New Approaches in the Management of Sudden Cardiac Death in Patients with Heart Failure—Targeting the Sympathetic Nervous System

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
1.1. Epidemiology of Sudden Cardiac Death
1.2. Treatment Indication
1.3. Prognosis
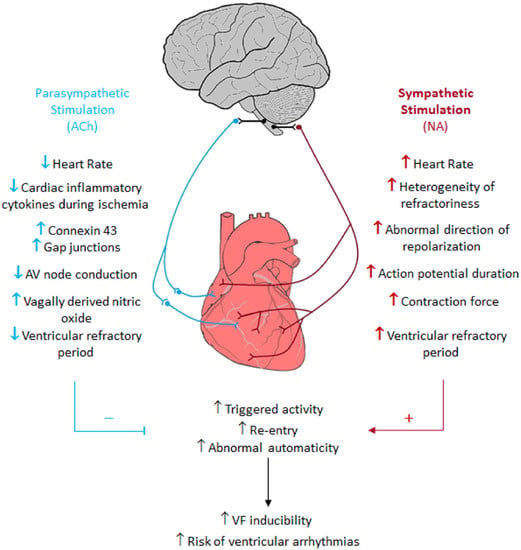
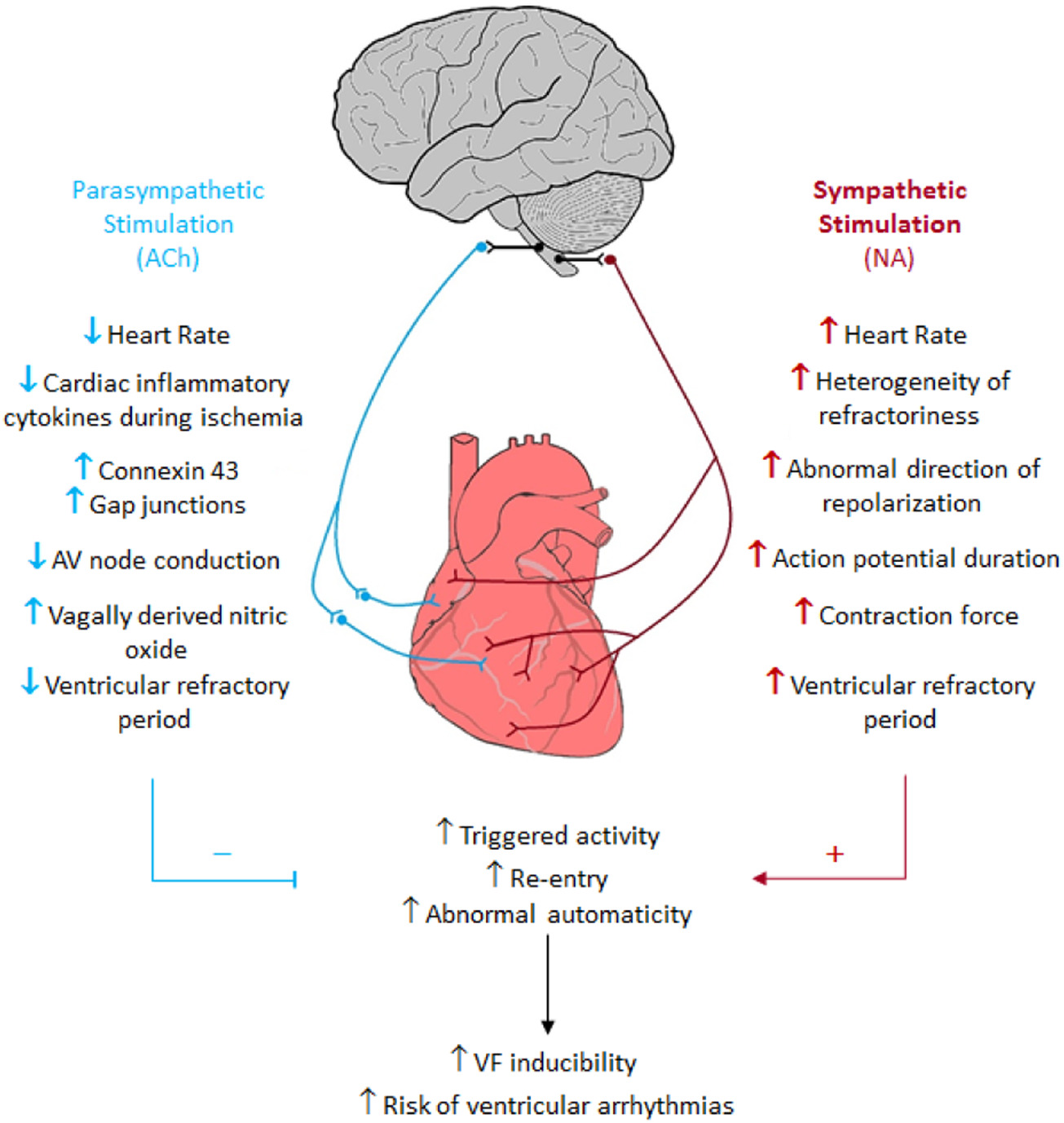
1.4. Pathophysiology of HF and Sympathetic Nerve Activity Synonym
1.5. Relevant Diagnostic Approaches: PET Scan and Cardiac SNS Imaging
2. Treatment Approaches Targeting the SNS: Renal Denervation
Cardiac Conduction System as a Target for RDN
3. SNS Management to Diagnose, Prevent and Treat HF, VAs, and SCD: Challenges and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clark, H. NCDs: A challenge to sustainable human development. Lancet 2013, 381, 510–511. [Google Scholar] [CrossRef]
- Alwan, A. Global status report on noncommunicable diseases 2010: Introduction. In Global Status Report on Noncommunicable Diseases 2010; World Health Organization: Geneva, Switzerland, 2011; pp. vii–ix. [Google Scholar]
- Bonita, R.; Magnusson, R.; Bovet, P.; Zhao, D.; Malta, D.C.; Geneau, R.; Suh, L.I.; Thankappan, K.R.; McKee, M.; FFPHM, J.H.; et al. Country actions to meet UN commitments on non-communicable diseases: A stepwise approach. Lancet 2013, 381, 575–584. [Google Scholar] [CrossRef]
- Sacco, R.L.; Roth, G.A.; Reddy, K.S.; Arnett, D.K.; Bonita, R.; Gaziano, T.A.; Heidenreich, P.A.; Huffman, M.D.; Mayosi, B.M.; Mendis, S.; et al. The Heart of 25 by 25: Achieving the Goal of Reducing Global and Regional Premature Deaths from Cardiovascular Diseases and Stroke: A Modeling Study from the American Heart Association and World Heart Federation. Circulation 2016, 133, e674–e690. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, M.N.; van den Berg, M.E.; Leening, M.J.; Hofman, A.; Franco, O.H.; Deckers, J.W.; Heeringa, J.; Rijnbeek, P.R.; Stricker, B.H.; Eijgelsheim, M.; et al. Declining incidence of sudden cardiac death from 1990-2010 in a general middle-aged and elderly population: The Rotterdam Study. Heart Rhythm 2015, 12, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Mendis, S.; Puska, P.; Norrving, B. Global Atlas on Cardiovascular Disease Prevention and Control; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Ishikawa, S.; Niwano, S.; Imaki, R.; Takeuchi, I.; Irie, W.; Toyooka, T.; Soma, K.; Kurihara, K.; Izumi, T. Usefulness of a simple prognostication score in prediction of the prognoses of patients with out-of-hospital cardiac arrests. Int. Heart J. 2013, 54, 362–370. [Google Scholar] [CrossRef]
- Marrugat, J.; Elosua, R.; Gil, M. Epidemiology of sudden cardiac death in Spain. Rev. Esp. Cardiol. 1999, 52, 717–725. [Google Scholar] [PubMed]
- Vaartjes, I.; Hendrix, A.; Hertogh, E.M.; Grobbee, D.E.; Doevendans, P.A.; Mosterd, A.; Bots, M.L. Sudden death in persons younger than 40 years of age: Incidence and causes. Eur. J. Cardiov. Prev. Rehabil. 2009, 16, 592–596. [Google Scholar] [CrossRef]
- Bengel, F.M. European perspective: Comparing the AHA/ACC and ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. J. Nucl. Cardiol. 2017, 24, 1909–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac Death. The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology. G. Ital. Cardiol. (Rome) 2016, 17, 108–170. [Google Scholar]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics--2012 update: A report from the American Heart Association. Circulation 2012, 125, e2–e220. [Google Scholar]
- Myerburg, R.J.; Junttila, M.J. Sudden cardiac death caused by coronary heart disease. Circulation 2012, 125, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar]
- Eckart, R.E.; Shry, E.A.; Burke, A.P.; McNear, J.A.; Appel, D.A.; Castillo-Rojas, L.M.; Avedissian, L.; Pearse, L.A.; Potter, R.N.; Tremaine, L.; et al. Sudden death in young adults: an autopsy-based series of a population undergoing active surveillance. J. Am. Coll. Cardiol. 2011, 58, 1254–1261. [Google Scholar] [CrossRef]
- Maron, B.J.; Gohman, T.E.; Aeppli, D. Prevalence of sudden cardiac death during competitive sports activities in Minnesota high school athletes. J. Am. Coll. Cardiol. 1998, 32, 1881–1884. [Google Scholar] [CrossRef]
- van der Werf, C.; Hendrix, A.; Birnie, E.; Bots, M.L.; Vink, A.; Bardai, A.; Blom, M.T.; Bosch, J.; Bruins, W.; Das, C.K.; et al. Improving usual care after sudden death in the young with focus on inherited cardiac diseases (the CAREFUL study): a community-based intervention study. Europace 2016, 18, 592–601. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomstrom-Lundqvist, C. 2015 European Society of Cardiology Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co-chairs. Eur. Heart J. 2015, 36, 2757–2759. [Google Scholar] [CrossRef]
- United Nations Economic Commission for Europe. UNECE Statistical Database. Available online: http://w3uneceorg/pxweb (accessed on 19 November 2015).
- Behr, E.; Ensam, B. New approaches to predicting the risk of sudden death. Clin. Med. (Lond.). 2016, 16, 283. [Google Scholar] [CrossRef]
- Moss, A.J.; Zareba, W.; Hall, W.J.; Klein, H.; Wilber, D.J.; Cannom, D.S.; Daubert, J.P.; Higgins, S.L.; Brown, M.W.; Andrews, M.L.; et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N. Engl. J. Med. 2002, 346, 877–883. [Google Scholar] [CrossRef]
