Association Analysis of Drought Tolerance and Associated Traits in Upland Cotton at the Seedling Stage

Abstract
1. Introduction
2. Results
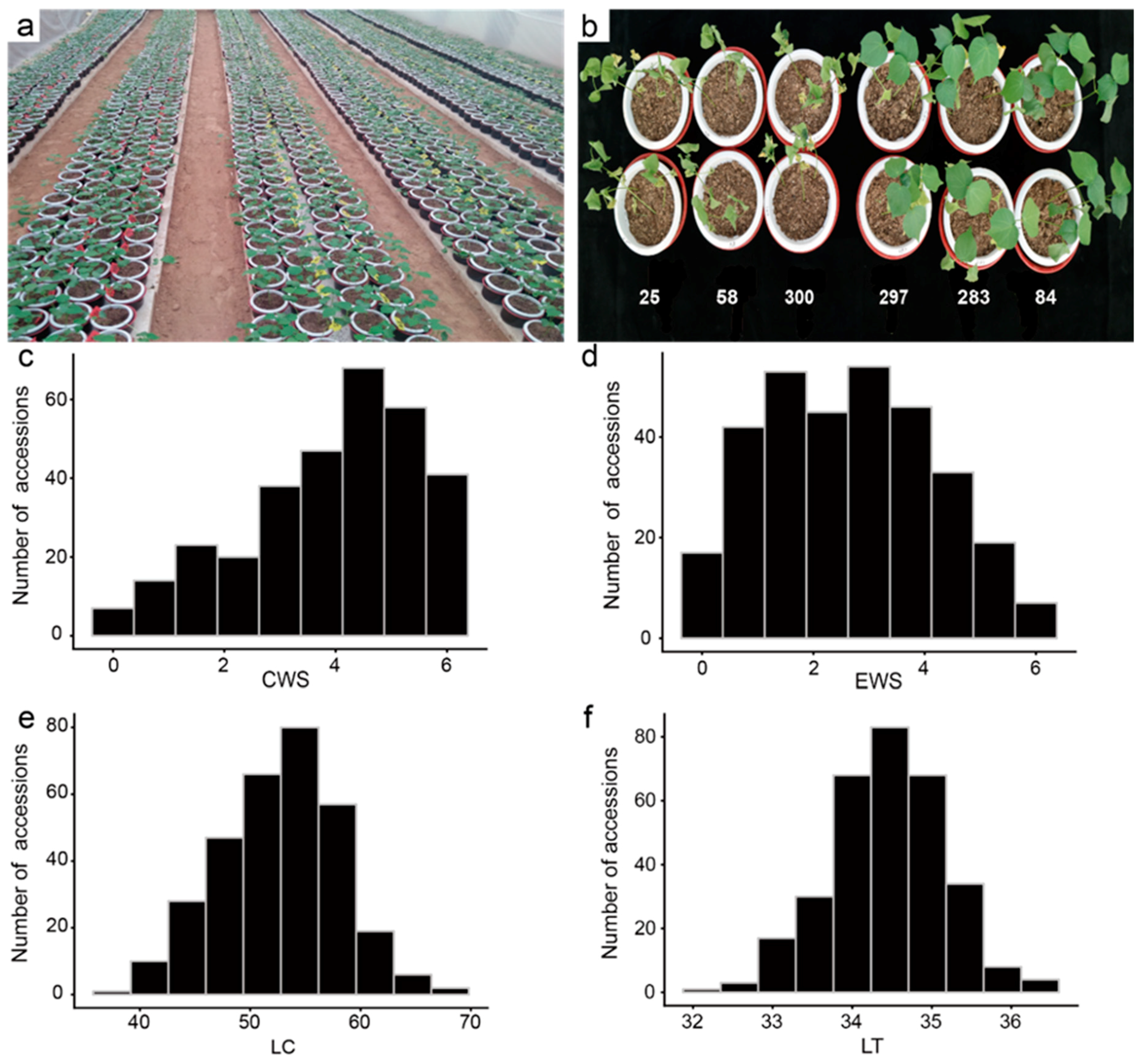
2.1. Analysis of Phenotypic Drought Resistance Traits
2.2. Phylogenetic Analyses and Linkage Disequilibrium
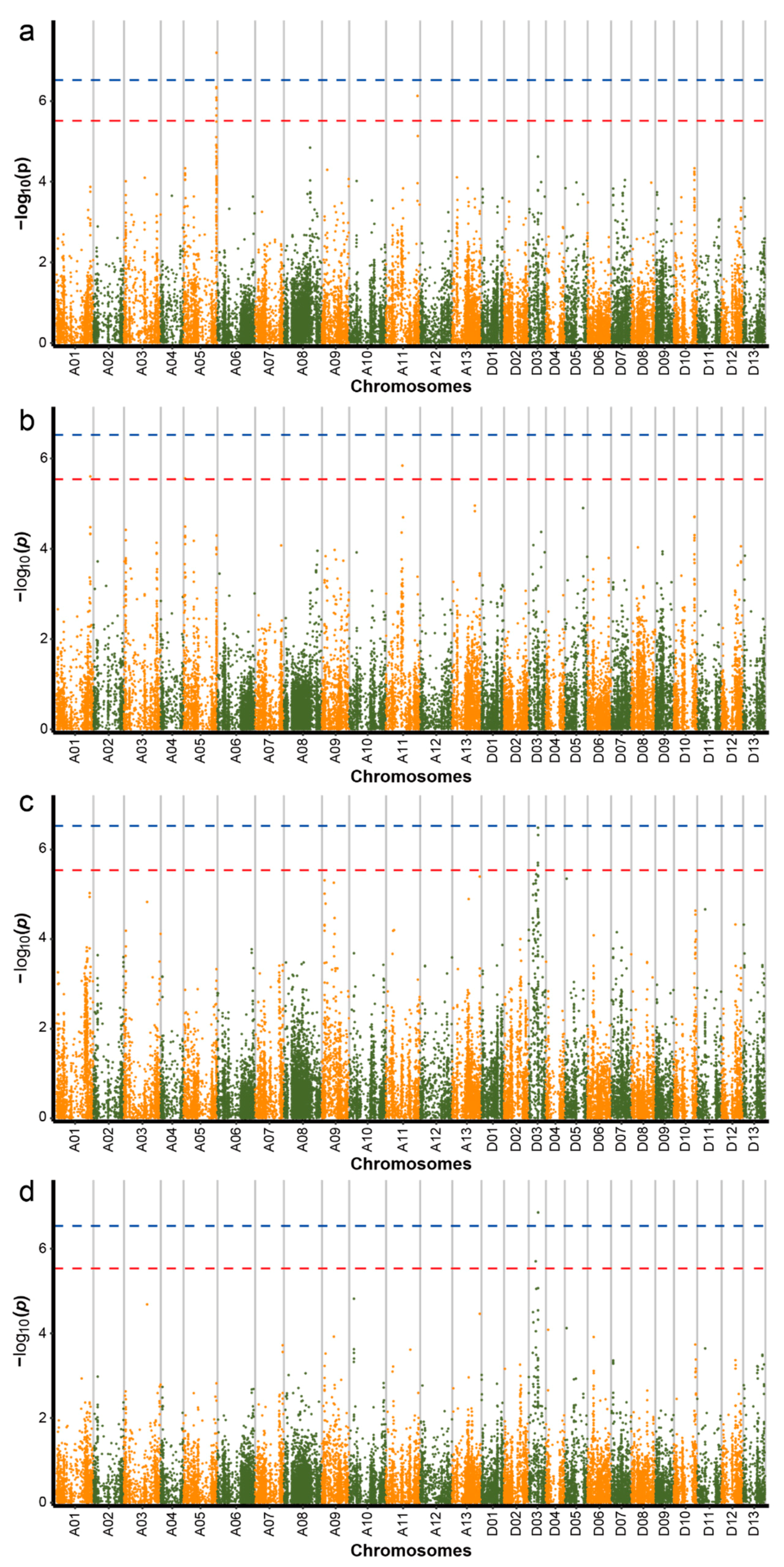
2.3. Genome-Wide Association Study
2.4. Prediction of Candidate Genes and Identification of Favorable Haplotypes
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Locations and Phenotyping
4.3. Genotyping and Phylogenetic Analysis
4.4. Population Structure and Linkage Disequilibrium (LD) Decay Analysis
4.5. Genome-Wide Association Study Analysis
4.6. Haplotype Analysis
4.7. RNA-seq Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association study |
| GLM | General linear model |
| FASTLMM | Factored spectrally transformed linear mixed model |
| EWS | Euphylla wilting score |
| CWS | Cotyledon wilting score |
| LT | Leaf temperature |
| LC | Leaf chlorophyll content |
| YZR | the Yangtze River basin region |
| YR | the Yellow River basin region |
| NEM | northern specific early maturation region |
