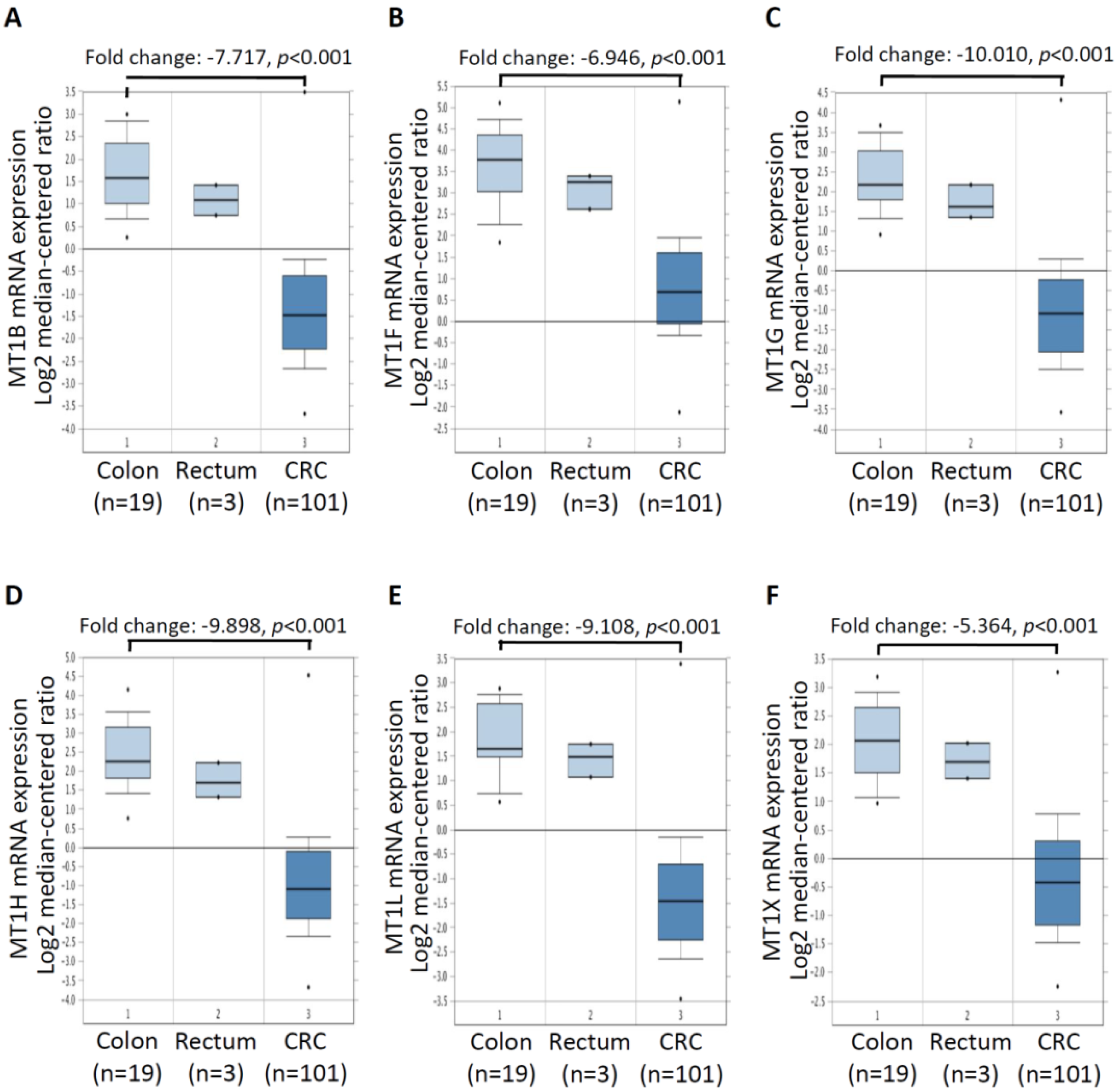
2.2. mRNA Expression of Six MT Genes in Pairs of Tumor (T) and Adjacent Non-Tumor (N) CRC Tissues
2.3. mRNA Expressionof Six MT Genes in Colorectal Cancer Tissues
2.4. Prognostic Relevance of the Six Investigated MT Genes in Colorectal Cancer Tissues
2.5. A Combination Four-Gene Signature Predicts Survival in Colorectal Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Tissue Samples and Ethics Statement
4.2. Quantitative Reverse-Transcription (RT)-(q)PCR
4.3. CellMiner Data Mining and Analysis
4.4. Oncomine Database Analysis
4.5. SurvExpress Database Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Linnekamp, J.F.; Wang, X.; Medema, J.P.; Vermeulen, L. Colorectal cancer heterogeneity and targeted therapy: A case for molecular disease subtypes. Cancer Res. 2015, 75, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig. Dis. Sci. 2015, 60, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Zarkavelis, G.; Boussios, S.; Papadaki, A.; Katsanos, K.H.; Christodoulou, D.K.; Pentheroudakis, G. Current and future biomarkers in colorectal cancer. Ann. Gastroenterol. 2017, 30, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xuan, Y.; Gao, B.; Sun, X.; Miao, S.; Lu, T.; Wang, Y.; Jiao, W. Identification of an eight-gene prognostic signature for lung adenocarcinoma. Cancer Manag. Res. 2018, 10, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Aran, V.; Victorino, A.P.; Thuler, L.C.; Ferreira, C.G. Colorectal Cancer: Epidemiology, Disease Mechanisms and Interventions to Reduce Onset and Mortality. Clin. Colorectal Cancer 2016, 15, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Grzadkowski, M.R.; Sendorek, D.H.; P’ng, C.; Huang, V.; Boutros, P.C. A comparative study of survival models for breast cancer prognostication revisited: The benefits of multi-gene models. BMC Bioinform. 2018, 19, 400. [Google Scholar] [CrossRef]
- Su, Y.H.; Tang, W.C.; Cheng, Y.W.; Sia, P.; Huang, C.C.; Lee, Y.C.; Jiang, H.Y.; Wu, M.H.; Lai, I.L.; Lee, J.W.; et al. Targeting of multiple oncogenic signaling pathways by Hsp90 inhibitor alone or in combination with berberine for treatment of colorectal cancer. Biochim. Biophys. Acta 2015, 1853 Pt A, 2261–2272. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S. Positive and negative regulators of the metallothionein gene (review). Mol. Med. Rep. 2015, 12, 795–799. [Google Scholar] [CrossRef]
- Vasak, M. Advances in metallothionein structure and functions. J. Trace Elem. Med. Biol. 2005, 19, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Krizkova, S.; Kepinska, M.; Emri, G.; Eckschlager, T.; Stiborova, M.; Pokorna, P.; Heger, Z.; Adam, V. An insight into the complex roles of metallothioneins in malignant diseases with emphasis on (sub)isoforms/isoforms and epigenetics phenomena. Pharmacol. Ther. 2018, 183, 90–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumulec, J.; Raudenska, M.; Adam, V.; Kizek, R.; Masarik, M. Metallothionein—Immunohistochemical cancer biomarker: A meta-analysis. PLoS ONE 2014, 9, e85346. [Google Scholar] [CrossRef] [PubMed]
