Targeting TGF-β Signaling in Kidney Fibrosis

Abstract
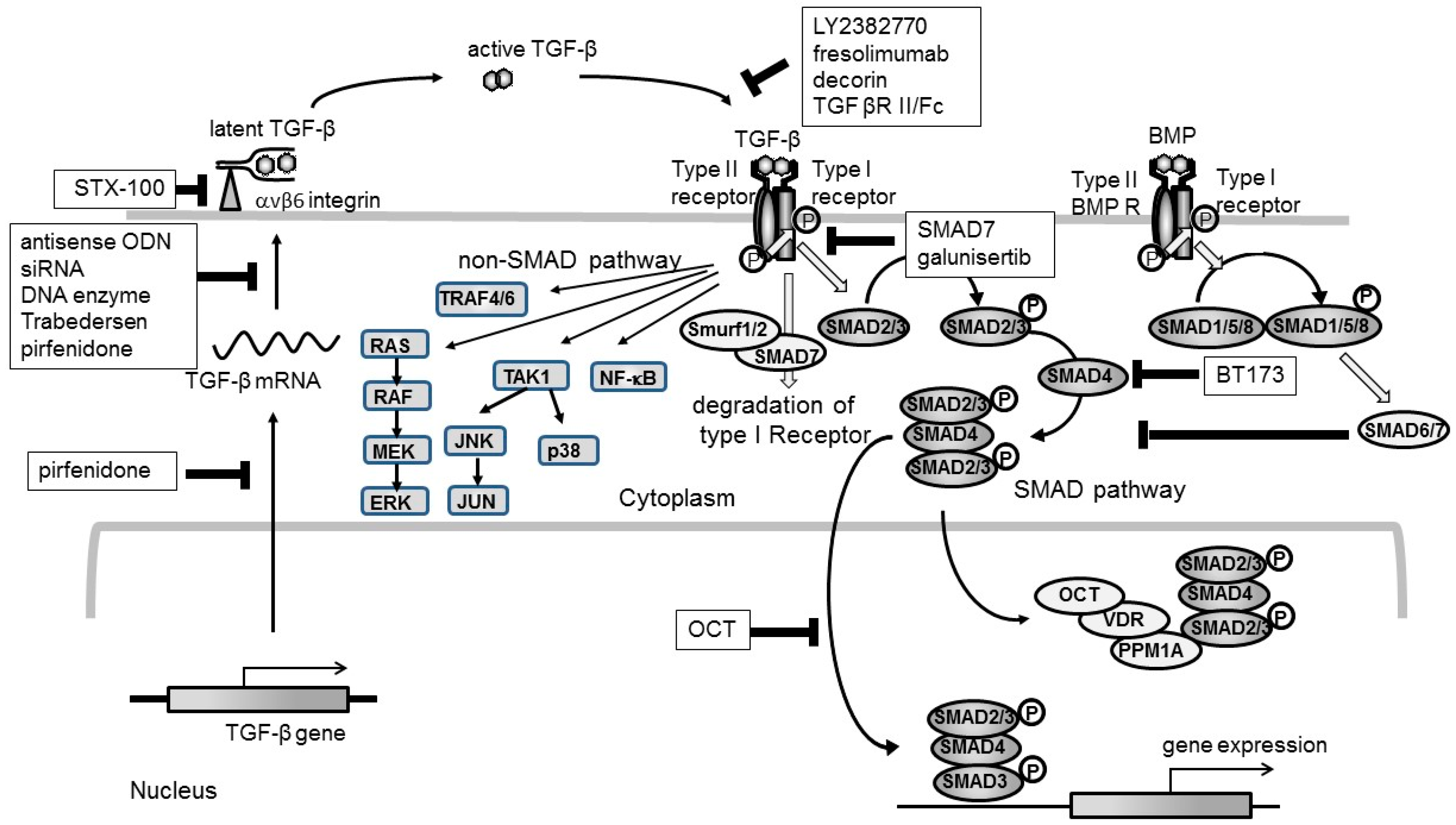
:1. TGF-β as a Target for Kidney Fibrosis
2. Clinical Trials Targeting TGF-β
3. Future Strategy Targeting TGF-β
4. Side Effect of Targeting TGF-β
Funding
Conflicts of Interest
References
- Border, W.; Noble, N. Transforming growth factor in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar]
- Okuda, S.; Languino, L.R.; Ruoslahti, E.; Border, W.A. Elevated expression of transforming growth factor-β and proteoglycan production in experimental glomerulonephritis. Possible role in expansion of the mesangial extracellular matrix. J. Clin. Investig. 1990, 86, 4534–4562. [Google Scholar] [CrossRef] [PubMed]
- Tomooka, S.; Border, W.A.; Marshall, B.C.; Noble, N.A. Glomerular matrix accumulation is linked to inhibition of the plasmin protease system. Kidney Int. 1992, 42, 1462–1469. [Google Scholar] [CrossRef]
- Kagami, S.; Border, W.A.; Ruoslahti, E.; Noble, N.A. Coordinated expression of β1 integrins and transforming growth factor-β-induced matrix proteins in glomerulonephritis. Lab. Investig. 1993, 69, 68–76. [Google Scholar] [PubMed]
- Wu, C.F.; Chiang, W.C.; Lai, C.F.; Chang, F.C.; Chen, Y.T.; Chou, Y.H.; Wu, T.H.; Linn, G.R.; Ling, H.; Wu, K.-D.; et al. Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am. J. Pathol. 2013, 182, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Takemura, T.; Murakami, K.; Okada, M.; Hino, S.; Miyamoto, H.; Maki, S. Transforming growth factor-β protein and mrna in glomeruli in normal and diseased human kidneys. Lab. Investig. 1993, 68, 154–163. [Google Scholar] [PubMed]
- Yamamoto, T.; Watanabe, T.; Ikegaya, N.; Fujigaki, Y.; Matsui, K.; Masaoka, H.; Nagase, M.; Hishida, A. Expression of types I, II, and III TGF-β receptors in human glomerulonephritis. J. Am. Soc. Nephrol. 1998, 9, 2253–2261. [Google Scholar] [PubMed]
- Iwano, M.; Akai, Y.; Fujii, Y.; Dohi, Y.; Matsumura, N.; Dohi, K. Intraglomerular expression of transforming growth factor-β1 (TGF-β1) mrna in patients with glomerulonephritis: Quantitative analysis by competitive polymerase chain reaction. Clin. Exp. Immunol. 1994, 97, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Noble, N.A.; Cohen, A.H.; Nast, C.C.; Hishida, A.; Gold, L.I.; Border, W.A. Expression of transforming growth factor-β isoforms in human glomerular diseases. Kidney Int. 1996, 49, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-β type I receptor through SMAD7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakamura, T.; Noble, N.A.; Ruoslahti, E.; Border, W.A. Expression of transforming growth factor β is elevated in human and experimental diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1993, 90, 1814–1818. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Noble, N.A.; Miller, D.E.; Border, W.A. Sustained expression of TGF-β1 underlies development of progressive kidney fibrosis. Kidney Int. 1994, 45, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ziyadeh, F.N.; Alzahabi, B.; McGowan, T.A.; Kapoor, S.; Kurnik, B.R.; Kurnik, P.B.; Weisberg, L.S. Increased renal production of transforming growth factor-β1 in patients with type II diabetes. Diabetes 1997, 46, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Terrell, T.G.; Working, P.K.; Chow, C.P.; Green, J.D. Pathology of recombinant human transforming growth factor-β1 in rats and rabbits. Int. Rev. Exp. Pathol. 1993, 34, 43–67. [Google Scholar] [PubMed]
