Importance of Hydrophilic Groups on Modulating the Structural, Mechanical, and Interfacial Properties of Bilayers: A Comparative Molecular Dynamics Study of Phosphatidylcholine and Ion Pair Amphiphile Membranes



Abstract
:
1. Introduction
2. Results and Discussion
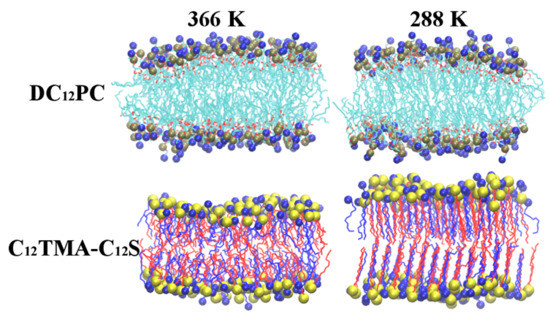
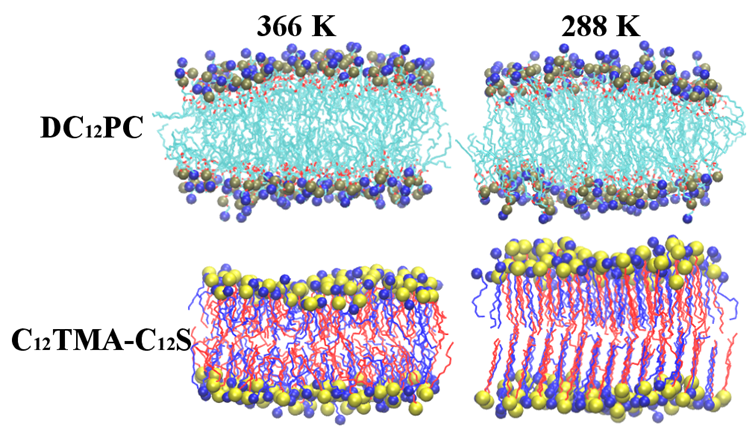
2.1. Bilayer Phases
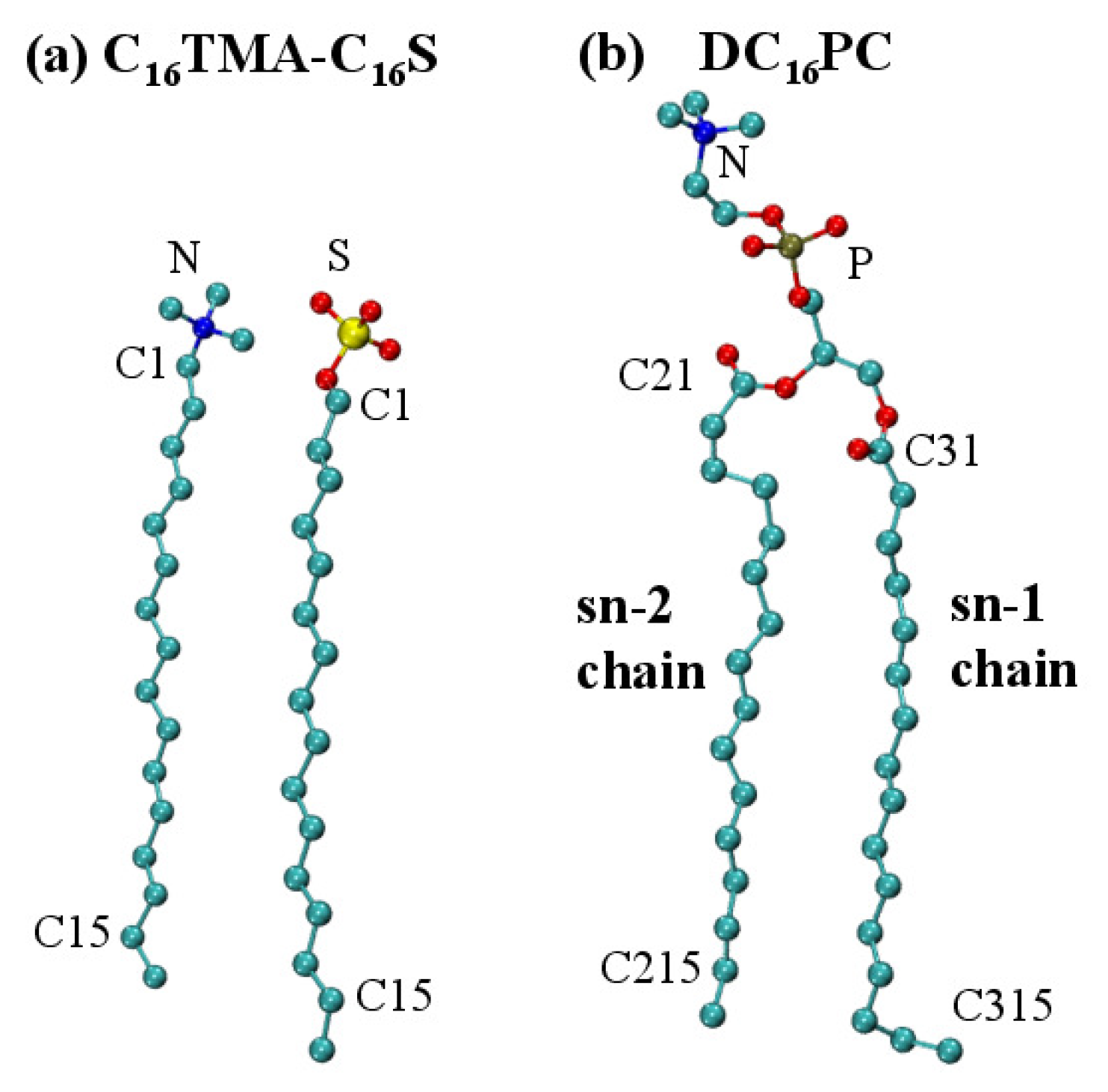
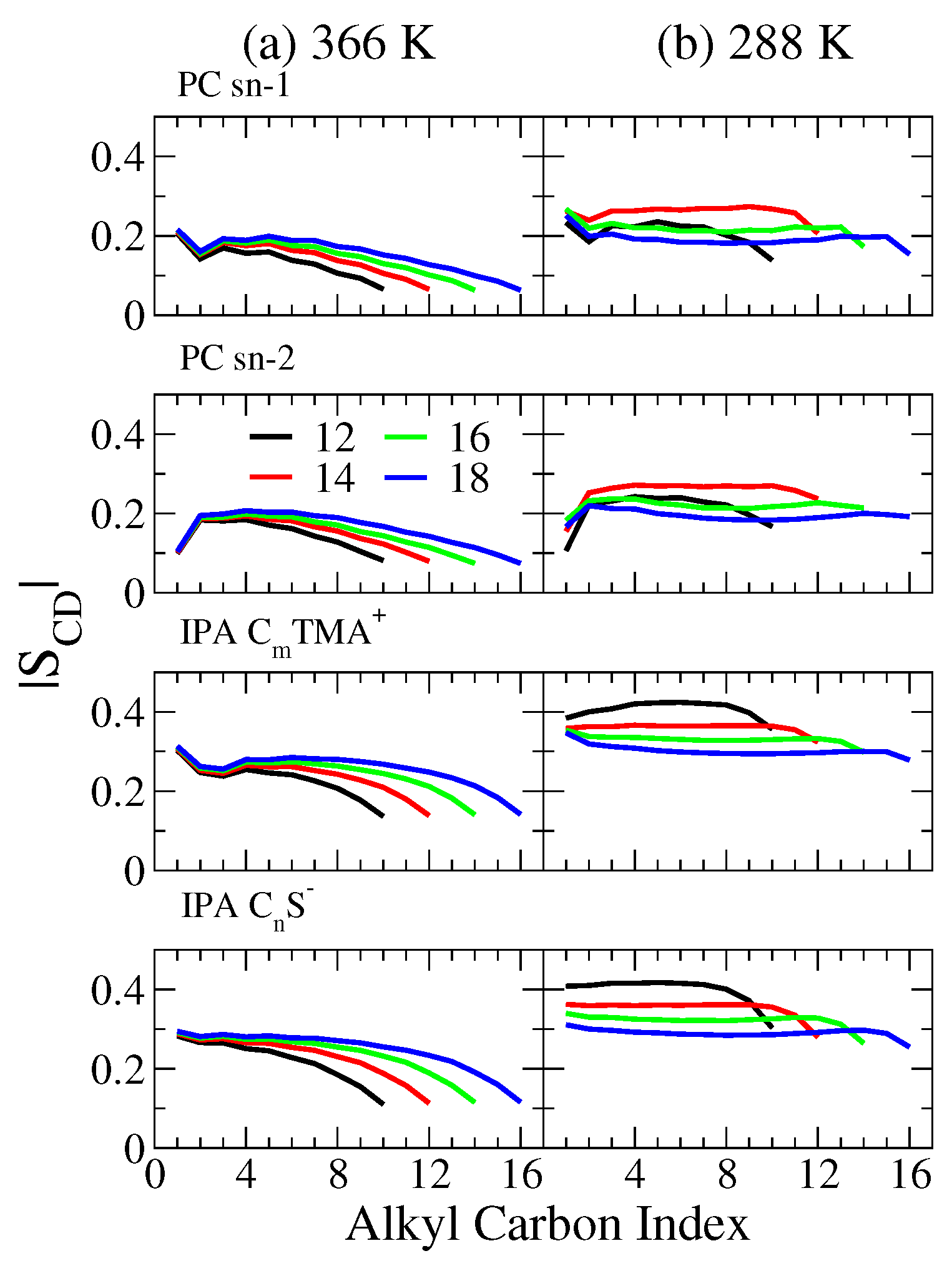
