The Role of Heme Oxygenase 1 in the Protective Effect of Caloric Restriction against Diabetic Cardiomyopathy

Abstract
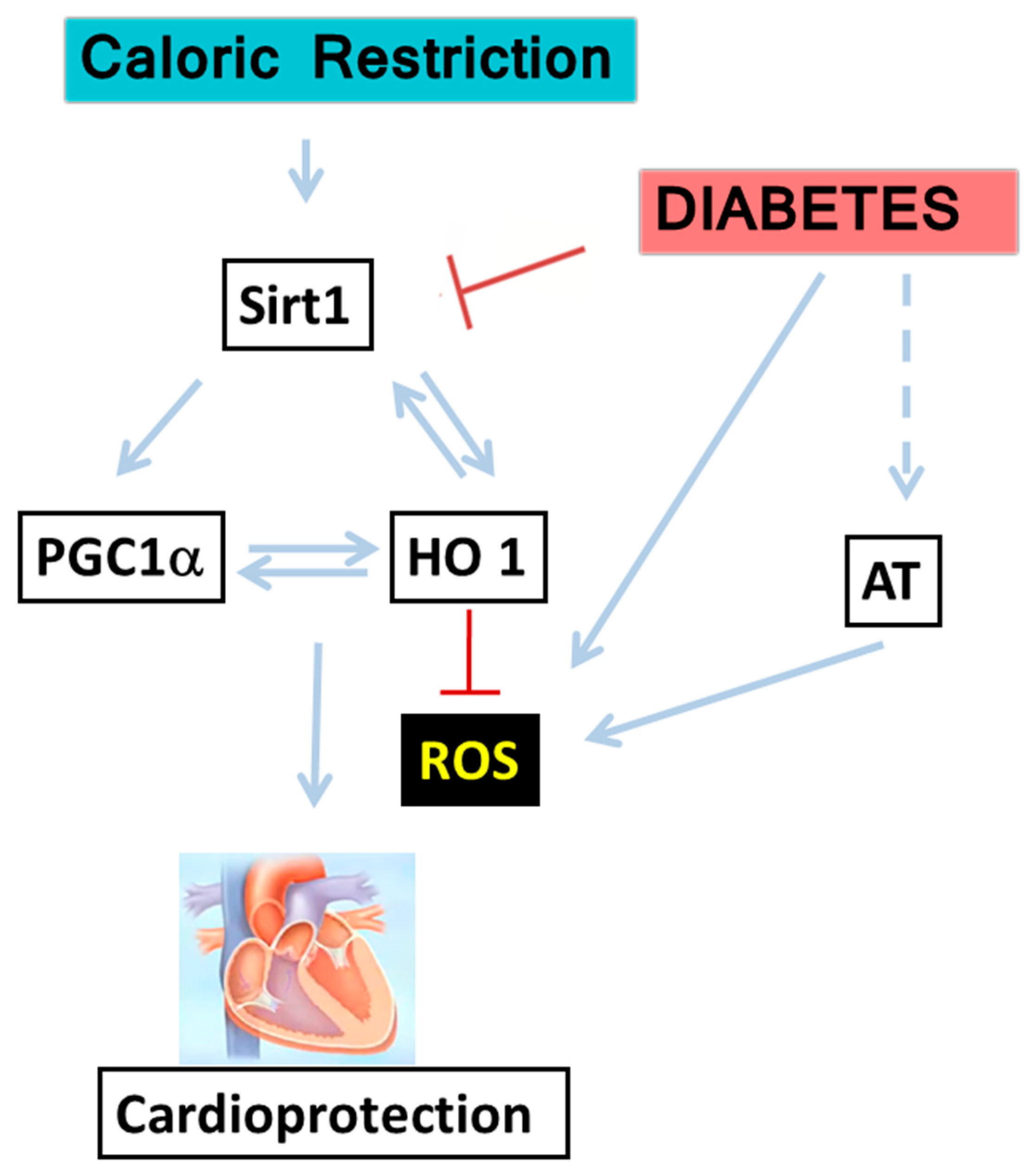
:1. Introduction
2. Results
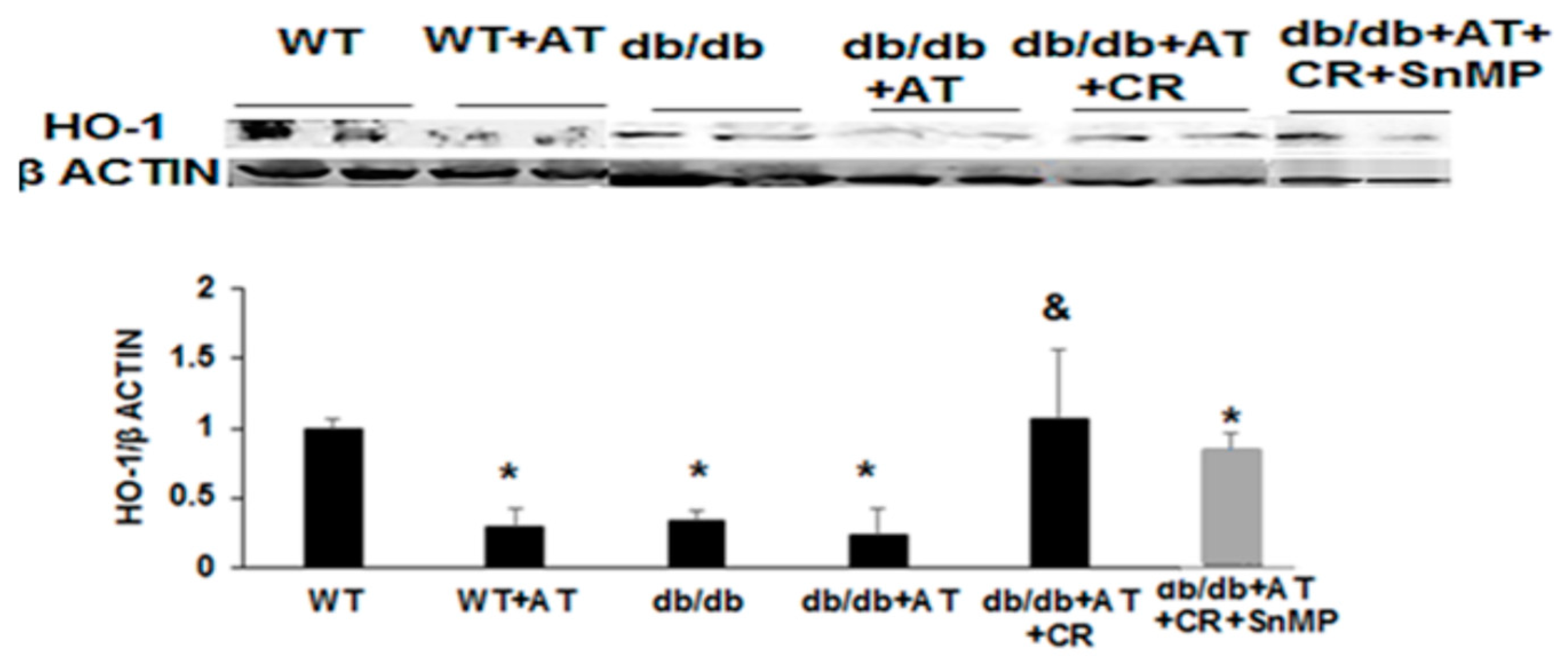
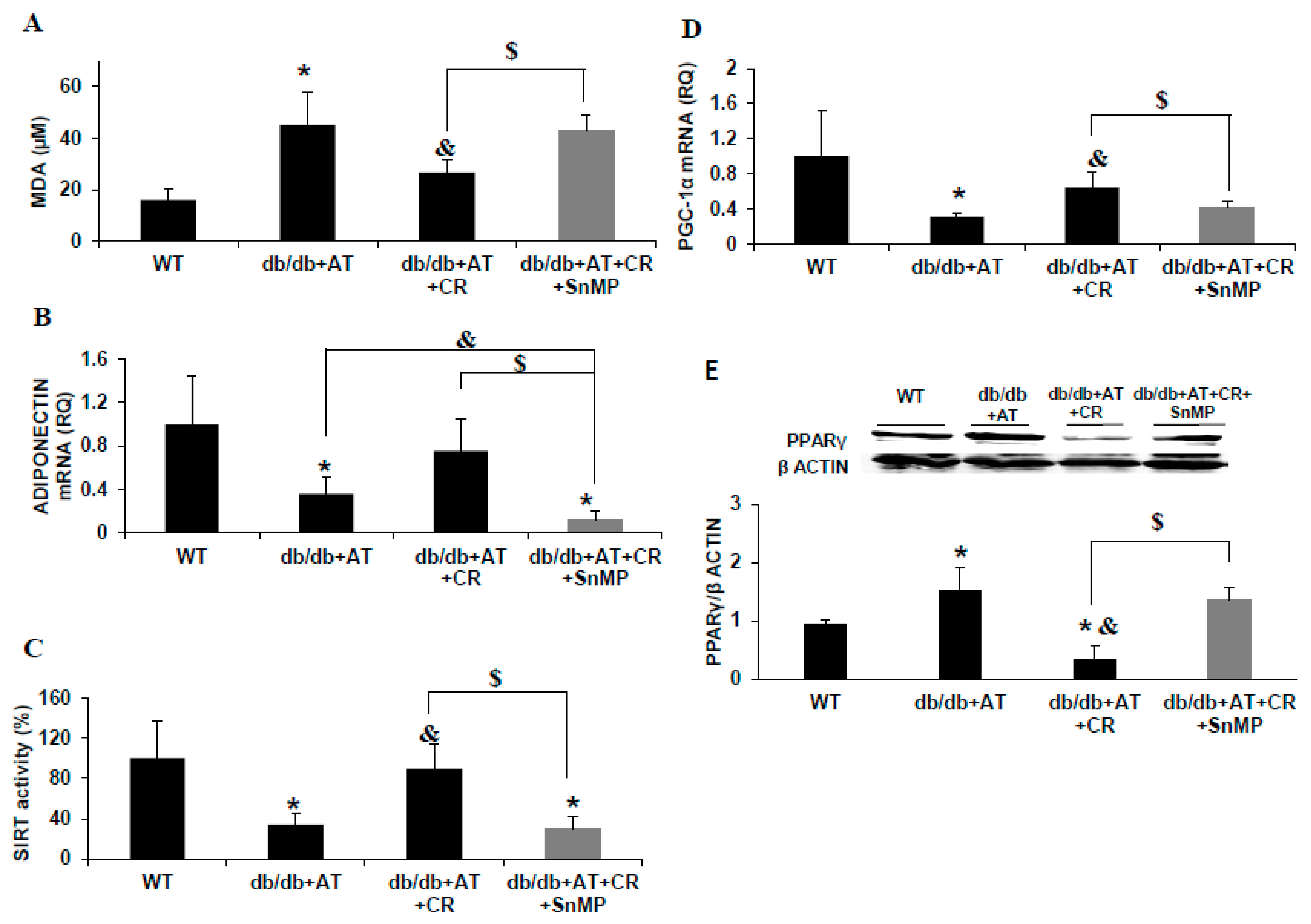
2.1. CR Reduced Oxidative Stress and Increased PGC-1α and HO-1 Levels
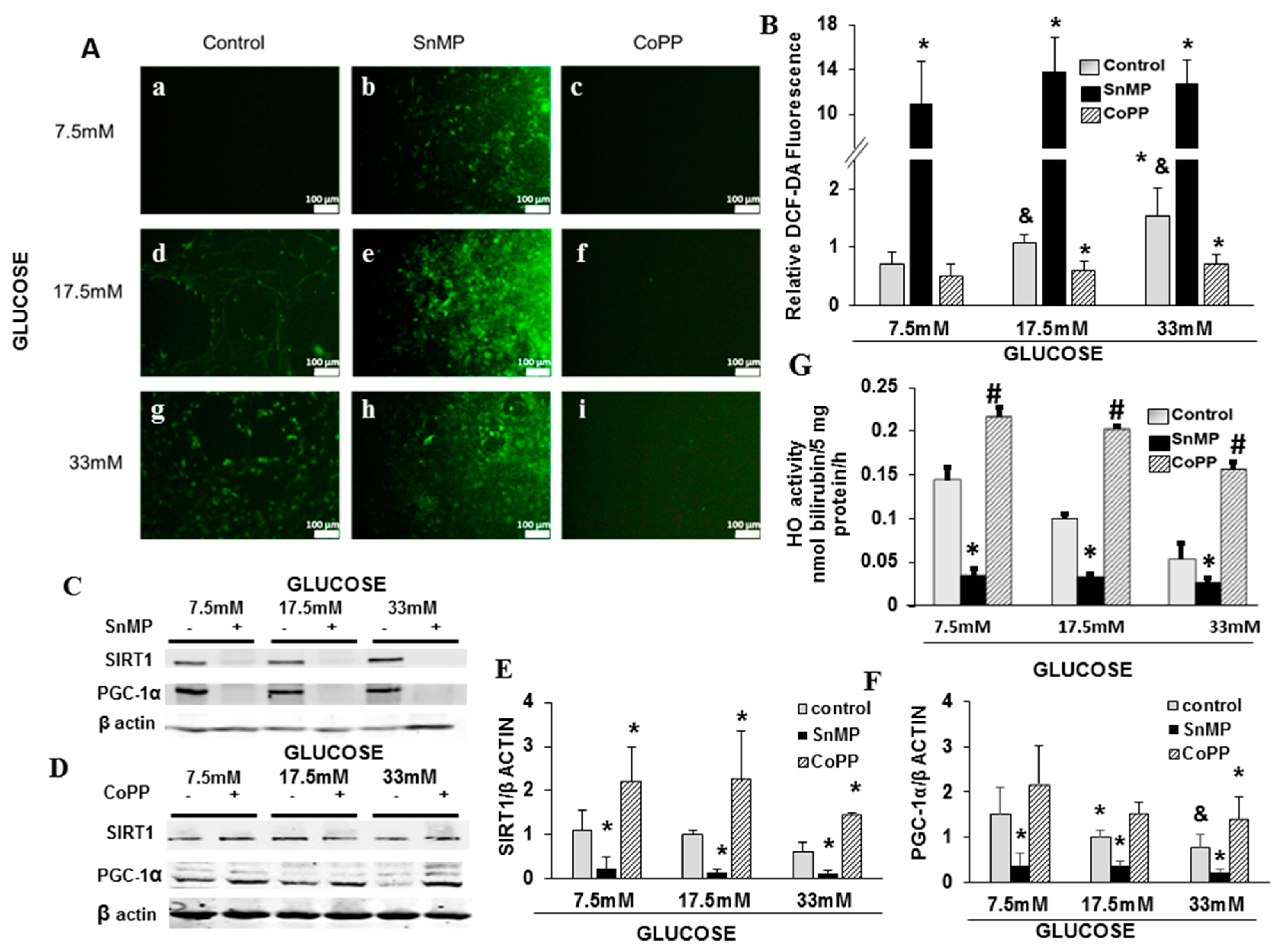
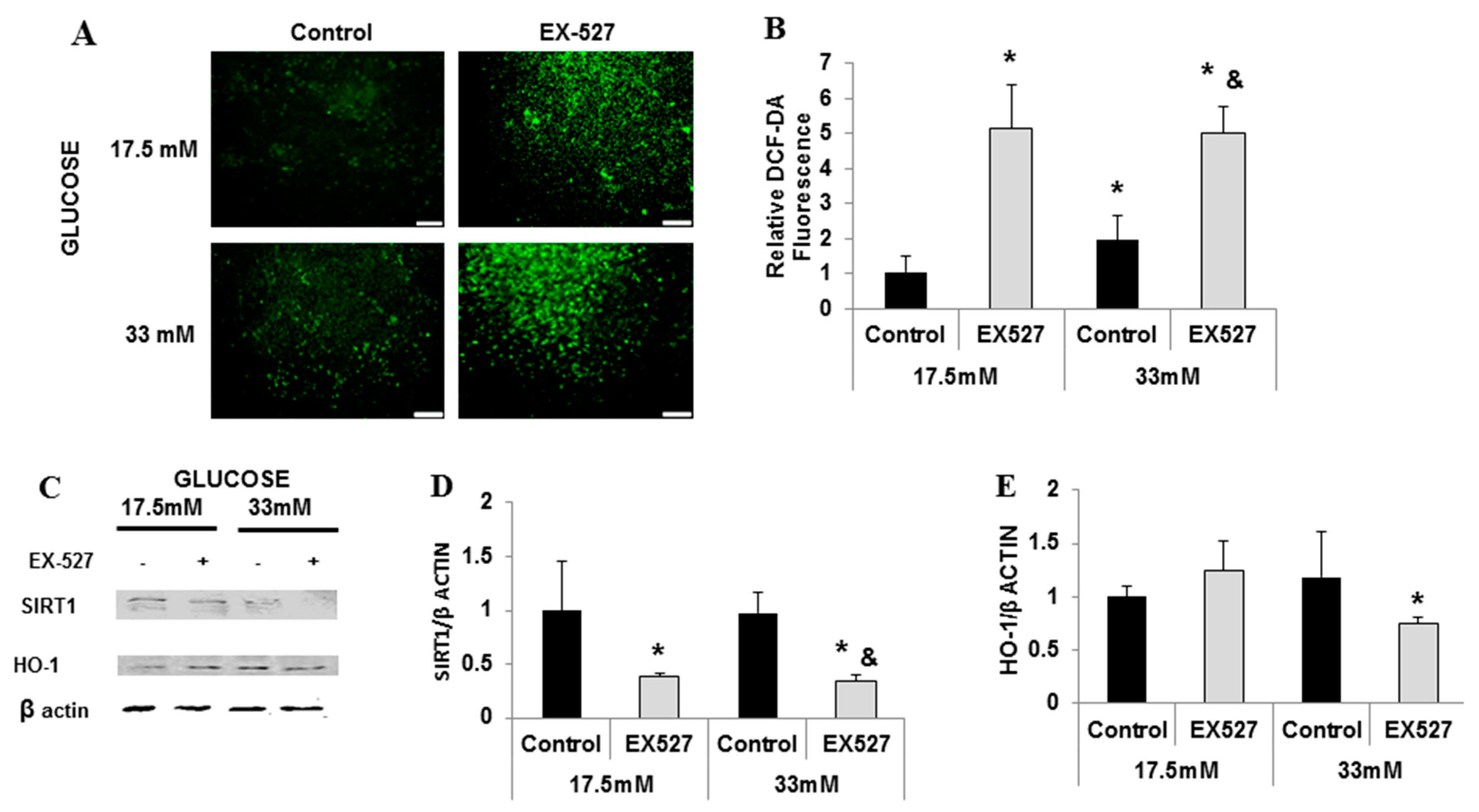
2.2. Cross-Talk between HO-1-SIRT1-and PGC-1α
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Angiotensin
4.3. Caloric Restriction
4.4. Cell Culture
4.5. Experiments with EX-527, CoPP, SnMP, and HO Activity
4.6. In Vitro ROS Production Measurement
4.7. Western Blotting
4.8. RT-PCR
| Gene | Assay ID |
| Tbp (TATA BOX) | Mm00446973 |
| Ppargc1 (PGC-1α) | Mm01208835 |
| Adipoq (adiponectin) | Mm00456425 |
4.9. Serum Thiobarbituric Acid Reactive Substances
4.10. SIRT Activity
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar]
- van Bilsen, M.; Daniels, A.; Brouwers, O.; Janssen, B.J.; Derks, W.J.; Brouns, A.E.; Munts, C.; Schalkwijk, C.G.; van der Vusse, G.J.; van Nieuwenhoven, F.A. Hypertension is a conditional factor for the development of cardiac hypertrophy in type 2 diabetic mice. Plos ONE 2014, 9, e85078. [Google Scholar] [CrossRef]
- Asbun, J.; Villarreal, F.J. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 47, 693–700. [Google Scholar] [CrossRef]
- Kai, H.; Kuwahara, F.; Tokuda, K.; Imaizumi, T. Diastolic dysfunction in hypertensive hearts: Roles of perivascular inflammation and reactive myocardial fibrosis. Hypertens. Res. 2005, 28, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Ansley, D.M.; Wang, B. Oxidative stress and myocardial injury in the diabetic heart. J. Pathol. 