| NW | northwestern inland region |
| FR | Foreign region |
| PCA | Principal component analysis |
| LD | Linkage disequilibrium |
| FH | Favorable haplotype |
| UFH | Unfavorable haplotype |
| HH | Heterozygous haplotype |
| MAF | Minor allele frequency |
| RWC | Relative water content |
| FDR | False discovery rate |
| DEG | Differential expression analysis |
References
- Fang, Y.; Xiong, L. General mechanisms of drought response and their application in drought resistance improvement in plants. Cell Mol. Life Sci. 2015, 72, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Farooq, J.; Sakhawat, G.; Mahmood, A.; Sadiq, M.A.; Yaseen, M. Genotypic variability for root/shoot parameters under water stress in some advanced lines of cotton (Gossypium hirsutum L.). Genet. Mol. Res. 2013, 12, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Rizza, F.; Badeck, F.W.; Cattivelli, L.; Lidestri, O.; Di Fonzo, N.; Stanca, A.M. Use of a water stress index to identify barley genotypes adapted to rainfed and irrigated conditions. Crop Sci. 2004, 44, 2127–2137. [Google Scholar] [CrossRef]
- Saranga, Y.; Menz, M.; Jiang, C.X.; Wright, R.J.; Yakir, D.; Paterson, A.H. Genomic dissection of genotype x environment interactions conferring adaptation of cotton to arid conditions. Genome Res. 2001, 11, 1988–1995. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.Z.; Guo, W.Z.; Chen, X.Y.; Stelly, D.V.; Rabinowica, P.D.; Town, C.D.; et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef]
- Fang, D.D.; Jenkins, J.N.; Deng, D.D.; McCarty, J.C.; Li, P.; Wu, J. Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in upland cotton (Gossypium hirsutum L.). BMC Genom. 2014, 15, 397. [Google Scholar] [CrossRef]
- Wang, R.H.; Cheng, H.L.; Song, X.X. Brief. History of Chinese Cotton; China Agricultural Science and Technology Press: Beijing, China, 2017; p. 15. [Google Scholar]
- Parida, A.K.; Dagaonkar, V.S.; Phalak, M.S.; Umalkar, G.V.; Aurangabadkar, L.P. Alterations in photosynthetic pigments, protein and osmotic components in cotton genotypes subjected to short-term drought stress followed by recovery. Plant. Biotechnol. Rep. 2007, 1, 37–48. [Google Scholar] [CrossRef]
- DAWN News. Cotton Production Plummets 34pc. 9 February 2016. Available online: https://www.dawn.com/news/1240448 (accessed on 19 February 2019).