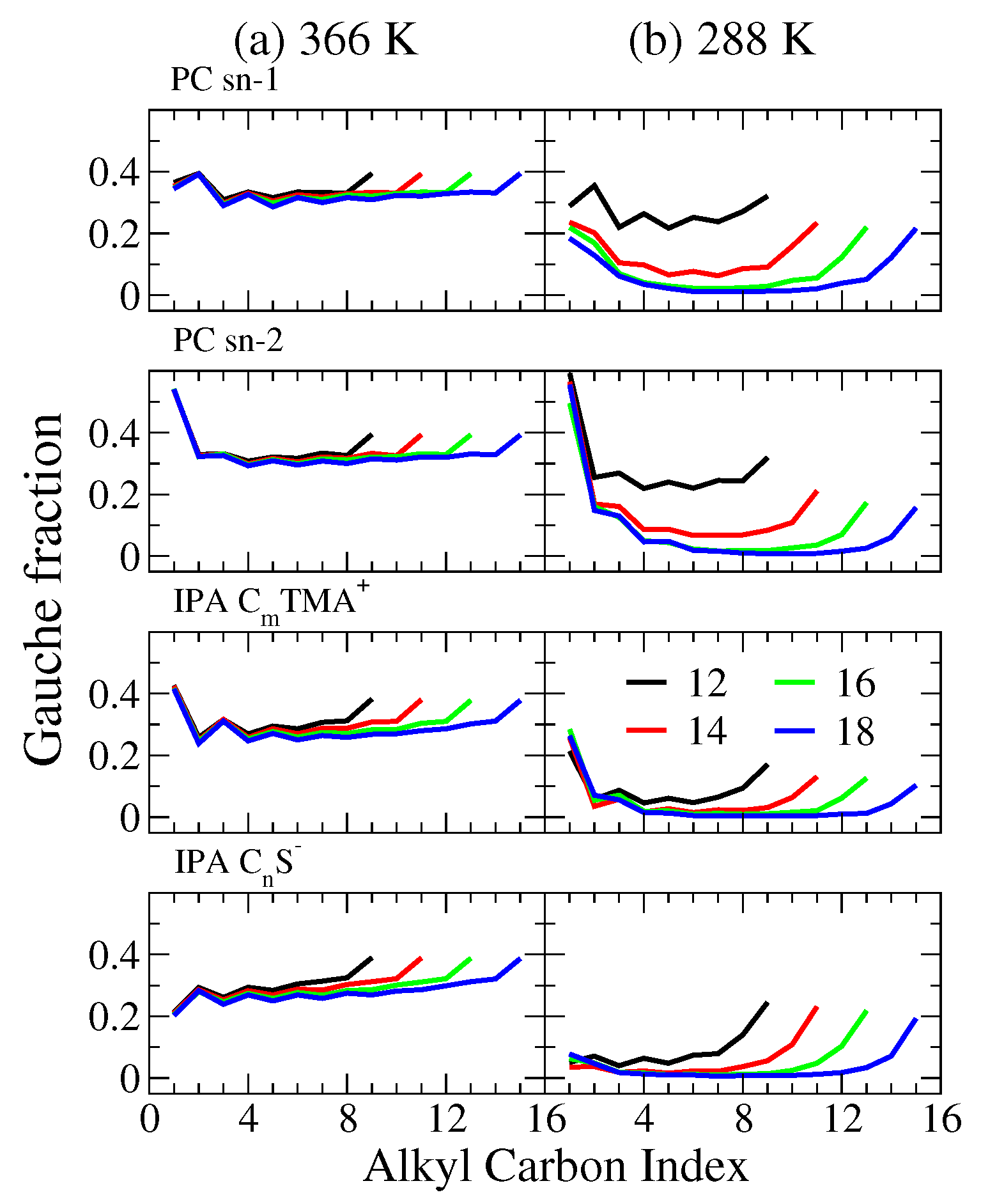
2.2. Bilayer Structures
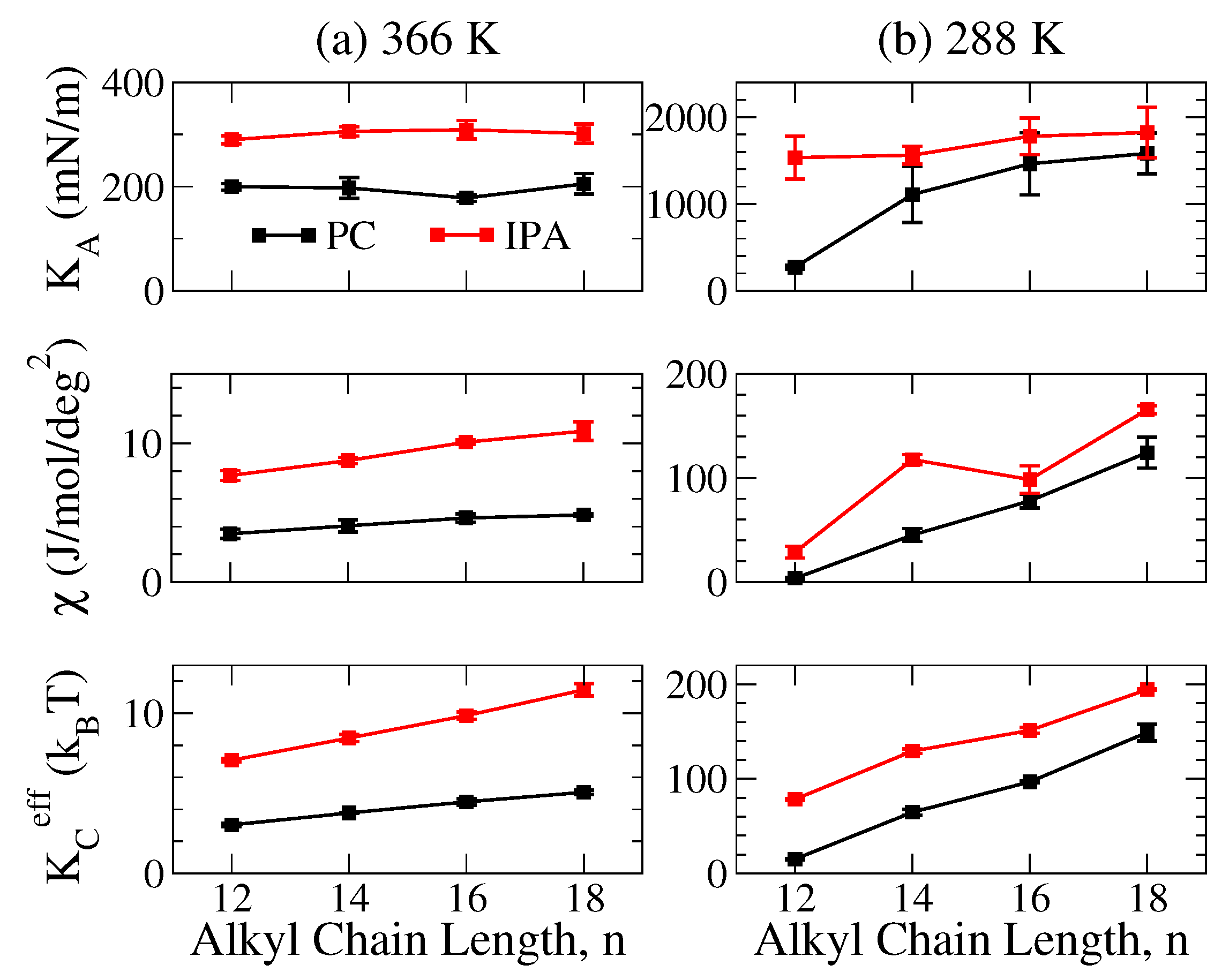
2.3. Mechanical Properties
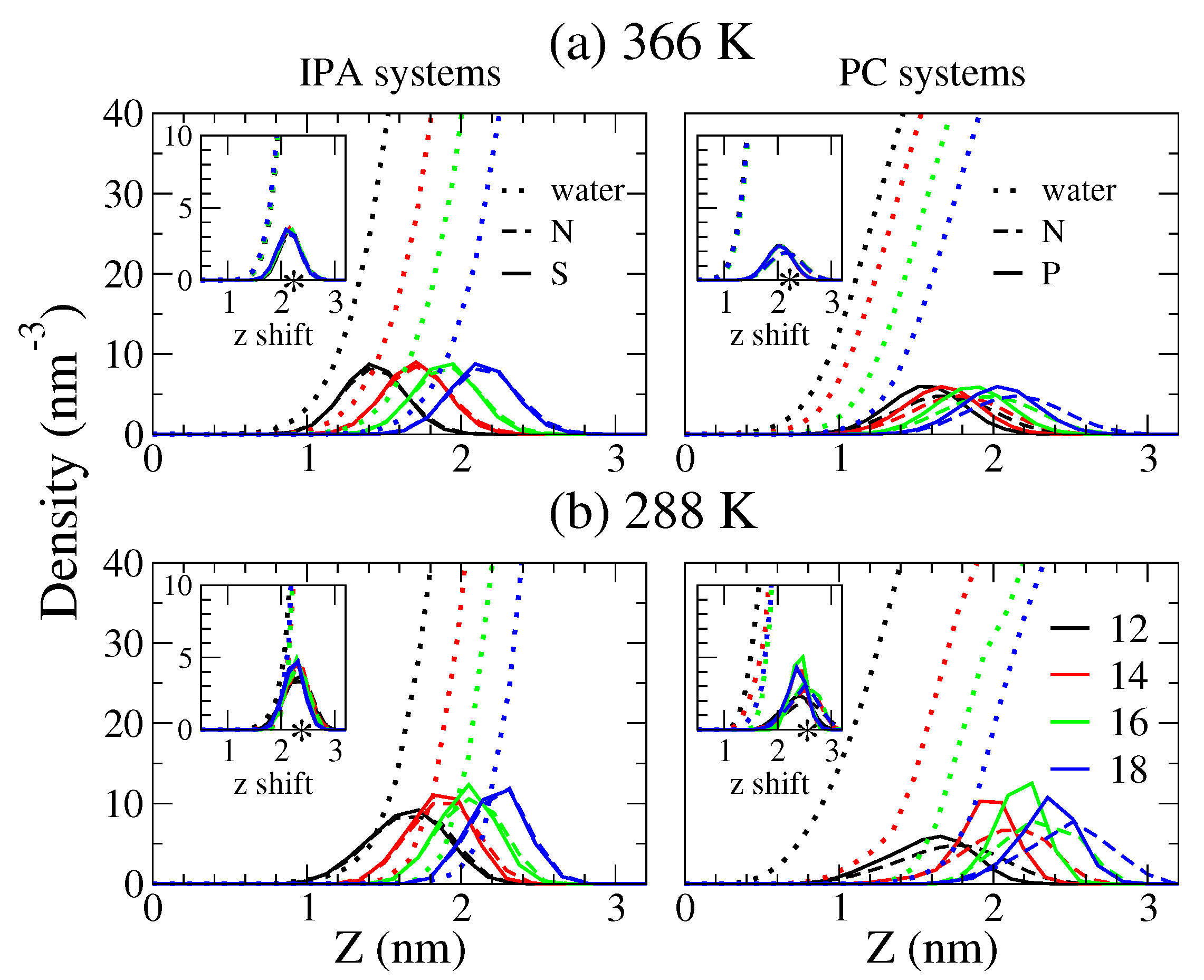
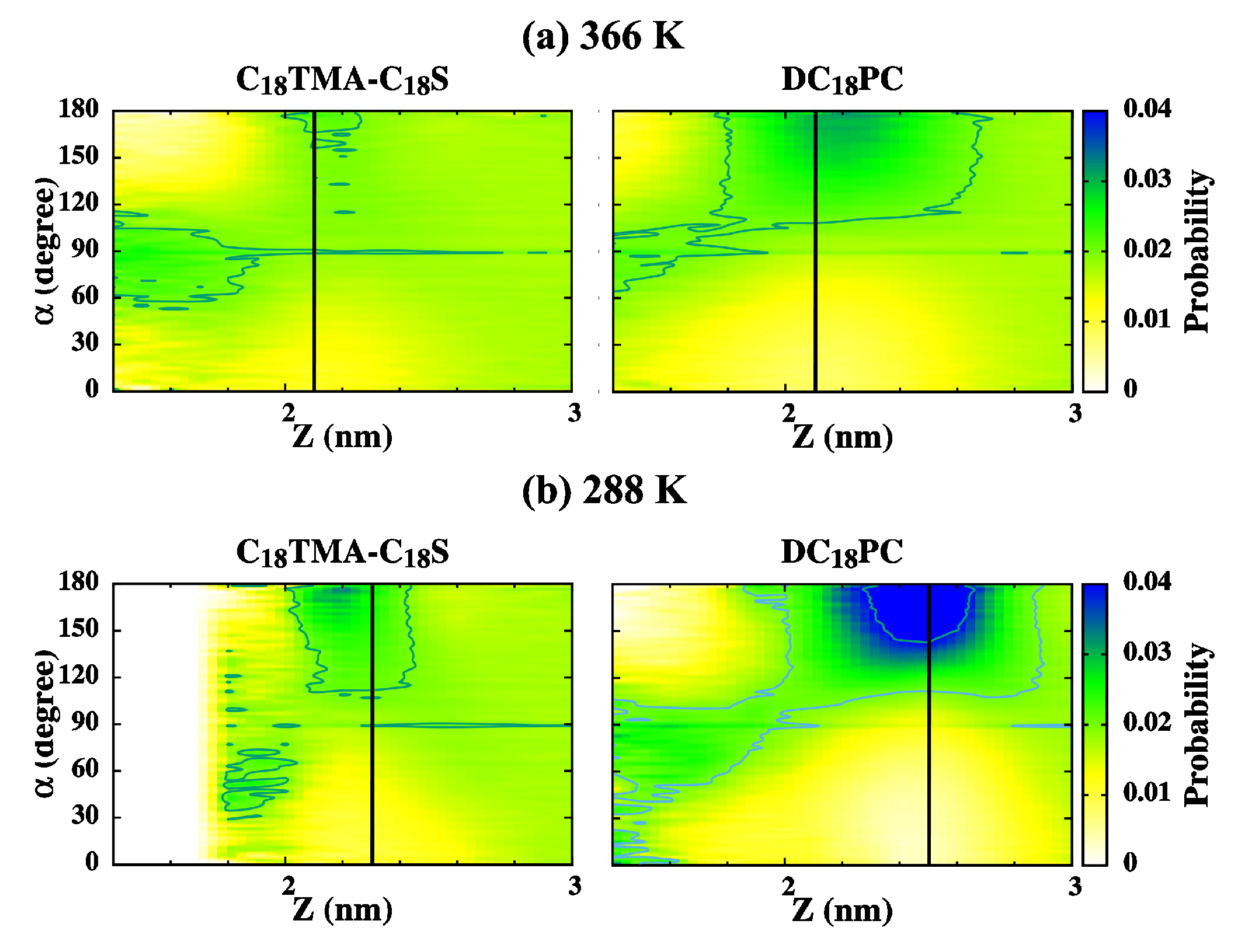
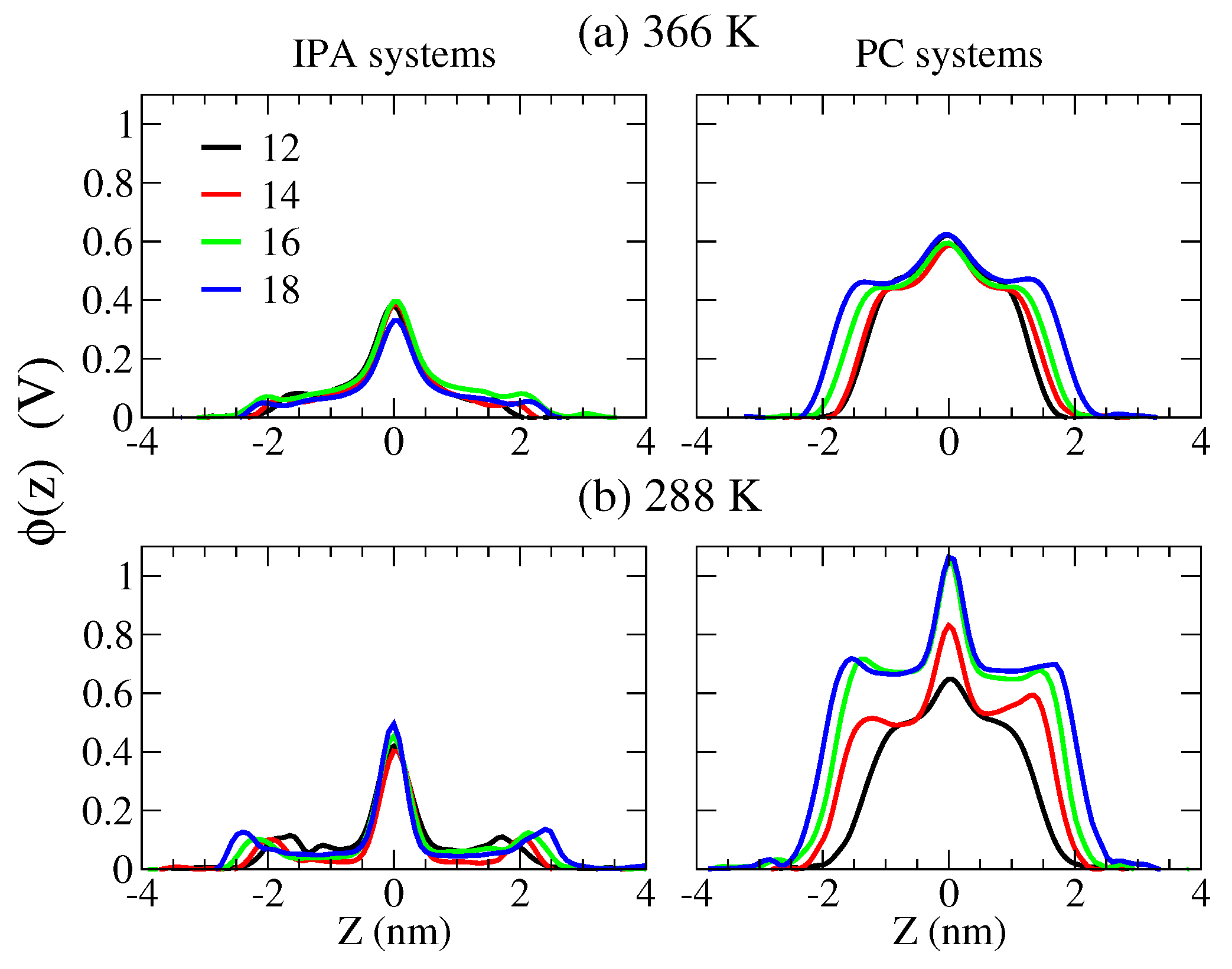
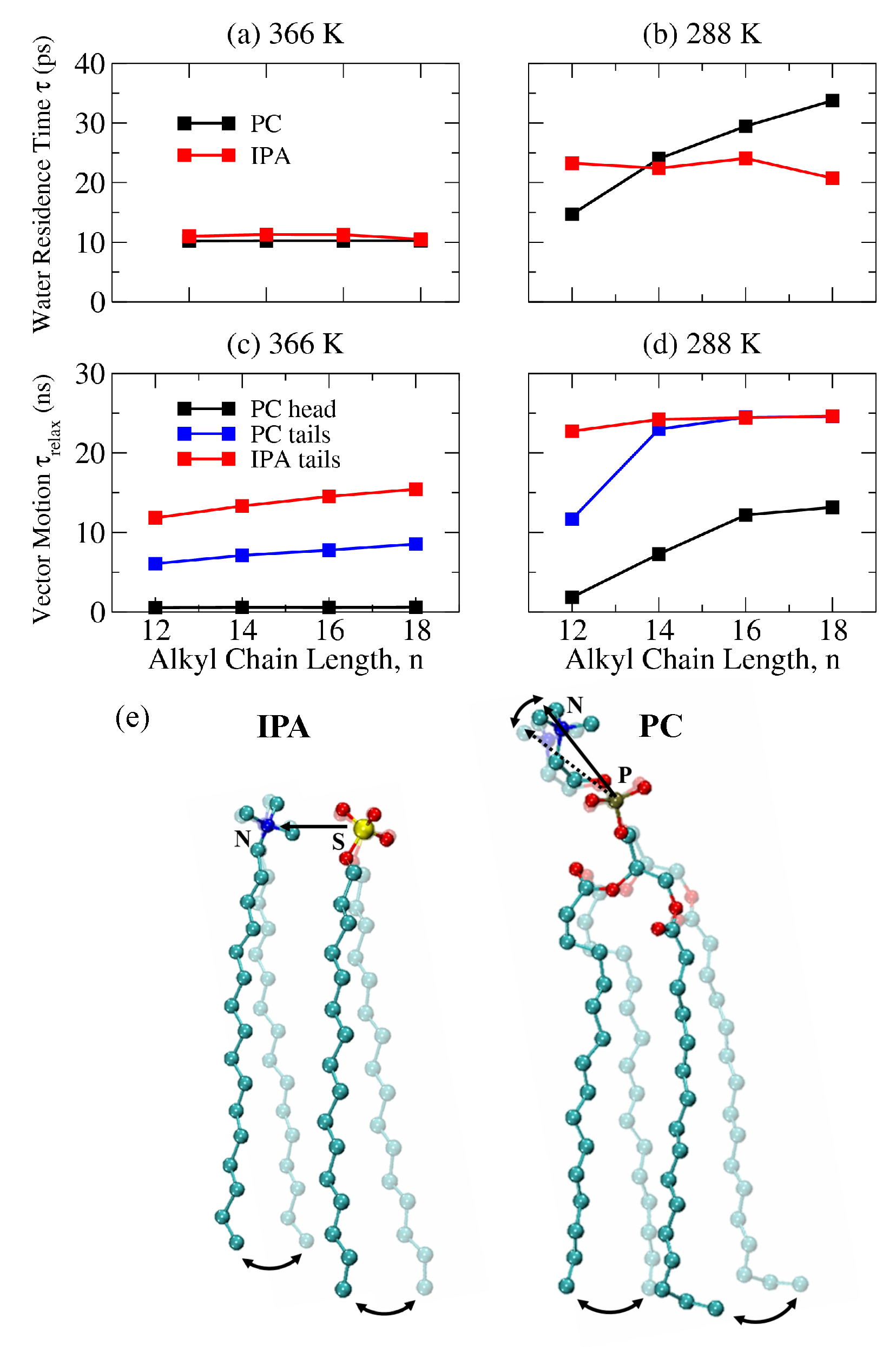
2.4. The Bilayer–Water Interface
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| IPA | ion pair amphiphile |
| PC | phosphatidylcholine |