2012, 229, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Jin, S.; Guo, W.; Li, C.; Li, X.; Rane, M.J.; Wang, G.; Cai, L. Attenuation of diabetes-induced cardiac inflammation and pathological remodeling by low-dose radiation. Radiat. Res. 2012, 175, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Laurant, P.; Schiffrin, E.L. Effect of magnesium on calcium responses to vasopressin in vascular smooth muscle cells of spontaneously hypertensive rats. J. Pharmacol. Exp. Ther. 1998, 284, 998–1005. [Google Scholar]
- Mazzolai, L.; Nussberger, J.; Aubert, J.F.; Brunner, D.B.; Gabbiani, G.; Brunner, H.R.; Pedrazzini, T. Blood pressure-independent cardiac hypertrophy induced by locally activated renin-angiotensin system. Hypertension 1998, 31, 1324–1330. [Google Scholar] [CrossRef]
- Tokuda, K.; Kai, H.; Kuwahara, F.; Yasukawa, H.; Tahara, N.; Kudo, H.; Takemiya, K.; Koga, M.; Yamamoto, T.; Imaizumi, T. Pressure-independent effects of angiotensin II on hypertensive myocardial fibrosis. Hypertension 2004, 43, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Soare, A.; Weiss, E.P.; Pozzilli, P. Benefits of caloric restriction for cardiometabolic health, including type 2 diabetes mellitus risk. Diabetes/Metab. Res. Rev. 2014, 30 (Suppl. 1), 41–47. [Google Scholar] [CrossRef]
- Speakman, J.R.; Mitchell, S.E. Caloric restriction. Mol. Asp. Med. 2011, 32, 159–221. [Google Scholar] [CrossRef]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef]
- Shinmura, K.; Tamaki, K.; Saito, K.; Nakano, Y.; Tobe, T.; Bolli, R. Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation 2007, 116, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Miura, J.; Lu, L.X.; Bernier, M.; DeCabo, R.; Lane, M.A.; Roth, G.S.; Ingram, D.K. Circulating adiponectin levels increase in rats on caloric restriction: The potential for insulin sensitization. Exp. Gerontol. 2004, 39, 1049–1059. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Landry, J.; Slama, J.T.; Sternglanz, R. Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem. Biophys. Res. Commun. 2000, 278, 685–690. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Predominant expression of Sir2alpha, an NAD-dependent histone deacetylase, in the embryonic mouse heart and brain. FEBS Lett. 2004, 556, 281–286. [Google Scholar] [CrossRef]