- Mao, S.C. Cotton Farming in China; Shanghai Scientific and Technical Press: Shanghai, China, 2013; pp. 884–898. [Google Scholar]
- Fu, X.Q.; Peng, J. Prospect and retrospection of national cotton regional test of China in last decade. Cotton Sci. 2017, 29, 113–117. [Google Scholar]
- Appiah, M.K.; Feike, T.; Wiredu, A.; Mamitimin, Y. Cotton production, land use change and resource competition in the Aksu-Tarim River Basin, Xinjiang, China. Q J. Int. Agric. 2014, 53, 243–261. [Google Scholar]
- Luo, X.X.; Yang, J.Q. Researches on the questions and countermeasures of sustainable utilization of water resources in the northwest area of China. Areal. Res. Dev. 2003, 22, 73–76. [Google Scholar]
- Shen, Y.; Chen, Y. Global perspective on hydrology, water balance, and water resources management in arid basins. Hydrol. Process. 2010, 24, 129–135. [Google Scholar] [CrossRef]
- Nakamura, A.; Osonoi, T.; Terauchi, Y. Relationship between urinary sodium excretion and pioglitazone-induced edema. J. Diabetes Investig. 2010, 1, 208–211. [Google Scholar] [CrossRef]
- Huang, X.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant. Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Pantaliao, G.F.; Narciso, M.; Guimaraes, C.; Castro, A.; Colombari, J.M.; Breseghello, F.; Rodrigues, L.; Vianello, R.P.; Borba, T.O.; Brondani, C. Genome wide association study (GWAS) for grain yield in rice cultivated under water deficit. Genetica 2016, 144, 651–664. [Google Scholar] [CrossRef]
- Rebolledo, M.C.; Dingkuhn, M.; Courtois, B.; Gibon, Y.; Clement-Vidal, A.; Cruz, D.F.; Duitama, J.; Lorieux, M.; Luquet, D. Phenotypic and genetic dissection of component traits for early vigour in rice using plant growth modelling, sugar content analyses and association mapping. J. Exp. Bot. 2015, 66, 5555–5566. [Google Scholar] [CrossRef] [PubMed]
- Clauw, P.; Coppens, F.; Korte, A.; Herman, D.; Slabbinck, B.; Dhondt, S.; Daele, T.V.; Milde, L.D.; Vermeersch, M.; Maleux, K.; et al. Leaf growth response to mild drought: natural variation in arabidopsis sheds light on trait architecture. Plant Cell 2016, 28, 2417–2434. [Google Scholar] [CrossRef]
- Kumar, V.; Singh, A.; Mithra, S.V.; Krishnamurthy, S.L.; Parida, S.K.; Jain, S.; Tiwari, K.K.; Kumar, P.; Rao, A.R.; Sharma, S.K.; et al. Genome-wide association mapping of salinity tolerance in rice (Oryza sativa). DNA Res. 2015, 22, 133–145. [Google Scholar]
- Wang, X.L.; Wang, H.W.; Liu, S.X.; Ferjani, A.; Li, J.S.; Yan, J.B.; Yang, X.H.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef]
- Edae, E.A.; Byrne, P.F.; Haley, S.D.; Lopes, M.S.; Reynolds, M.P. Genome-wide association mapping of yield and yield components of spring wheat under contrasting moisture regimes. Appl. Genet. 2014, 127, 791–807. [Google Scholar] [CrossRef]
- Wang, M.J.; Tu, L.L.; Lin, M.; Lin, Z.G.; Wang, P.C.; Yang, Q.Y.; Ye, Z.X.; Shen, C.; Li, J.Y.; Zhang, L.; et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef]