- Bardy, G.H. Amiodarone or an implantable cardioverter-defibrillator for congestive heart. N. Engl. J. Med. 2005, 352, 225–237. [Google Scholar] [CrossRef]
- Moss, A.J.; Hall, W.J.; Cannom, D.S.; Daubert, J.P.; Higgins, S.L.; Klein, H.; Levine, J.H.; Saksena, S.; Waldo, A.L.; Wilber, D.; et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N. Engl. J. Med. 1996, 335, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Landis, J.R.; Leighton, R.; Ritter, G.; Vasu, C.M.; Wolfe, R.A.; Acheson, A.; VanderBrug Medendorp, S. Predictive survival models for resuscitated victims of out-of-hospital cardiac arrest with coronary heart disease. Circulation 1985, 71, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.G.; McDonagh, T.; Rigby, A.S.; Yassin, A.; Whittaker, T.; Dargie, H.J.; National Heart Failure Audit Team for England and Wales. The national heart failure audit for England and Wales 2008–2009. Heart 2011, 97, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Harjola, V.P.; Follath, F.; Nieminen, M.S.; Brutsaert, D.; Dickstein, K.; Drexler, H.; Hochadel, M.; Komajda, M.; Lopez-Sendon, J.L.; Ponikowski, P.; et al. Characteristics, outcomes, and predictors of mortality at 3 months and 1 year in patients hospitalized for acute heart failure. Eur. J. Heart Fail. 2010, 12, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Swedberg, K.; Komajda, M.; Bohm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L.; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B.; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.G.; Freemantle, N.; Erdmann, E.; Gras, D.; Kappenberger, L.; Tavazzi, L.; Daubert, J.C. Long-term mortality with cardiac resynchronization therapy in the Cardiac Resynchronization-Heart Failure (CARE-HF) trial. Eur. J. Heart Fail. 2012, 14, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Cohn, J.N.; Archibald, D.G.; Ziesche, S.; Franciosa, J.A.; Harston, W.E.; Tristani, F.E.; Dunkman, W.B.; Jacobs, W.; Francis, G.S.; Flohr, K.H.; et al. Effect of Vasodilator Therapy on Mortality in Chronic Congestive-Heart-Failure—Results of a Veterans-Administration Cooperative Study. N. Engl. J. Med. 1986, 314, 1547–1552. [Google Scholar] [CrossRef]
- Cleland, J.G.F.; Daubert, J.C.; Erdmann, E.; Freemantle, N.; Gras, D.; Kappenberger, L.; Tavazzi, L. Longer-term effects of cardiac resynchronization therapy on mortality in heart failure [the CArdiac REsynchronization-Heart Failure (CARE-HF) trial extension phase]. Eur. Heart J. 2006, 27, 1928–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogale, N.; Witte, K.; Priori, S.; Cleland, J.; Auricchio, A.; Gadler, F.; Gitt, A.; Limbourg, T.; Linde, C.; Dickstein, K.; et al. The European Cardiac Resynchronization Therapy Survey: comparison of outcomes between de novo cardiac resynchronization therapy implantations and upgrades. Eur. J. Heart Fail. 2011, 13, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Parati, G.; Esler, M. The human sympathetic nervous system: Its relevance in hypertension and heart failure. Eur. Heart J. 2012, 33, 1058–1066. [Google Scholar] [CrossRef]
- Florea, V.G.; Cohn, J.N. The autonomic nervous system and heart failure. Circ. Res. 2014, 114, 1815–1826. [Google Scholar] [CrossRef]
- Wang, Y.; Seto, S.W.; Golledge, J. Angiotensin II, sympathetic nerve activity and chronic heart failure. Heart Fail. Rev. 2014, 19, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T. Heart failure as an autonomic nervous system dysfunction. J. Cardiol. 2012, 59, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Floras, J.S. Sympathetic nervous system activation in human heart failure: clinical implications of an updated model. J. Am. Coll. Cardiol. 2009, 54, 375–385. [Google Scholar] [CrossRef]
- Dzau, V.J.; Colucci, W.S.; Hollenberg, N.K.; Williams, G.H. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation 1981, 63, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Pepper, G.S.; Lee, R.W. Sympathetic activation in heart failure and its treatment with beta-blockade. Arch. Intern. Med. 1999, 159, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Cox, H.S.; Lambert, G.W.; Kaye, D.M.; Jennings, G.L.; Meredith, I.T.; Esler, M.D. Region-specific neuropeptide Y overflows at rest and during sympathetic activation in humans. Hypertension 1997, 29, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Regitz, V.; Leuchs, B.; Bossaller, C.; Sehested, J.; Rappolder, M.; Fleck, E. Myocardial catecholamine concentrations in dilated cardiomyopathy and heart failure of different origins. Eur. Heart J. 1991, 12 (Suppl. D), 171–174. [Google Scholar] [CrossRef] [PubMed]
- Backs, J.; Haunstetter, A.; Gerber, S.H.; Metz, J.; Borst, M.M.; Strasser, R.H.; Kübler, W.; Haass, M. The neuronal norepinephrine transporter in experimental heart failure: Evidence for a posttranscriptional downregulation. J. Mol. Cell. Cardiol. 2001, 33, 461–472. [Google Scholar] [CrossRef]
- Todd, G.L.; Baroldi, G.; Pieper, G.M.; Clayton, F.C.; Eliot, R.S. Experimental catecholamine-induced myocardial necrosis. II. Temporal development of isoproterenol-induced contraction band lesions correlated with ECG, hemodynamic and biochemical changes. J. Mol. Cell. Cardiol. 1985, 17, 647–656. [Google Scholar] [CrossRef]