- Kondo, M.; Shibata, R.; Miura, R.; Shimano, M.; Kondo, K.; Li, P.; Ohashi, T.; Kihara, S.; Maeda, N.; Walsh, K.; et al. Caloric restriction stimulates revascularization in response to ischemia via adiponectin-mediated activation of endothelial nitric-oxide synthase. J. Biol. Chem. 2009, 284, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Romashko, M.; Schragenheim, J.; Abraham, N.G.; McClung, J.A. Epoxyeicosatrienoic acid as therapy for diabetic and ischemic cardiomyopathy. Trends Pharmacol. Sci. 2016, 37, 945–962. [Google Scholar] [CrossRef]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme Oxygenases in Cardiovascular Health and Disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef] [Green Version]
- Lever, J.M.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Abraham, N.G.; Junge, J.M.; Drummond, G.S. Translational Significance of Heme Oxygenase in Obesity and Metabolic Syndrome. Trends Pharmacol. Sci. 2016, 37, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, A.R.; Rajala, M.W.; Tomas, E.; Pajvani, U.B.; Saha, A.K.; Trumbauer, M.E.; Pang, Z.; Chen, A.S.; Ruderman, N.B.; Chen, H.; et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J. Biol. Chem. 2006, 281, 2654–2660. [Google Scholar] [CrossRef]
- Cohen, K.; Waldman, M.; Abraham, N.G.; Laniado-Schwartzman, M.; Gurfield, D.; Aravot, D.; Arad, M.; Hochhauser, E. Caloric restriction ameliorates cardiomyopathy in animal model of diabetes. Exp. Cell. Res. 2016, 350, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Waldman, M.; Cohen, K.; Yadin, D.; Nudelman, V.; Gorfil, D.; Laniado-Schwartzman, M.; Kornwoski, R.; Aravot, D.; Abraham, N.G.; Arad, M.; et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving ‘SIRT1 and PGC-1alpha’. Cardiovasc. Diabetol. 2018, 17, 111. [Google Scholar] [CrossRef] [PubMed]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1alpha axis. Exp. Cell Res. 2018. [Google Scholar] [CrossRef]