- Isaka, Y.; Fujiwara, Y.; Ueda, N.; Kaneda, Y.; Kamada, T.; Imai, E. Glomerulosclerosis induced by in vivo transfection of transforming growth factor-β or platelet-derived growth factor gene into the rat kidney. J. Clin. Investig. 1993, 92, 2597–2601. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.; Factor, V.; Mozes, M.; Nagy, P.; Sanderson, N.; Bottinger, E.; Klotman, P.; Thorgeirsson, S. Transgenic mice with increased plasma levels of TGF-β1 develop progressive renal disease. Lab. Investig. 1996, 74, 991–1003. [Google Scholar] [PubMed]
- Koesters, R.; Kaissling, B.; Lehir, M.; Picard, N.; Theilig, F.; Gebhardt, R.; Glick, A.B.; Hahnel, B.; Hosser, H.; Grone, H.J.; et al. Tubular overexpression of transforming growth factor-β1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am. J. Pathol. 2010, 177, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Border, W.A.; Okuda, S.; Languino, L.R.; Sporn, M.B.; Ruoslahti, E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor-β1. Nature 1990, 346, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, H.; Ito, Y.; Sakamoto, S.; Kawachi, H.; Shimizu, F.; Yuzawa, Y.; Matsuo, S. Effects of anti-TGF-β type II receptor antibody on experimental glomerulonephritis. Kidney Int. 2001, 60, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akagi, Y.; Isaka, Y.; Arai, M.; Kaneko, T.; Takenaka, M.; Moriyama, T.; Kaneda, Y.; Ando, A.; Orita, Y.; Kamada, T.; et al. Inhibition of TGF-β1 expression by antisense oligonucleotides suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1996, 50, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Iglesias-de la Cruz, M.C.; Jim, B.; Hong, S.W.; Isono, M.; Ziyadeh, F.N. Reversibility of established diabetic glomerulopathy by anti-TGF-β antibodies in db/db mice. Biochem. Biophys. Res. Commun. 2003, 300, 16–22. [Google Scholar] [CrossRef]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-De La Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, A.; Chen, J.; Lawrence, C.; Ledbetter, S.; Soslow, R.A.; Stern, J.; Jha, S.; Pigato, J.; Lemer, M.L.; Poppas, D.P.; et al. Antibody to transforming growth factor-β ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 2000, 58, 2301–2313. [Google Scholar] [CrossRef] [PubMed]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-β1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A phase 2, double-blind, placebo-controlled, randomized study of fresolimumab in patients with steroid-resistant primary focal segmental glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Trachtman, H.; Fervenza, F.C.; Gipson, D.S.; Heering, P.; Jayne, D.R.; Peters, H.; Rota, S.; Remuzzi, G.; Rump, L.C.; Sellin, L.K.; et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-β antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011, 79, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- McGaraughty, S.; Davis-Taber, R.A.; Zhu, C.Z.; Cole, T.B.; Nikkel, A.L.; Chhaya, M.; Doyle, K.J.; Olson, L.M.; Preston, G.M.; Grinnell, C.M.; et al. Targeting anti-TGF-β therapy to fibrotic kidneys with a dual specificity antibody approach. J. Am. Soc. Nephrol. 