| MD | molecular dynamics |
| REMD | replica-exchange molecular dynamics |
| DCPC | dilauroyl phosphatidylcholine, DLPC |
| DCPC | dimyristoyl phosphatidylcholine, DMPC |
| DCPC | dipalmitoyl phosphatidylcholine, DPPC |
| DCPC | distearoyl phosphatidylcholine, DSPC |
| CTMA-CS | dodecyltrimethylammonium-dodecylsulfate |
| CTMA-CS | tetradecyltrimthylammonium-tetradecylsulfate |
| CTMA-CS | hexadecyltrimethylammonium-hexadecylsulfate |
| CTMA-CS | octadecyltrimthylammonium-octadecyllsulfate |
References
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G.; Florence, A.T. Lipsomes in Drug Delivery. Drugs 1993, 45, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Kunjappu, J.T. Surfactants and Interfacial Phenomena, 4th ed.; Wiley-Interscience: Hoboken, NJ, USA, 2004. [Google Scholar]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Nanomedicine: Current status and future prospects. FASEB J. 2005, 19, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Lasic, D.D. Novel applications of liposomes. Trends Biotechnol. 1998, 16, 307–321. [Google Scholar] [CrossRef]
- Ahmad, I.; Longenecker, M.; Samue, J.; Allen, T.M. Antibody-targeted Delivery of Doxorubicin Entrapped in Sterically Stabilized Liposomes Can Eradicate Lung Cancer in Mice. Cancer Res. 1993, 53, 1484–1488. [Google Scholar] [PubMed]