- Leviner, D.B.; Hochhauser, E.; Arad, M. Inherited cardiomyopathies--Novel therapies. Pharmacol. Ther. 2015, 155, 36–48. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, W.Y.; Liao, H.H.; Yang, Z.; Hu, C.; Wang, S.S.; Deng, W.; Tang, Q.Z. AdipoRon, an adiponectin receptor agonist, attenuates cardiac remodeling induced by pressure overload. J. Mol. Med. (Berl) 2018, 96, 1345–1357. [Google Scholar] [CrossRef]
- Fang, X.; Stroud, M.J.; Ouyang, K.; Fang, L.; Zhang, J.; Dalton, N.D.; Gu, Y.; Wu, T.; Peterson, K.L.; Huang, H.D.; et al. Adipocyte-specific loss of PPARgamma attenuates cardiac hypertrophy. JCI Insight 2016, 1, e89908. [Google Scholar] [CrossRef]
- L’Abbate, A.; Neglia, D.; Vecoli, C.; Novelli, M.; Ottaviano, V.; Baldi, S.; Barsacchi, R.; Paolicchi, A.; Masiello, P.; Drummond, G.S.; et al. Beneficial effect of heme oxygenase-1 expression on myocardial ischemia-reperfusion involves an increase in adiponectin in mildly diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3532–H3541. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, B.; Riches, K.; Bageghni, S.A.; O’Regan, D.J.; Porter, K.E.; Turner, N.A. Investigating inherent functional differences between human cardiac fibroblasts cultured from nondiabetic and Type 2 diabetic donors. Cardiovasc. Pathol. 2014, 23, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Waldman, M.; Bellner, L.; Vanella, L.; Schragenheim, J.; Sodhi, K.; Singh, S.P.; Lin, D.; Lakhkar, A.; Li, J.; Hochhauser, E.; et al. Epoxyeicosatrienoic Acids Regulate Adipocyte Differentiation of Mouse 3T3 Cells, Via PGC-1alpha Activation, Which Is Required for HO-1 Expression and Increased Mitochondrial Function. Stem Cells Dev. 2016, 25, 1084–1094. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Turdi, S.; Hu, N.; Guo, R.; Zhang, Y.; Ren, J. Influence of long-term caloric restriction on myocardial and cardiomyocyte contractile function and autophagy in mice. J. Nutr. Biochem. 2012, 23, 1592–1599. [Google Scholar] [CrossRef]
- de Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Dalla Man, C.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Takemori, K.; Kimura, T.; Shirasaka, N.; Inoue, T.; Masuno, K.; Ito, H. Food restriction improves glucose and lipid metabolism through Sirt1 expression: A study using a new rat model with obesity and severe hypertension. Life Sci. 2011, 88, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Peterson, S.J.; Sodhi, K.; Vanella, L.; Barbagallo, I.; Rodella, L.F.; Schwartzman, M.L.; Abraham, N.G.; Kappas, A. Heme oxygenase gene targeting to adipocytes attenuates adiposity and vascular dysfunction in mice fed a high-fat diet. Hypertension 2012, 60, 467–475. [Google Scholar] [CrossRef]
- Calay, D.; Mason, J.C. The multifunctional role and therapeutic potential of HO-1 in the vascular endothelium. Antioxid. Redox Signal. 2014, 20, 1789–1809. [Google Scholar] [CrossRef]
- Ares-Carrasco, S.; Picatoste, B.; Benito-Martin, A.; Zubiri, I.; Sanz, A.B.; Sanchez-Nino, M.D.; Ortiz, A.; Egido, J.; Tunon, J.; Lorenzo, O. Myocardial fibrosis and apoptosis, but not inflammation, are present in long-term experimental diabetes. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2109–H2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombrita, C.; Lombardo, G.; Scapagnini, G.; Abraham, N.G. Heme oxygenase-1 expression levels are cell cycle dependent. Biochem. Biophys. Res. Commun. 2003, 308, 1001–1008. [Google Scholar] [CrossRef]