- Ma, Z.Y.; He, S.P.; Wang, X.F.; Sun, J.L.; Zhang, Y.; Zhang, G.Y.; Wu, L.Q.; Li, Z.K.; Liu, Z.H.; Sun, G.F.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.H.; Chen, J.D.; Liu, B.L.; Zhang, Z.Y.; Guan, X.Y.; Chen, S.Q.; Zhou, B.L.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Du, X.M.; Huang, G.; He, S.P.; Yang, Z.E.; Sun, G.F.; Ma, X.F.; Li, N.; Zhang, X.Y.; Sun, J.L.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Li, F.G.; Fan, G.Y.; Lu, C.R.; Xiao, G.H.; Zou, C.S.; Kohel, R.J.; Ma, Z.Y.; Shang, H.H.; Ma, X.F.; Wu, J.Y.; et al. Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Z.; Hu, Y.; Jiang, W.K.; Fang, L.; Guan, X.Y.; Chen, J.D.; Zhang, J.B.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- Lippert, C.; Listgarten, J.; Liu, Y.; Kadie, C.M.; Davidson, R.I.; Heckerman, D. FaST linear mixed models for genome-wide association studies. Nat. Methods 2011, 8, 833. [Google Scholar] [CrossRef]
- Hou, S.; Zhu, G.Z.; Li, Y.; Li, W.X.; Fu, J.; Niu, E.L.; Li, L.C.; Zhang, D.Y.; Guo, W.Z. Genome-wide association studies reveal genetic variation and candidate genes of drought stress related traits in cotton (Gossypium hirsutum L.). Front. Plant Sci. 2018, 9, 1276. [Google Scholar] [CrossRef]
- Islam, M.S.; Wang, M.H. Expression of dehydration responsive element-binding protein-3 (DREB3) under different abiotic stresses in tomato. BMB Rep. 2009, 42, 611–616. [Google Scholar] [CrossRef]
- Xu, Z.S.; Ni, Z.Y.; Li, Z.Y.; Li, L.C.; Chen, M.; Gao, D.Y.; Yu, X.D.; Liu, P.; Ma, Y.Z. Isolation and functional characterization of HvDREB1-a gene encoding a dehydration-responsive element binding protein in Hordeum vulgare. J. Plant Res. 2009, 122, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Sakiroglu, M.; Krom, N.; Stanton-Geddes, J.; Wang, M.Y.; Lee, Y.C.; Young, N.D.; Udvardi, M. Genome-wide association of drought-related and biomass traits with HapMap SNPs in Medicago truncatula. Plant Cell Env. 2015, 38, 1997–2011. [Google Scholar] [CrossRef] [PubMed]
- Biscarini, F.; Marini, S.; Stevanato, P.; Broccanello, C.; Bellazzi, R.; Nazzicari, N. Developing a parsimonius predictor for binary traits in sugar beet (Beta vulgaris). Mol. Breed. 2015, 35, 10. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys Acta 2012, 1819, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.Z.; Chen, Z.Z.; Liu, Y.; Zhang, H.R.; Zhang, M.; Liu, Q.; Hong, X.H.; Zhu, J.K.; Gong, Z.Z. ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant. J. 2010, 63, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Besseau, S.; Toronen, P.; Sipari, N.; Kollist, H.; Holm, L.; Palva, E.T. Defense-related transcription factors WRKY70 and WRKY54 modulate osmotic stress tolerance by regulating stomatal aperture in Arabidopsis. New Phytol. 2013, 200, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Nolan, T.M.; Ye, H.; Zhang, M.C.; Tong, H.N.; Xin, P.Y.; Chu, J.F.; Chu, C.C.; Li, Z.H.; Yin, Y.H. Arabidopsis WRKY46, WRKY54, and WRKY70 transcription factors are involved in brassinosteroid-regulated plant growth and drought responses. Plant Cell 2017, 29, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Q.Q.; Zhou, L.; Ren, F.; Li, D.D.; Li, X.B. Arabidopsis CBL-interacting protein kinase (CIPK6) is involved in plant response to salt/osmotic stress and ABA. Mol. Biol. Rep. 2013, 40, 4759–4767. [Google Scholar] [CrossRef]
- Chen, L.; Ren, F.; Zhou, L.; Wang, Q.Q.; Zhong, H.; Li, X.B. The Brassica napus calcineurin B-Like 1/CBL-interacting protein kinase 6 (CBL1/CIPK6) component is involved in the plant response to abiotic stress and ABA signaling. J. Exp. Bot. 2012, 63, 6211–6222. [Google Scholar] [CrossRef]