- Mann, D.L.; Kent, R.L.; Parsons, B.; Cooper, G.T. Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 1992, 85, 790–804. [Google Scholar] [CrossRef]
- Osadchii, O.E.; Norton, G.R.; McKechnie, R.; Deftereos, D.; Woodiwiss, A.J. Cardiac dilatation and pump dysfunction without intrinsic myocardial systolic failure following chronic beta-adrenoreceptor activation. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H1898–H1905. [Google Scholar] [CrossRef]
- Communal, C.; Singh, K.; Pimentel, D.R.; Colucci, W.S. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation 1998, 98, 1329–1334. [Google Scholar] [CrossRef]
- Fu, Y.C.; Chi, C.S.; Yin, S.C.; Hwang, B.T.; Chiu, Y.T.; Hsu, S.L. Norepinephrine induces apoptosis in neonatal rat cardiomyocytes through a reactive oxygen species-TNF alpha-caspase signaling pathway. Cardiovasc. Res. 2004, 62, 558–567. [Google Scholar] [CrossRef]
- Mori, H.; Ishikawa, S.; Kojima, S.; Hayashi, J.; Watanabe, Y.; Hoffman, J.I.; Okino, H. Increased responsiveness of left ventricular apical myocardium to adrenergic stimuli. Cardiovasc. Res. 1993, 27, 192–198. [Google Scholar] [CrossRef]
- Lyon, A.R.; Rees, P.S.C.; Prasad, S.; Poole-Wilson, P.A.; Harding, S.E. Stress (Takotsubo) cardiomyopathy—A novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat. Clin. Pract. Card. 2008, 5, 22–29. [Google Scholar] [CrossRef]
- Kuniyoshi, R.R.; Martinelli, M.; Negrao, C.E.; Siqueira, S.F.; Rondon, M.U.; Trombetta, I.C.; Kuniyoshi, F.H.; Laterza, M.C.; Nishioka, S.A.; Costa, R.; et al. Effects of cardiac resynchronization therapy on muscle sympathetic nerve activity. Pacing Clin. Electrophysiol. 2014, 37, 11–18. [Google Scholar] [CrossRef]
- Grassi, G.; Vincenti, A.; Brambilla, R.; Trevano, F.Q.; Dell’Oro, R.; Ciro, A.; Trocino, G.; Vincenzi, A.; Mancia, G. Sustained sympathoinhibitory effects of cardiac resynchronization therapy in severe heart failure. Hypertension 2004, 44, 727–731. [Google Scholar] [CrossRef]
- Waldo, A.L.; Plumb, V.J.; Arciniegas, J.G.; MacLean, W.A.; Cooper, T.B.; Priest, M.F.; James, T.N. Transient entrainment and interruption of the atrioventricular bypass pathway type of paroxysmal atrial tachycardia. A model for understanding and identifying reentrant arrhythmias. Circulation 1983, 67, 73–83. [Google Scholar] [CrossRef]
- Schmidt, A.; Azevedo, C.F.; Cheng, A.; Gupta, S.N.; Bluemke, D.A.; Foo, T.K.; Gerstenblith, G.; Weiss, R.G.; Marbán, E.; Tomaselli, G.F.; et al. Infarct tissue heterogeneity by magnetic resonance imaging identifies enhanced cardiac arrhythmia susceptibility in patients with left ventricular dysfunction. Circulation 2007, 115, 2006–2014. [Google Scholar] [CrossRef]
- Yan, A.T.; Shayne, A.J.; Brown, K.A.; Gupta, S.N.; Chan, C.W.; Luu, T.M.; Di Carli, M.F.; Reynolds, H.G.; Stevenson, W.G.; Kwong, R.Y. Characterization of the peri-infarct zone by contrast-enhanced cardiac magnetic resonance imaging is a powerful predictor of post-myocardial infarction mortality. Circulation 2006, 114, 32–39. [Google Scholar] [CrossRef]
- Roes, S.D.; Borleffs, C.J.; van der Geest, R.J.; Westenberg, J.J.; Marsan, N.A.; Kaandorp, T.A.; Reiber, J.H.; Zeppenfeld, K.; Lamb, H.J.; de Roos, A.; et al. Infarct tissue heterogeneity assessed with contrast-enhanced MRI predicts spontaneous ventricular arrhythmia in patients with ischemic cardiomyopathy and implantable cardioverter-defibrillator. Circ. Cardiovasc. Imaging 2009, 2, 183–190. [Google Scholar] [CrossRef]
- Boutagy, N.E.; Sinusas, A.J. Recent Advances and Clinical Applications of PET Cardiac Autonomic Nervous System Imaging. Curr. Cardiol. Rep. 2017, 19, 33. [Google Scholar] [CrossRef]
- Henneman, M.M.; Bengel, F.M.; van der Wall, E.E.; Knuuti, J.; Bax, J.J. Cardiac neuronal imaging: Application in the evaluation of cardiac disease. J. Nucl. Cardiol. 2008, 15, 442–455. [Google Scholar] [CrossRef]
- Higuchi, T.; Schwaiger, M. Imaging cardiac neuronal function and dysfunction. Curr. Cardiol. Rep. 2006, 8, 131–138. [Google Scholar] [CrossRef]
- Fallavollita, J.A.; Heavey, B.M.; Luisi, A.J.; Michalek, S.M.; Baldwa, S.; Mashtare, T.L.; Hutson, A.D.; de Kempet, R.A.; Haka, M.S.; Sajjad, M.; et al. Regional Myocardial Sympathetic Denervation Predicts the Risk of Sudden Cardiac Arrest in Ischemic Cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 141–149. [Google Scholar] [CrossRef]
- Lambert, E.; Eikelis, N.; Esler, M.; Dawood, T.; Schlaich, M.; Bayles, R.; Socratous, F.; Agrotis, A.; Jennings, G.; Lambert, G.; et al. Altered sympathetic nervous reactivity and norepinephrine transporter expression in patients with postural tachycardia syndrome. Circ. Arrhythm. Electrophysiol. 2008, 1, 103–109. [Google Scholar] [CrossRef]
- Esler, M.; Alvarenga, M.; Pier, C.; Richards, J.; El-Osta, A.; Barton, D.; Haikerwal, D.; Kaye, D.; Schlaich, M.; Guo, L.; et al. The neuronal noradrenaline transporter, anxiety and cardiovascular disease. J. Psychopharmacol. 2006, 20 (Suppl. 4), 60–66. [Google Scholar] [CrossRef]