2017, 28, 3616–3626. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on procollagen gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 289, 211–218. [Google Scholar] [PubMed]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Pirfenidone treatment decreases transforming growth factor-β1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am. J. Transplant. 2002, 2, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fukagawa, M.; Kuroda, T.; Hata, S.; Iwasaki, Y.; Nemoto, M.; Shirai, K.; Yamauchi, S.; Margolin, S.B.; Shimizu, F.; et al. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int. 1997, 63, S239–243. [Google Scholar]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Eng. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; et al. Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Tsujie, M.; Isaka, Y.; Ando, Y.; Akagi, Y.; Kaneda, Y.; Ueda, N.; Imai, E.; Hori, M. Gene transfer targeting interstitial fibroblasts by the artificial viral envelope-type hemagglutinating virus of japan liposome method. Kidney Int. 2000, 57, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Isaka, Y.; Tsujie, M.; Rupprecht, H.D.; Akagi, Y.; Ueda, N.; Imai, E.; Hori, M. Introduction of DNA enzyme for Egr-1 into tubulointerstitial fibroblasts by electroporation reduced interstitial α-smooth muscle actin expression and fibrosis in unilateral ureteral obstruction (UUO) rats. Gene Ther. 2002, 9, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaka, Y.; Nakamura, H.; Mizui, M.; Takabatake, Y.; Horio, M.; Kawachi, H.; Shimizu, F.; Imai, E.; Hori, M. Dnazyme for TGF-β suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 2004, 66, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Takabatake, Y.; Isaka, Y.; Mizui, M.; Kawachi, H.; Shimizu, F.; Ito, T.; Hori, M.; Imai, E. Exploring rna interference as a therapeutic strategy for renal disease. Gene Ther. 2005, 12, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y.; Tsujie, M.; Ando, Y.; Nakamura, H.; Kaneda, Y.; Imai, E.; Hori, M. Transforming growth factor-β1 antisense oligodeoxynucleotides block interstitial fibrosis in unilateral ureteral obstruction. Kidney Int. 2000, 58, 1885–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Ogasawara, T.; Tamura, Y.; Saito, T.; Ikeda, T.; Suzuki, N.; Shimosawa, T.; Shibata, S.; Chung, U.I.; Nangaku, M.; et al. Targeting gene expression to specific cells of kidney tubules in vivo, using adenoviral promoter fragments. PLoS ONE 2017, 12, e0168638. [Google Scholar] [CrossRef] [PubMed]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro-oncology 2011, 13, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGF-β activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.J.; Yang, H.; Gaspert, A.; Carlesso, G.; Barty, M.M.; Davidson, J.M.; Sheppard, D.; Fogo, A.B. Transforming growth factor-β-dependent and -independent pathways of induction of tubulointerstitial fibrosis in β6−/− mice. Am. J. Pathol. 2003, 163, 1261–1273. [Google Scholar] [CrossRef]