- He, L.R.; Yang, X.Y.; Wang, L.C.; Zhu, L.F.; Zhou, T.; Deng, J.W.; Zhang, X.L. Molecular cloning and functional characterization of a novel cotton CBL-interacting protein kinase gene (GhCIPK6) reveals its involvement in multiple abiotic stress tolerance in transgenic plants. Biochem. Biophys. Res. Commun. 2013, 435, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.; Wang, H.; Yu, D. The sucrose non-fermenting-1-related protein kinases SAPK1 and SAPK2 function collaboratively as positive regulators of salt stress tolerance in rice. BMC Plant Biol. 2018, 18, 203. [Google Scholar] [CrossRef] [PubMed]
- Furihata, T.; Maruyama, K.; Fujita, Y.; Umezawa, T.; Yoshida, R.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Abscisic acid-dependent multisite phosphorylation regulates the activity of a transcription activator AREB1. Proc. Natl. Acad. Sci. USA 2006, 103, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Sugiyama, N.; Mizoguchi, M.; Hayashi, S.; Myouga, F.; Yamaguchi-Shinozaki, K.; Ishihama, Y.; Hirayama, T.; Shinozaki, K. Type 2C protein phosphatases directly regulate abscisic acid-activated protein kinases in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 17588–17593. [Google Scholar] [CrossRef]
- Fujii, H.; Chinnusamy, V.; Rodrigues, A.; Rubio, S.; Antoni, R.; Park, S.Y.; Cutler, S.R.; Sheen, J.; Rodriguez, P.L.; Zhu, J.-K. In vitro reconstitution of an abscisic acid signalling pathway. Nature 2009, 462, 660. [Google Scholar] [CrossRef]
- Jiang, M.; Niu, C.; Cao, J.; Ni, D.A.; Chu, Z. In silico-prediction of protein-protein interactions network about MAPKs and PP2Cs reveals a novel docking site variants in Brachypodium distachyon. Sci. Rep. 2018, 8, 15083. [Google Scholar] [CrossRef]
- Li, C.Q.; Wang, C.B.; Dong, N.; Wang, X.Y.; Zhao, H.H.; Converse, R.; Xia, Z.; Wang, R.; Wang, Q.L. QTL detection for node of first fruiting branch and its height in upland cotton (Gossypium hirsutum L.). Euphytica 2012, 188, 441–451. [Google Scholar] [CrossRef]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef]
- Ren, C.; Zhu, X.; Zhang, P.; Gong, Q. Arabidopsis COP1-interacting protein 1 is a positive regulator of ABA response. Biochem. Biophys. Res. Commun. 2016, 477, 847–853. [Google Scholar] [CrossRef]
- Levi, A.; Paterson, A.H.; Barak, V.; Yakir, D.; Wang, B.; Chee, P.W.; Saranga, Y. Field evaluation of cotton near-isogenic lines introgressed with QTLs for productivity and drought related traits. Mol. Breed. 2009, 23, 179–195. [Google Scholar] [CrossRef]
- Chen, X.B.; Wang, J.; Zhu, M.; Jia, H.H.; Liu, D.D.; Hao, L.L.; Guo, X.Q. A cotton Raf-like MAP3K gene, GhMAP3K40, mediates reduced tolerance to biotic and abiotic stress in Nicotiana benthamiana by negatively regulating growth and development. Plant Sci. 2015, 240, 10–24. [Google Scholar] [CrossRef]
- Chen, T.; Li, W.; Hu, X.; Guo, J.; Liu, A.; Zhang, B. A cotton MYB transcription factor, GbMYB5, is positively involved in plant adaptive response to drought stress. Plant Cell Physiol. 2015, 56, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.M.; Ecker, J.R. Moving forward in reverse: Genetic technologies to enable genome-wide phenomic screens in Arabidopsis. Nat. Rev. Genet. 2006, 7, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Su, J.J.; Pang, C.Y.; Wei, H.L.; Li, L.B.; Wang, C.X.; Song, M.Z.; Wang, H.T.; Zhao, S.Q.; Jia, X.Y.; Mao, G.Z.; et al. Identification of favorable SNP alleles and candidate genes for traits related to early maturity via GWAS in upland cotton. BMC Genom. 2016, 17, 687. [Google Scholar] [CrossRef] [PubMed]