- Yu, M.; Bozek, J.; Lamoy, M.; Guaraldi, M.; Silva, P.; Kagan, M.; Yalamanchili, P.; Onthank, D.; Mistry, M.; Lazewatsky, J.; et al. Evaluation of LMI1195, a novel 18F-labeled cardiac neuronal PET imaging agent, in cells and animal models. Circ. Cardiovasc. Imaging 2011, 4, 435–443. [Google Scholar] [CrossRef]
- DiBona, G.F.; Kopp, U.C. Neural control of renal function. Physiol. Rev. 1997, 77, 75–197. [Google Scholar] [CrossRef]
- Bohm, M.; Linz, D.; Ukena, C.; Esler, M.; Mahfoud, F. Renal denervation for the treatment of cardiovascular high risk-hypertension or beyond? Circ. Res. 2014, 115, 400–409. [Google Scholar] [CrossRef]
- Sakakura, K.; Ladich, E.; Cheng, Q.; Otsuka, F.; Yahagi, K.; Fowler, D.R.; Kolodgie, F.D.; Virmani, R.; Joner, M. Anatomic assessment of sympathetic peri-arterial renal nerves in man. J. Am. Coll. Cardiol. 2014, 64, 635–643. [Google Scholar] [CrossRef]
- Stella, A.; Zanchetti, A. Functional role of renal afferents. Physiol. Rev. 1991, 71, 659–682. [Google Scholar] [CrossRef]
- Bradley, T.; Hjemdahl, P. Influence of afferent renal nerve activity on contralateral renal overflow of noradrenaline and dopamine to plasma in the dog. Acta Physiol. Scand. 1986, 128, 119–120. [Google Scholar] [CrossRef]
- Larsen, R.; Thorp, A.; Schlaich, M. Regulation of the sympathetic nervous system by the kidney. Curr. Opin. Nephrol. Hypertens. 2014, 23, 61–68. [Google Scholar] [CrossRef]
- Patel, K.P.; Knuepfer, M.M. Effect of afferent renal nerve stimulation on blood pressure, heart rate and noradrenergic activity in conscious rats. J. Auton. Nerv. Syst. 1986, 17, 121–130. [Google Scholar] [CrossRef]
- Rogenes, P.R. Single-unit and multiunit analyses of renorenal reflexes elicited by stimulation of renal chemoreceptors in the rat. J. Auton. Nerv. Syst. 1982, 6, 143–156. [Google Scholar] [CrossRef]
- Linz, D.; Hohl, M.; Schutze, J.; Mahfoud, F.; Speer, T.; Linz, B.; Hübschle, T.; Juretschke, H.P.; Dechend, R.; Geisel, J.; et al. Progression of kidney injury and cardiac remodeling in obese spontaneously hypertensive rats: the role of renal sympathetic innervation. Am. J. Hypertens. 2015, 28, 256–265. [Google Scholar] [CrossRef]
- Mahfoud, F.; Moon, L.B.; Pipenhagen, C.A.; Jensen, J.A.; Pathak, A.; Papademetriou, V.; Ewen, S.; Linz, D.; Böhm, M. Catheter-based radio-frequency renal nerve denervation lowers blood pressure in obese hypertensive swine model. J. Hypertens. 2016, 34, 1854–1862. [Google Scholar] [CrossRef]
- Hohl, M.; Linz, D.; Fries, P.; Muller, A.; Stroeder, J.; Urban, D.; Speer, T.; Geisel, J.; Hummel, B.; Laufs, U.; et al. Modulation of the sympathetic nervous system by renal denervation prevents reduction of aortic distensibility in atherosclerosis prone ApoE-deficient rats. J. Transl. Med. 2016, 14, 167. [Google Scholar] [CrossRef] [Green Version]
- Clayton, S.C.; Haack, K.K.; Zucker, I.H. Renal denervation modulates angiotensin receptor expression in the renal cortex of rabbits with chronic heart failure. Am. J. Physiol. Ren. Physiol. 2011, 300, F31–F39. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F.; Sawin, L.L. Effect of renal denervation on dynamic autoregulation of renal blood flow. Am. J. Physiol. Ren. Physiol. 2004, 286, F1209–F1218. [Google Scholar] [CrossRef] [Green Version]
- Kon, V.; Yared, A.; Ichikawa, I. Role of renal sympathetic nerves in mediating hypoperfusion of renal cortical microcirculation in experimental congestive heart failure and acute extracellular fluid volume depletion. J. Clin. Investig. 1985, 76, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Schiller, A.M.; Pellegrino, P.R.; Zucker, I.H. The renal nerves in chronic heart failure: Efferent and afferent mechanisms. Front. Physiol. 2015, 6, 224. [Google Scholar] [CrossRef] [PubMed]
- Witty, R.T.; Davis, J.O.; Shade, R.E.; Johnson, J.A.; Prewitt, R.L. Mechanisms regulating renin release in dogs with thoracic caval constriction. Circ. Res. 1972, 31, 339–347. [Google Scholar] [CrossRef]
- Foss, J.D.; Fink, G.D.; Osborn, J.W. Differential role of afferent and efferent renal nerves in the maintenance of early- and late-phase Dahl S hypertension. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2016, 310, R262–R267. [Google Scholar] [CrossRef]
- Osborn, J.W.; Foss, J.D. Renal Nerves and Long-Term Control of Arterial Pressure. Compr. Physiol. 2017, 7, 263–320. [Google Scholar]
- Linz, D.; Ukena, C.; Mahfoud, F.; Neuberger, H.R.; Bohm, M. Atrial autonomic innervation: A target for interventional antiarrhythmic therapy? J. Am. Coll. Cardiol. 2014, 63, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; van Hunnik, A.; Ukena, C.; Ewen, S.; Mahfoud, F.; Schirmer, S.H.; Lenski, M.; Neuberger, H.R.; Schotten, U.; Böhm, M. Renal denervation: effects on atrial electrophysiology and arrhythmias. Clin. Res. Cardiol. 2014, 103, 765–774. [Google Scholar] [CrossRef]
- Ripplinger, C.M.; Noujaim, S.F.; Linz, D. The nervous heart. Prog. Biophys. Mol. Biol. 2016, 120, 199–209. [Google Scholar] [CrossRef]
- Yu, L.L.; Huang, B.; Wang, Z.; Wang, S.Y.; Wang, M.L.; Li, X.F.; Zhou, L.; Meng, G.; Yuan, S.; Zhou, X.; et al. Impacts of Renal Sympathetic Activation on Atrial Fibrillation: The Potential Role of the Autonomic Cross Talk Between Kidney and Heart. J. Am. Heart Assoc. 2017, 6, e004716. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Chan, Y.H.; Chinda, K.; Chen, Z.; Patel, J.; Shen, C.; Zhao, Y.; Jiang, Z.; Yuan, Y.; Ye, M.; et al. Effects of renal sympathetic denervation on the stellate ganglion and brain stem in dogs. Heart Rhythm 2017, 14, 255–262. [Google Scholar] [CrossRef]