- Horan, G.S.; Wood, S.; Ona, V.; Li, D.J.; Lukashev, M.E.; Weinreb, P.H.; Simon, K.J.; Hahm, K.; Allaire, N.E.; Rinaldi, N.J.; et al. Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 2008, 177, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Border, W.A.; Huang, Y.; Noble, N.A. TGF-β isoform in renal fibrogenesis. Kidney Int. 2003, 64, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Moustakas, A.; Knaus, P.; Wells, R.G.; Henis, Y.I.; Lodish, H.F. The soluble exoplasmic domain of the type II transforming growth factor (TGF)-β receptor. A heterogeneously glycosylated protein with high affinity and selectivity for TGF-β ligands. J. Biol. Chem. 1995, 270, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Komesli, S.; Vivien, D.; Dutartre, P. Chimeric extracellular domain type II transforming growth factor -β receptor fused to the Fc region of human immunoglobulin as a TGF-β antagonist. Eur. J. Biochem. 1998, 254, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Babelova, A.; Moreth, K.; Xin, C.; Eberhardt, W.; Doller, A.; Pavenstädt, H.; Schaefer, L.; Pfeilschifter, J.; Huwiler, A. Transforming growth factor-β2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-β2 by impeding CTGF expression. Kidney Int. 2009, 76, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaka, Y.; Akagi, Y.; Ando, Y.; Tsujie, M.; Sudo, T.; Ohno, N.; Border, W.A.; Noble, N.A.; Kaneda, Y.; Hori, M.; et al. Gene therapy by transforming growth factor-β receptor-IgG Fc chimera suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1999, 55, 465–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Border, W.A.; Noble, N.A.; Yamamoto, T.; Harper, J.R.; Yamaguchi, Y.; Pierschbacher, M.D.; Ruoslahti, E. Natural inhibitor of transforming growth factor-β protects against scarring in experimental kidney disease. Nature 1992, 360, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y.; Brees, D.K.; Ikegaya, K.; Kaneda, Y.; Imai, E.; Noble, N.A.; Border, W.A. Gene therapy by skeletal muscle expression of decorin prevents fibrotic disease in rat kidney. Nat. Med. 1996, 2, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.-i.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Mulder, K.M. Activation of the mitogen-activated protein kinase pathway by transforming growth factor-β. Methods Mol. Biol. 2000, 142, 125–131. [Google Scholar] [PubMed]
- Kim, S.I.; Kwak, J.H.; Zachariah, M.; He, Y.; Wang, L.; Choi, M.E. TGF-β-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-β1-induced MKK3-p38 MAPK activation and stimulation of type I. collagen. Am. J. Physiol. Renal. Physiol. 2007, 292, F1471–F1478. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-β1/SMAD3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Luangmonkong, T.; Suriguga, S.; Bigaeva, E.; Boersema, M.; Oosterhuis, D.; de Jong, K.P.; Schuppan, D.; Mutsaers, H.A.M.; Olinga, P. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharmacol. 2017, 174, 3107–3117. [Google Scholar] [CrossRef] [PubMed]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. SMAD7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF-β receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. TRAF6 ubiquitinates TGF-β type I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef] [PubMed]