- Thirumoorthy, N.; Shyam Sunder, A.; Manisenthil Kumar, K.; Senthil Kumar, M.; Ganesh, G.; Chatterjee, M. A review of metallothionein isoforms and their role in pathophysiology. World J. Surg. Oncol. 2011, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.G.; Trevino, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef]
- Zhao, K.; Li, Z.; Tian, H. Twenty-gene-based prognostic model predicts lung adenocarcinoma survival. Onco Targets Ther. 2018, 11, 3415–3424. [Google Scholar] [CrossRef]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9–14. [Google Scholar]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. Arch. Pathol. Lab. Med. 2017, 141, 625–657. [Google Scholar] [CrossRef]
- Kocarnik, J.M.; Shiovitz, S.; Phipps, A.I. Molecular phenotypes of colorectal cancer and potential clinical applications. Gastroenterol. Rep. (Oxf.) 2015, 3, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Pamplona, R.; Berenguer, A.; Cordero, D.; Riccadonna, S.; Sole, X.; Crous-Bou, M.; Guino, E.; Sanjuan, X.; Biondo, S.; Soriano, A.; et al. Clinical value of prognosis gene expression signatures in colorectal cancer: A systematic review. PLoS ONE 2012, 7, e48877. [Google Scholar] [CrossRef]
- Hong, S.N. Genetic and epigenetic alterations of colorectal cancer. Intest. Res. 2018, 16, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Iwaya, T.; Yokobori, T.; Nishida, N.; Kogo, R.; Sudo, T.; Tanaka, F.; Shibata, K.; Sawada, G.; Takahashi, Y.; Ishibashi, M.; et al. Downregulation of miR-144 is associated with colorectal cancer progression via activation of mTOR signaling pathway. Carcinogenesis 2012, 33, 2391–2397. [Google Scholar] [CrossRef] [Green Version]
- Aibar, S.; Fontanillo, C.; Droste, C.; Roson-Burgo, B.; Campos-Laborie, F.J.; Hernandez-Rivas, J.M.; De Las Rivas, J. Analyse multiple disease subtypes and build associated gene networks using genome-wide expression profiles. BMC Genom. 2015, 16 (Suppl. 5), S3. [Google Scholar] [CrossRef]
- Aibar, S.; Abaigar, M.; Campos-Laborie, F.J.; Sanchez-Santos, J.M.; Hernandez-Rivas, J.M.; De Las Rivas, J. Identification of expression patterns in the progression of disease stages by integration of transcriptomic data. BMC Bioinform. 2016, 17 (Suppl. 15), 432. [Google Scholar] [CrossRef]
- Ambler, G.; Seaman, S.; Omar, R.Z. An evaluation of penalised survival methods for developing prognostic models with rare events. Stat. Med. 2012, 31, 1150–1161. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, K.-C.; Huang, T.-C.; Cheng, C.-H.; Cheng, Y.-W.; Lin, D.-Y.; Fan, J.-J.; Lee, K.-H. The Expression Profile and Prognostic Significance of Metallothionein Genes in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 3849. https://doi.org/10.3390/ijms20163849
Hung K-C, Huang T-C, Cheng C-H, Cheng Y-W, Lin D-Y, Fan J-J, Lee K-H. The Expression Profile and Prognostic Significance of Metallothionein Genes in Colorectal Cancer. International Journal of Molecular Sciences. 2019; 20(16):3849. https://doi.org/10.3390/ijms20163849
Chicago/Turabian StyleHung, Kuo-Chen, Tsui-Chin Huang, Chia-Hsiung Cheng, Ya-Wen Cheng, Ding-Yen Lin, Jhen-Jia Fan, and Kuen-Haur Lee. 2019. "The Expression Profile and Prognostic Significance of Metallothionein Genes in Colorectal Cancer" International Journal of Molecular Sciences 20, no. 16: 3849. https://doi.org/10.3390/ijms20163849