- Berukstis, A.; Vajauskas, D.; Gargalskaite, U.; Misonis, N.; Burneikaite, G.; Zakarkaite, D.; Miglinas, M.; Laucevicius, A. Impact of renal sympathetic denervation on cardiac sympathetic nerve activity evaluated by cardiac MIBG imaging. EuroIntervention 2016, 11, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Donazzan, L.; Mahfoud, F.; Ewen, S.; Ukena, C.; Cremers, B.; Kirsch, C.M.; Hellwig, D.; Eweiwi, T.; Ezziddin, S.; Esler, M.; et al. Effects of catheter-based renal denervation on cardiac sympathetic activity and innervation in patients with resistant hypertension. Clin. Res. Cardiol. 2016, 105, 364–371. [Google Scholar] [CrossRef]
- Brandt, M.C.; Mahfoud, F.; Reda, S.; Schirmer, S.H.; Erdmann, E.; Bohm, M.; Hoppe, U.C. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J. Am. Coll. Cardiol. 2012, 59, 901–909. [Google Scholar] [CrossRef]
- Mahfoud, F.; Urban, D.; Teller, D.; Linz, D.; Stawowy, P.; Hassel, J.H.; Fries, P.; Dreysse, S.; Wellnhofer, E.; Schneider, G.; et al. Effect of renal denervation on left ventricular mass and function in patients with resistant hypertension: Data from a multi-centre cardiovascular magnetic resonance imaging trial. Eur. Heart J. 2014, 35, 2224–2231. [Google Scholar] [CrossRef]
- Doltra, A.; Messroghli, D.; Stawowy, P.; Hassel, J.H.; Gebker, R.; Leppanen, O.; Gräfe, M.; Schneeweis, C.; Schnackenburg, B.; Fleck, E.; et al. Potential reduction of interstitial myocardial fibrosis with renal denervation. J. Am. Heart Assoc. 2014, 3, e001353. [Google Scholar] [CrossRef]
- Perlini, S.; Palladini, G.; Ferrero, I.; Tozzi, R.; Fallarini, S.; Facoetti, A.; Nano, R.; Clari, F.; Busca, G.; Fogari, R.; et al. Sympathectomy or doxazosin, but not propranolol, blunt myocardial interstitial fibrosis in pressure-overload hypertrophy. Hypertension 2005, 46, 1213–1218. [Google Scholar] [CrossRef]
- McLellan, A.J.; Schlaich, M.P.; Taylor, A.J.; Prabhu, S.; Hering, D.; Hammond, L.; Marusic, P.; Duval, J.; Sata, Y.; Ellims, A.; et al. Reverse cardiac remodeling after renal denervation: Atrial electrophysiologic and structural changes associated with blood pressure lowering. Heart Rhythm 2015, 12, 982–990. [Google Scholar] [CrossRef]
- Lau, D.H.; Mackenzie, L.; Kelly, D.J.; Psaltis, P.J.; Brooks, A.G.; Worthington, M.; Rajendram, A.; Kelly, D.R.; Zhang, Y.; Kuklik, P.; et al. Hypertension and atrial fibrillation: Evidence of progressive atrial remodeling with electrostructural correlate in a conscious chronically instrumented ovine model. Heart Rhythm 2010, 7, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Dorr, O.; Liebetrau, C.; Mollmann, H.; Gaede, L.; Troidl, C.; Morczeck, K.; Wiebe, J.; Hoffmann, J.; Voss, S.; Bauer, T.; et al. Influence of Renal Sympathetic Denervation on Cardiac Extracellular Matrix Turnover and Cardiac Fibrosis. Am. J. Hypertens. 2015, 28, 1285–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krum, H.; Schlaich, M.P.; Sobotka, P.A.; Bohm, M.; Mahfoud, F.; Rocha-Singh, K.; Katholi, R.; Esler, M.D. Percutaneous renal denervation in patients with treatment-resistant hypertension: final 3-year report of the Symplicity HTN-1 study. Lancet 2014, 383, 622–629. [Google Scholar] [CrossRef]
- Mahfoud, F.; Luscher, T.F.; Andersson, B.; Baumgartner, I.; Cifkova, R.; Dimario, C.; Doevendans, P.; Fagard, R.; Fajadet, J.; Komajda, M.; et al. Expert consensus document from the European Society of Cardiology on catheter-based renal denervation. Eur Heart J. 2013, 34, 2149–2157. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.Y.; Chen, C.; Huo, J.Y.; Lu, D.S.; Jiang, Z.X.; Geng, J.; Xu, H.; Shan, Q. Comparison between renal denervation and metoprolol on the susceptibility of ventricular arrhythmias in rats with myocardial infarction. Sci. Rep.-UK 2018, 8, 10206. [Google Scholar] [CrossRef]
- Huang, B.; Yu, L.; Scherlag, B.J.; Wang, S.; He, B.; Yang, K.; Liao, K.; Lu, Z.; He, W.; Zhang, L.; et al. Left renal nerves stimulation facilitates ischemia-induced ventricular arrhythmia by increasing nerve activity of left stellate ganglion. J. Cardiovasc. Electrophysiol. 2014, 25, 1249–1256. [Google Scholar] [CrossRef]
- Linz, D.; Wirth, K.; Ukena, C.; Mahfoud, F.; Poss, J.; Linz, B.; Böhm, M.; Neuberger, H.R. Renal denervation suppresses ventricular arrhythmias during acute ventricular ischemia in pigs. Heart Rhythm 2013, 10, 1525–1530. [Google Scholar] [CrossRef]
- Jackson, N.; Gizurarson, S.; Azam, M.A.; King, B.; Ramadeen, A.; Zamiri, N.; Porta-Sánchez, A.; Al-Hesayen, A.; Graham, J.; Kusha, M.; et al. Effects of Renal Artery Denervation on Ventricular Arrhythmias in a Postinfarct Model. Circ. Cardiovasc. Interv. 2017, 10, e004172. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.N.; Chang, S.H.; Yu, C.C.; Wu, C.K.; Lai, L.P.; Chiang, F.T.; Hwang, J.J.; Lin, J.L.; Tsai, C.T. Renal Denervation Decreases Susceptibility to Arrhythmogenic Cardiac Alternans and Ventricular Arrhythmia in a Rat Model of Post-Myocardial Infarction Heart Failure. JACC Basic Transl. Sci. 2017, 2, 184–193. [Google Scholar] [CrossRef]