- Gudey, S.K.; Sundar, R.; Mu, Y.; Wallenius, A.; Zang, G.; Bergh, A.; Heldin, C.-H.; Landström, M. TRAF6 stimulates the tumor-promoting effects of TGF-β type I receptor through polyubiquitination and activation of presenilin 1. Sci. Signal. 2014, 7, ra2. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Warrior, R.A. A new Smurf in the village. Dev. Cell. 2001, 1, 441–442. [Google Scholar] [CrossRef]
- Jin, Y.; Ratnam, K.; Chuang, P.Y.; Fan, Y.; Zhong, Y.; Dai, Y.; Mazloom, A.R.; Chen, E.Y.; D’Agati, V.; Xiong, H.; et al. A systems approach identifies HIPK2 as a key regulator of kidney fibrosis. Nat. Med. 2012, 18, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Das, B.; Xiao, W.; Li, Z.; Li, H.; Lee, K.; He, J.C. A novel inhibitor of homeodomain interacting protein kinase 2 mitigates kidney fibrosis through inhibition of the TGF-β1/SMAD3 pathway. J. Am. Soc. Nephrol. 2017, 28, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Duan, X.; Liang, Y.Y.; Su, Y.; Wrighton, K.H.; Long, J.; Hu, M.; Davis, C.M.; Wang, J.; Brunicardi, F.C.; et al. PPM1A functions as a Smad phosphatase to terminate TGF-β signaling. Cell 2016, 166, 1597. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Matsui, I.; Hamano, T.; Fujii, N.; Shimomura, A.; Nakano, C.; Kusunoki, Y.; Takabatake, Y.; Hirata, M.; Nishiyama, A.; et al. Maxacalcitol ameliorates tubulointerstitial fibrosis in obstructed kidneys by recruiting PPM1A/VDR complex to pSMAD3. Lab. Investig. 2012, 92, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chen, H.Y.; Zhong, X.; Chung, A.C.; Lan, H.Y. Diverse roles of TGF-β receptor ii in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 2012, 227, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Target | Disease | Results | Reference |
|---|---|---|---|---|
| LY2382770 | TGF-β1 | diabetic nephropathy | No efficacy on change in SCr, eGFR, and proteinuria | [24] |
| fresolimumab | TGF-β1,2,3 | FSGS | less eGFR decline (not significant) | [25,26] |
| pirfenidone | TGF-β1,2,3 | FSGS | slower eGFR decline, no effect on BP or proteinuria | [30] |
| diabetic nephropathy | Increased eGFR | [32] | ||
| pulmonary fibrosis | reduced disease progression and death | [33] | ||
| STX-100 | αvβ6 integrin | pulmonary fibrosis | Ongoing phase 2 trial (NCT01371305) | |
| galunisertib | Type I receptor kinase | cancer | Ongoing clinical trial on glioblastoma, pancreatic cancer, and so on | |
| trabedersen | TGF-β2 | glioma | Superiority of tumor control and survival over chemotherapy | [40] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. https://doi.org/10.3390/ijms19092532
Isaka Y. Targeting TGF-β Signaling in Kidney Fibrosis. International Journal of Molecular Sciences. 2018; 19(9):2532. https://doi.org/10.3390/ijms19092532
Chicago/Turabian StyleIsaka, Yoshitaka. 2018. "Targeting TGF-β Signaling in Kidney Fibrosis" International Journal of Molecular Sciences 19, no. 9: 2532. https://doi.org/10.3390/ijms19092532
APA StyleIsaka, Y. (2018). Targeting TGF-β Signaling in Kidney Fibrosis. International Journal of Molecular Sciences, 19(9), 2532. https://doi.org/10.3390/ijms19092532