- Chang, S.H.; Barbosa-Tessmann, I.; Chen, C.; Kilberg, M.S.; Agarwal, A. Glucose deprivation induces heme oxygenase-1 gene expression by a pathway independent of the unfolded protein response. J. Biol. Chem. 2002, 277, 1933–1940. [Google Scholar] [CrossRef]
- Quan, S.; Kaminski, P.M.; Yang, L.; Morita, T.; Inaba, M.; Ikehara, S.; Goodman, A.I.; Wolin, M.S.; Abraham, N.G. Heme oxygenase-1 prevents superoxide anion-associated endothelial cell sloughing in diabetic rats. Biochem. Biophys. Res. Commun. 2004, 315, 509–516. [Google Scholar] [CrossRef]
- Cao, J.; Singh, S.P.; McClung, J.A.; Joseph, G.; Vanella, L.; Barbagallo, I.; Jiang, H.; Falck, J.R.; Arad, M.; Shapiro, J.I.; et al. EET intervention on Wnt1, NOV, and HO-1 signaling prevents obesity-induced cardiomyopathy in obese mice. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H368–H380. [Google Scholar] [CrossRef]
- Singh, S.P.; McClung, J.A.; Bellner, L.; Cao, J.; Waldman, M.; Schragenheim, J.; Arad, M.; Hochhauser, E.; Falck, J.R.; Weingarten, J.A.; et al. CYP-450 Epoxygenase Derived Epoxyeicosatrienoic Acid Contribute To Reversal of Heart Failure in Obesity-Induced Diabetic Cardiomyopathy via PGC-1 alpha Activation. Cardiovasc. Pharm. Open Access 2018, 7, 233. [Google Scholar]
- Raffaele, M.; Li Volti, G.; Barbagallo, I.A.; Vanella, L. Therapeutic Efficacy of Stem Cells Transplantation in Diabetes: Role of Heme Oxygenase. Front. Cell Dev. Biol. 2016, 4, 80. [Google Scholar] [CrossRef]
- Rodella, L.F.; Vanella, L.; Peterson, S.J.; Drummond, G.; Rezzani, R.; Falck, J.R.; Abraham, N.G. Heme oxygenase-derived carbon monoxide restores vascular function in type 1 diabetes. Drug Metab. Lett. 2008, 2, 290–300. [Google Scholar] [CrossRef]
- Cao, J.; Sodhi, K.; Inoue, K.; Quilley, J.; Rezzani, R.; Rodella, L.; Vanella, L.; Germinario, L.; Stec, D.E.; Abraham, N.G.; et al. Lentiviral-human heme oxygenase targeting endothelium improved vascular function in angiotensin II animal model of hypertension. Hum. Gene 2011, 22, 271–282. [Google Scholar] [CrossRef]