- Guo, Z.; Zhao, Q.; Deng, H.; Tang, Y.; Wang, X.; Dai, Z.; Xiao, J.; Wan, P.; Wang, X.; Huang, H.; et al. Renal sympathetic denervation attenuates the ventricular substrate and electrophysiological remodeling in dogs with pacing-induced heart failure. Int. J. Cardiol. 2014, 175, 185–186. [Google Scholar] [CrossRef]
- Dai, Z.; Yu, S.; Zhao, Q.; Meng, Y.; He, H.; Tang, Y.; Wang, X.; Xiao, J.; Wang, X.; Huang, C. Renal sympathetic denervation suppresses ventricular substrate remodelling in a canine high-rate pacing model. EuroIntervention 2014, 10, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; Denner, A.; Illing, S.; Hohl, M.; Ukena, C.; Mahfoud, F.; Ewen, S.; Reil, J.C.; Wirth, K.; Böhm, M. Impact of obstructive and central apneas on ventricular repolarisation: Lessons learned from studies in man and pigs. Clin. Res. Cardiol. 2016, 105, 639–647. [Google Scholar] [CrossRef]
- Yu, L.; Huang, B.; Zhou, X.; Wang, S.; Wang, Z.; Wang, M.; Li, X.; Zhou, L.; Meng, G.; Yuan, S.; et al. Renal sympathetic stimulation and ablation affect ventricular arrhythmia by modulating autonomic activity in a cesium-induced long QT canine model. Heart Rhythm 2017, 14, 912–919. [Google Scholar] [CrossRef]
- Linz, D.; Hohl, M.; Elliott, A.D.; Lau, D.H.; Mahfoud, F.; Esler, M.D.; Sanders, P.; Böhm, M. Modulation of renal sympathetic innervation: Recent insights beyond blood pressure control. Clin. Auton. Res. 2018, 28, 375–384. [Google Scholar] [CrossRef]
- Ceia, F.; Fonseca, C.; Mota, T.; Morais, H.; Matias, F.; de Sousa, A.; Oliveira, A.; EPICA Investigators. Prevalence of chronic heart failure in Southwestern Europe: The EPICA study. Eur. J. Heart Fail. 2002, 4, 531–539. [Google Scholar] [CrossRef]
- Stevens, T.L.; Rasmussen, T.E.; Wei, C.M.; Kinoshita, M.; Matsuda, Y.; Burnett, J.C., Jr. Renal role of the endogenous natriuretic peptide system in acute congestive heart failure. J. Card. Fail. 1996, 2, 119–125. [Google Scholar] [CrossRef]
- da Silva, P.M.; Aguiar, C. Sacubitril/valsartan: An important piece in the therapeutic puzzle of heart failure. Rev. Port. Cardiol. 2017, 36, 655–668. [Google Scholar] [Green Version]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Bohm, M.; Ewen, S.; Wolf, M. Renal Denervation Halts Left Ventricular Remodeling and Dysfunction in Heart Failure: New Shores Ahead. J. Am. Coll. Cardiol. 2018, 72, 2622–2624. [Google Scholar] [CrossRef]
- Cohn, J.N.; Levine, T.B.; Olivari, M.T.; Garberg, V.; Lura, D.; Francis, G.S.; Simon, A.B.; Rector, T. Plasma Norepinephrine as a Guide to Prognosis in Patients with Chronic Congestive Heart-Failure. N. Engl. J. Med. 1984, 311, 819–823. [Google Scholar] [CrossRef]
- Hasking, G.J.; Esler, M.D.; Jennings, G.L.; Burton, D.; Johns, J.A.; Korner, P.I. Norepinephrine Spillover to Plasma in Patients with Congestive-Heart-Failure—Evidence of Increased Overall and Cardiorenal Sympathetic Nervous Activity. Circulation 1986, 73, 615–621. [Google Scholar] [CrossRef]
- Petersson, M.; Friberg, P.; Eisenhofer, G.; Lambert, G.; Rundqvist, B. Long-term outcome in relation to renal sympathetic activity in patients with chronic heart failure. Eur. Heart J. 2005, 26, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Ukena, C.; Bauer, A.; Mahfoud, F.; Schreieck, J.; Neuberger, H.R.; Eick, C.; Sobotka, P.A.; Gawaz, M.; Böhm, M. Renal sympathetic denervation for treatment of electrical storm: First-in-man experience. Clin. Res. Cardiol. 2012, 101, 63–67. [Google Scholar] [CrossRef]
- Armaganijan, L.V.; Staico, R.; Moreira, D.A.; Lopes, R.D.; Medeiros, P.T.; Habib, R.; Melo Neto, J.; Katz, M.; Armaganijan, D.; Sousa, A.G.; et al. 6-Month Outcomes in Patients with Implantable Cardioverter-Defibrillators Undergoing Renal Sympathetic Denervation for the Treatment of Refractory Ventricular Arrhythmias. JACC Cardiovasc. Interv. 2015, 8, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Remo, B.F.; Preminger, M.; Bradfield, J.; Mittal, S.; Boyle, N.; Gupta, A.; Shivkumar, K.; Steinberg, J.S.; Dickfeld, T. Safety and efficacy of renal denervation as a novel treatment of ventricular tachycardia storm in patients with cardiomyopathy. Heart Rhythm 2014, 11, 541–546. [Google Scholar] [CrossRef]
- Ukena, C.; Mahfoud, F.; Ewen, S.; Bollmann, A.; Hindricks, G.; Hoffmann, B.A.; Linz, D.; Musat, D.; Pavlicek, V.; Scholz, E.; et al. Renal denervation for treatment of ventricular arrhythmias: Data from an International Multicenter Registry. Clin. Res. Cardiol. 2016, 105, 873–879. [Google Scholar] [CrossRef]
- Hopper, I.; Gronda, E.; Hoppe, U.C.; Rundqvist, B.; Marwick, T.H.; Shetty, S.; Hayward, C.; Lambert, T.; Hering, D.; Esler, M.; et al. Sympathetic Response and Outcomes Following Renal Denervation in Patients with Chronic Heart Failure: 12-Month Outcomes from the Symplicity HF Feasibility Study. J. Card. Fail. 2017, 23, 702–707. [Google Scholar] [CrossRef]
- Kiuchi, M.G.; Chen, S.J.; Paz, L.M.R.; Prerfellner, H. Renal sympathetic denervation guided by renal nerve stimulation to treat ventricular arrhythmia in CKD patients with ICD. Oncotarget 2017, 8, 37296–37307. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Kanazawa, H.; Aizawa, Y.; Ardell, J.L.; Shivkumar, K. Cardiac innervation and sudden cardiac death. Circ. Res. 2015, 116, 2005–2019. [Google Scholar] [CrossRef]