- Mukeshimana, G.; Lasley, A.L.; Loescher, W.H.; Kelly, J.D. Identification of shoot traits related to drought tolerance in common bean seedlings. J. Am. Soc. Hortic. Sci. 2014, 139, 299–309. [Google Scholar] [CrossRef]
- Su, J.J.; Li, L.; Pang, C.Y.; Wei, H.L.; Wang, C.X.; Song, M.Z.; Wang, H.T.; Zhao, S.Q.; Zhang, C.; Mao, G.Z.; et al. Two genomic regions associated with fiber quality traits in Chinese upland cotton under apparent breeding selection. Sci. Rep. 2016, 6, 38496. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyze spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; Bakker, P.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | SNP Locus | Chromosome | P Value | Major Allele | Minor Allele | Minor Allele Freq. |
|---|---|---|---|---|---|---|
| EWS | A05:89580589 | A05 | 2.67 × 10−6 | C | A | 0.10 |
| EWS | A05:89717711 | A05 | 5.28 × 10−7 | T | C | 0.19 |
| EWS | A05:89762730 | A05 | 5.14 × 10−7 | C | G | 0.20 |
| EWS | A05:89762936 | A05 | 8.70 × 10−7 | G | A | 0.19 |
| EWS | A05:89984532 | A05 | 1.62 × 10−6 | A | G | 0.19 |
| EWS | A05:90061632 | A05 | 7.20 × 10−6 | G | A | 0.17 |
| EWS | A05:90150044 | A05 | 1.05 × 10−6 | A | G | 0.21 |
| EWS | A11:86279835 | A11 | 7.42 × 10−7 | A | G | 0.11 |
| CWS | A01:92213571 | A01 | 2.31 × 10−6 | A | G | 0.18 |
| CWS | A05:4580354 | A05 | 2.80 × 10−6 | T | A | 0.42 |
| CWS | A11:45127798 | A11 | 1.46 × 10−6 | T | C | 0.06 |
| LT | D03:20786242 | D03 | 2.65 × 10−6 | T | C | 0.24 |
| LT | D03:25327187 | D03 | 1.71 × 10−6 | A | T | 0.18 |
| LT | D03:25327414 | D03 | 1.45 × 10−6 | T | C | 0.18 |
| LT | D03:25669014 | D03 | 2.66 × 10−6 | A | C | 0.22 |
| LT | D03:25729410 | D03 | 3.96 × 10−7 | A | G | 0.21 |
| LT | D03:25991794 | D03 | 4.04 × 10−7 | A | G | 0.19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.-M.; Liu, S.-D.; Ge, C.-W.; Zhang, X.-M.; Zhang, S.-P.; Chen, J.; Shen, Q.; Ju, F.-Y.; Yang, Y.-F.; Li, Y.; et al. Association Analysis of Drought Tolerance and Associated Traits in Upland Cotton at the Seedling Stage. Int. J. Mol. Sci. 2019, 20, 3888. https://doi.org/10.3390/ijms20163888
Li H-M, Liu S-D, Ge C-W, Zhang X-M, Zhang S-P, Chen J, Shen Q, Ju F-Y, Yang Y-F, Li Y, et al. Association Analysis of Drought Tolerance and Associated Traits in Upland Cotton at the Seedling Stage. International Journal of Molecular Sciences. 2019; 20(16):3888. https://doi.org/10.3390/ijms20163888
Chicago/Turabian StyleLi, Hai-Ming, Shao-Dong Liu, Chang-Wei Ge, Xiao-Meng Zhang, Si-Ping Zhang, Jing Chen, Qian Shen, Fei-Yan Ju, Yong-Fei Yang, Yang Li, and et al. 2019. "Association Analysis of Drought Tolerance and Associated Traits in Upland Cotton at the Seedling Stage" International Journal of Molecular Sciences 20, no. 16: 3888. https://doi.org/10.3390/ijms20163888
APA StyleLi, H.-M., Liu, S.-D., Ge, C.-W., Zhang, X.-M., Zhang, S.-P., Chen, J., Shen, Q., Ju, F.-Y., Yang, Y.-F., Li, Y., Liu, R.-H., Ma, H.-J., Zhao, X.-H., Li, C.-D., & Pang, C.-Y. (2019). Association Analysis of Drought Tolerance and Associated Traits in Upland Cotton at the Seedling Stage. International Journal of Molecular Sciences, 20(16), 3888. https://doi.org/10.3390/ijms20163888