- Daniels, A.; van Bilsen, M.; Janssen, B.J.; Brouns, A.E.; Cleutjens, J.P.; Roemen, T.H.; Schaart, G.; van der Velden, J.; van der Vusse, G.J.; van Nieuwenhoven, F.A. Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiol. (Oxf.) 2010, 200, 11–22. [Google Scholar] [CrossRef]
- Levy, E.; Kornowski, R.; Gavrieli, R.; Fratty, I.; Greenberg, G.; Waldman, M.; Birk, E.; Shainberg, A.; Akirov, A.; Miskin, R.; et al. Long-Lived alphaMUPA Mice Show Attenuation of Cardiac Aging and Leptin-Dependent Cardioprotection. PloS ONE 2015, 10, e0144593. [Google Scholar] [CrossRef]
- Avlas, O.; Srara, S.; Shainberg, A.; Aravot, D.; Hochhauser, E. Silencing cardiomyocyte TLR4 reduces injury following hypoxia. Exp. Cell Res. 2016, 348, 115–122. [Google Scholar] [CrossRef]
- Issan, Y.; Kornowski, R.; Aravot, D.; Shainberg, A.; Laniado-Schwartzman, M.; Sodhi, K.; Abraham, N.G.; Hochhauser, E. Heme oxygenase-1 induction improves cardiac function following myocardial ischemia by reducing oxidative stress. PloS ONE 2014, 9, e92246. [Google Scholar] [CrossRef]
- Da Silva, J.L.; Tiefenthaler, M.; Park, E.; Escalante, B.; Schwartzman, M.L.; Levere, R.D.; Abraham, N.G. Tin-mediated heme oxygenase gene activation and cytochrome P450 arachidonate hydroxylase inhibition in spontaneously hypertensive rats. Am. J. Med. Sci 1994, 307, 173–181. [Google Scholar] [CrossRef]
- Waldman, M.; Hochhauser, E.; Fishbein, M.; Aravot, D.; Shainberg, A.; Sarne, Y. An ultra-low dose of tetrahydrocannabinol provides cardioprotection. Biochem. Pharmacol. 2013, 85, 1626–1633. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT n = 8 | db/db n = 14 | db/db + AT n = 14 | db/db + AT + CR n = 8 | db/db+AT+ CR + SnMP n = 5 | |
|---|---|---|---|---|---|
| IVS (mm) | 0.8 ± 0.1 | 0.9 ± 0.1 | 1.1 ± 0.1 # | 1 ± 0.1 & | 1.3 ± 0.1 $ |