- Zipes, D.P. Antiarrhythmic therapy in 2014: Contemporary approaches to treating arrhythmias. Nat. Rev. Cardiol. 2015, 12, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.J.; Zipes, D.P. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ. Res. 2014, 114, 1004–1021. [Google Scholar] [CrossRef] [PubMed]
- Ardell, J.L.; Andresen, M.C.; Armour, J.A.; Billman, G.E.; Chen, P.S.; Foreman, R.D.; Herring, N.; O’Leary, D.S.; Sabbah, H.N.; Schultz, H.D.; et al. Translational neurocardiology: Preclinical models and cardioneural integrative aspects. J. Physiol. 2016, 594, 3877–3909. [Google Scholar] [CrossRef] [PubMed]
- Ng, G.A. Neuro-cardiac interaction in malignant ventricular arrhythmia and sudden cardiac death. Auton. Neurosci. 2016, 199, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Franciosi, S.; Perry, F.K.G.; Roston, T.M.; Armstrong, K.R.; Claydon, V.E.; Sanatani, S. The role of the autonomic nervous system in arrhythmias and sudden cardiac death. Auton. Neurosci. 2017, 205, 1–11. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Progressive damage after brain and spinal cord injury: Pathomechanisms and treatment strategies. Prog. Brain Res. 2007, 161, 125–141. [Google Scholar] [PubMed]
- Sripairojthikoon, W.; Wyss, J.M. Cells of origin of the sympathetic renal innervation in rat. Am. J. Physiol. 1987, 252 (Pt 2), F957–F963. [Google Scholar] [CrossRef]
- Gattone, V.H., 2nd; Marfurt, C.F.; Dallie, S. Extrinsic innervation of the rat kidney: A retrograde tracing study. Am. J. Physiol. 1986, 250 (Pt 2), F189–F196. [Google Scholar] [CrossRef]
- Ferguson, M.; Ryan, G.B.; Bell, C. Localization of sympathetic and sensory neurons innervating the rat kidney. J. Auton. Nerv. Syst. 1986, 16, 279–288. [Google Scholar] [CrossRef]
- Pilowsky, P.; Llewellynsmith, I.J.; Minson, J.; Chalmers, J. Sympathetic Preganglionic Neurons in Rabbit Spinal-Cord That Project to the Stellate or the Superior Cervical-Ganglion. Brain Res. 1992, 577, 181–188. [Google Scholar] [CrossRef]
- Campese, V.M.; Kogosov, E. Renal afferent denervation prevents hypertension in rats with chronic renal failure. Hypertension 1995, 25, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.S.; Wessendorf, M.W.; Loewy, A.D. Transneuronal labeling of CNS neuropeptide and monoamine neurons after pseudorabies virus injections into the stellate ganglion. Brain Res. 1995, 683, 1–24. [Google Scholar] [CrossRef]
- Leimbach, W.N., Jr.; Wallin, B.G.; Victor, R.G.; Aylward, P.E.; Sundlof, G.; Mark, A.L. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 1986, 73, 913–919. [Google Scholar] [CrossRef]
- Feldman, J.L.; Del Negro, C.A.; Gray, P.A. Understanding the Rhythm of Breathing: So Near, Yet So Far. Annu. Rev. Physiol. 2013, 75, 423–452. [Google Scholar] [CrossRef]
- Guyenet, P.G.; Stornetta, R.L.; Bochorishvili, G.; Depuy, S.D.; Burke, P.G.; Abbott, S.B. C1 neurons: The body’s EMTs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R187–R204. [Google Scholar] [CrossRef]
- La Rovere, M.T.; Specchia, G.; Mortara, A.; Schwartz, P.J. Baroreflex sensitivity, clinical correlates, and cardiovascular mortality among patients with a first myocardial infarction. A prospective study. Circulation 1988, 78, 816–824. [Google Scholar] [CrossRef]
- La Rovere, M.T.; Bigger, J.T., Jr.; Marcus, F.I.; Mortara, A.; Schwartz, P.J. Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators. Lancet 1998, 351, 478–484. [Google Scholar] [CrossRef]
- Billman, G.E.; Schwartz, P.J.; Stone, H.L. The effects of daily exercise on susceptibility to sudden cardiac death. Circulation 1984, 69, 1182–1189. [Google Scholar] [CrossRef]
- Schmidt, G.; Malik, M.; Barthel, P.; Schneider, R.; Ulm, K.; Rolnitzky, L.; Camm, A.J.; Bigger, J.T., Jr.; Schömig, A. Heart-rate turbulence after ventricular premature beats as a predictor of mortality after acute myocardial infarction. Lancet 1999, 353, 1390–1396. [Google Scholar] [CrossRef] [Green Version]
- Ghuran, A.; Reid, F.; La Rovere, M.T.; Schmidt, G.; Bigger, J.T., Jr.; Camm, A.J.; Schwartz, P.J.; Malik, M.; ATRAMI Investigators. Heart rate turbulence-based predictors of fatal and nonfatal cardiac arrest (The Autonomic Tone and Reflexes After Myocardial Infarction substudy). Am. J. Cardiol. 2002, 89, 184–190. [Google Scholar] [CrossRef]
- Bauer, A.; Kantelhardt, J.W.; Barthel, P.; Schneider, R.; Makikallio, T.; Ulm, K.; Hnatkova, K.; Schömig, A.; Huikuri, H.; Bunde, A.; et al. Deceleration capacity of heart rate as a predictor of mortality after myocardial infarction: cohort study. Lancet 2006, 367, 1674–1681. [Google Scholar] [CrossRef]
- Ikeda, T.; Yoshino, H.; Sugi, K.; Tanno, K.; Shimizu, H.; Watanabe, J.; Kasamaki, Y.; Yoshida, A.; Kato, T. Predictive value of microvolt T-wave alternans for sudden cardiac death in patients with preserved cardiac function after acute myocardial infarction: Results of a collaborative cohort study. J. Am. Coll. Cardiol. 2006, 48, 2268–2274. [Google Scholar] [CrossRef]
- Lombardi, F.; Malliani, A.; Pagani, M.; Cerutti, S. Heart rate variability and its sympatho-vagal modulation. Cardiovasc. Res. 1996, 32, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.W.; Kadish, A.H.; Parker, M.A.; Goldberger, J.J. Effect of physiologic and pharmacologic adrenergic stimulation on heart rate variability. J. Am. Coll. Cardiol. 1994, 24, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Vanderlaan, R.D.; Conway, J.; Manlhiot, C.; McCrindle, B.W.; Dipchand, A.I. Enhanced exercise performance and survival associated with evidence of autonomic reinnervation in pediatric heart transplant recipients. Am. J. Transplant. 2012, 12, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.M.; Leslie, S.J. Risk factors for sudden cardiac death to determine high risk patients in specific patient populations that may benefit from a wearable defibrillator. World J. Cardiol. 2019, 11, 103–119. [Google Scholar] [CrossRef]
- Barold, S.S.; Ilercil, A.; Herweg, B. Echocardiographic optimization of the atrioventricular and interventricular intervals during cardiac resynchronization. Europace 2008, 10 (Suppl 3), iii88–iii95. [Google Scholar] [CrossRef] [Green Version]
- Abraham, W.T.; Fisher, W.G.; Smith, A.L.; Delurgio, D.B.; Leon, A.R.; Loh, E.; Kocovic, D.Z.; Packer, M.; Clavell, A.L.; Hayes, D.L.; et al. Cardiac resynchronization in chronic heart failure. N. Engl. J. Med. 2002, 346, 1845–1853. [Google Scholar] [CrossRef]
- Moss, A.J.; Hall, W.J.; Cannom, D.S.; Klein, H.; Brown, M.W.; Daubert, J.P.; Estes, N.A., III; Foster, E.; Greenberg, H.; Higgins, S.L.; et al. Cardiac-resynchronization therapy for the prevention of heart-failure events. N. Engl. J. Med. 2009, 361, 1329–1338. [Google Scholar] [CrossRef]
- Vernooy, K.; Verbeek, X.A.; Peschar, M.; Crijns, H.J.; Arts, T.; Cornelussen, R.N.; Prinzen, F.W. Left bundle branch block induces ventricular remodeling and functional septal hypoperfusion. Eur. Heart J. 2005, 26, 91–98. [Google Scholar] [CrossRef]
- Chen, L.S.; Zhou, S.; Fishbein, M.C.; Chen, P.S. New perspectives on the role of autonomic nervous system in the genesis of arrhythmias. J. Cardiovasc. Electrophysiol. 2007, 18, 123–127. [Google Scholar] [CrossRef]
- Goshima, Y.; Sasaki, Y.; Yamashita, N.; Nakamura, F. Class 3 semaphorins as a therapeutic target. Expert Opin. Ther. Targets 2012, 16, 933–944. [Google Scholar] [CrossRef]
- Kimura, K.; Ieda, M.; Fukuda, K. Development, maturation, and transdifferentiation of cardiac sympathetic nerves. Circ. Res. 2012, 110, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Shusterman, V.; Aysin, B.; Gottipaty, V.; Weiss, R.; Brode, S.; Schwartzman, D.; Anderson, K.P. Autonomic nervous system activity and the spontaneous initiation of ventricular tachycardia. ESVEM Investigators. Electrophysiologic Study Versus Electrocardiographic Monitoring Trial. J. Am. Coll. Cardiol. 1998, 32, 1891–1899. [Google Scholar] [CrossRef]
- Zhou, S.; Jung, B.C.; Tan, A.Y.; Trang, V.Q.; Gholmieh, G.; Han, S.W.; Lin, S.F.; Fishbein, M.C.; Chen, P.S.; Chen, L.S. Spontaneous stellate ganglion nerve activity and ventricular arrhythmia in a canine model of sudden death. Heart Rhythm. 2008, 5, 131–139. [Google Scholar] [CrossRef]
- Cao, J.M.; Fishbein, M.C.; Han, J.B.; Lai, W.W.; Lai, A.C.; Wu, T.J.; Czer, L.; Wolf, P.L.; Denton, T.A.; Shintaku, I.P.; et al. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation 2000, 101, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.B.; Wu, C.C.; Lu, L.S.; Su, M.J.; Lin, C.W.; Lin, S.F.; Chen, L.S.; Fishbein, M.C.; Chen, P.S.; Lee, Y.T. Sympathetic nerve sprouting, electrical remodeling, and increased vulnerability to ventricular fibrillation in hypercholesterolemic rabbits. Circ. Res. 2003, 92, 1145–1152. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Univariate | Multivariate | ||
|---|---|---|---|---|
| HR (95% CI) | p-Value | HR (95% CI) | p-Value | |
| Denervated myocardium | 1.057 (1.023–1.092) | 0.001 | 1.069 (1.023–1.117) | 0.003 |
| Viable denervated myocardium | 1.067 (1.008–1.130) | 0.025 | ||
| Infarcted myocardium | 1.029 (0.990–1.069) | 0.15 | ||
| Hibernating myocardium | 0.950 (0.822–1.099) | 0.49 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiuchi, M.G.; Nolde, J.M.; Villacorta, H.; Carnagarin, R.; Chan, J.J.S.-Y.; Lugo-Gavidia, L.M.; Ho, J.K.; Matthews, V.B.; Dwivedi, G.; Schlaich, M.P. New Approaches in the Management of Sudden Cardiac Death in Patients with Heart Failure—Targeting the Sympathetic Nervous System. Int. J. Mol. Sci. 2019, 20, 2430. https://doi.org/10.3390/ijms20102430
Kiuchi MG, Nolde JM, Villacorta H, Carnagarin R, Chan JJS-Y, Lugo-Gavidia LM, Ho JK, Matthews VB, Dwivedi G, Schlaich MP. New Approaches in the Management of Sudden Cardiac Death in Patients with Heart Failure—Targeting the Sympathetic Nervous System. International Journal of Molecular Sciences. 2019; 20(10):2430. https://doi.org/10.3390/ijms20102430
Chicago/Turabian StyleKiuchi, Márcio Galindo, Janis Marc Nolde, Humberto Villacorta, Revathy Carnagarin, Justine Joy Su-Yin Chan, Leslie Marisol Lugo-Gavidia, Jan K. Ho, Vance B. Matthews, Girish Dwivedi, and Markus P. Schlaich. 2019. "New Approaches in the Management of Sudden Cardiac Death in Patients with Heart Failure—Targeting the Sympathetic Nervous System" International Journal of Molecular Sciences 20, no. 10: 2430. https://doi.org/10.3390/ijms20102430