| LVPW (mm) | 0.9 ± 0.1 | 0.9 ± 0.1 | 1.1 ± 0.2 # | 0.9 ± 0.2 & | 1.3 ± 0.3 $ |
| LVEDD (mm) | 3.6 ± 0.7 | 3.9 ± 0.2 | 3.5 ± 0.05 # | 4.1 ± 0.4 & | 3 ± 1.2 $ |
| LVESD (mm) | 2.9 ± 0.2 | 2.6 ± 0.3 | 2.4 ± 0.6 | 2.5 ± 0.5 | 2.1 ± 0.4 |
| FS (%) | 33 ± 14 | 34 ± 7 | 34 ± 7 | 41 ± 10 & | 40 ± 4 & |
| Body Weight (g) | 26 ± 3 | 41 ± 10 * | 40 ± 5 | 33 ± 7 & | 36 ± 6 |
| Systolic Blood Pressure (mmHg) | 95 ± 21 | 99 ± 30 | 148 ± 15 # | 114 ± 11 & | 138 ± 9 $ |
| Glucose (mg/dL) | 137 ± 44 | 617 ± 93 * | 658 ± 107 | 531 ± 127 & | 427 ± 195 & |
| AST (U/L) | 62 ± 25 | 127 ± 53 * | 226 ± 149 | 99 ± 21 & | 138 ± 122 |
| ALT (U/L) | 126 ± 42 | 182 ± 134 | 281 ± 176 | 117 ± 32 & | 194 ± 198 |
| Cholesterol (mg/dL) | 79 ± 24 | 112 ± 21 * | 199 ± 91 # | 118 ± 25 & | 156 ± 29 $ |
| Triglycerides (mg/dL) | 124 ± 57 | 185 ± 66 * | 208 ± 75 | 127 ± 35 & | 188 ± 19 $ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waldman, M.; Nudelman, V.; Shainberg, A.; Zemel, R.; Kornwoski, R.; Aravot, D.; Peterson, S.J.; Arad, M.; Hochhauser, E. The Role of Heme Oxygenase 1 in the Protective Effect of Caloric Restriction against Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2427. https://doi.org/10.3390/ijms20102427
Waldman M, Nudelman V, Shainberg A, Zemel R, Kornwoski R, Aravot D, Peterson SJ, Arad M, Hochhauser E. The Role of Heme Oxygenase 1 in the Protective Effect of Caloric Restriction against Diabetic Cardiomyopathy. International Journal of Molecular Sciences. 2019; 20(10):2427. https://doi.org/10.3390/ijms20102427
Chicago/Turabian StyleWaldman, Maayan, Vadim Nudelman, Asher Shainberg, Romy Zemel, Ran Kornwoski, Dan Aravot, Stephen J. Peterson, Michael Arad, and Edith Hochhauser. 2019. "The Role of Heme Oxygenase 1 in the Protective Effect of Caloric Restriction against Diabetic Cardiomyopathy" International Journal of Molecular Sciences 20, no. 10: 2427. https://doi.org/10.3390/ijms20102427