- Kaler, E.; Murthy, A.; Rodriguez, B.; Zasadzinski, J. Spontaneous Vesicle Formation in Aqueous Mixtures of Single-Tailed Surfactants. Science 1989, 245, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Tondre, C.; Caillet, C. Properties of the amphiphilic films in mixeded cationic-anionis vesicles. Adv. Colloid Interface Sci. 2001, 93, 115–134. [Google Scholar] [CrossRef]
- Marques, E.F.; Regev, O.; Khan, A.; Lindman, B. Self-organization of double-chained and pseudodouble-chained surfactants: Counterion and geometry effects. Adv. Colloid Interface Sci. 2003, 100, 83–104. [Google Scholar] [CrossRef]
- Kuo, A.T.; Chang, C.H. Recent Strategies in the Development of Catanionic Vesicles. J. Oleo Sci. 2016, 65, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Kawata, K.; Okuda, H.; Regen, S.L. Bilayer-forming ion pair amphiphiles from single-chain surfactants. J. Am. Chem. Soc. 1990, 112, 1635–1637. [Google Scholar] [CrossRef]
- Hirano, K.; Fukuda, H.; Regen, S.L. Polymerizable ion pair amphiphiles. J. Am. Chem. Soc. 1991, 112, 1045–1047. [Google Scholar]
- Yeh, S.J.; Yang, Y.M.; Chang, C.H. Cosolvent Effects on the Stability of Catanionic Vesicles Formed from Ion-Pair Amphiphiles. Langmuir 2005, 21, 6179–6184. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Kuo, A.T.; Lee, C.H.; Yang, Y.M.; Chang, C.H. Fabrication of positively charged catanionic vesicles from ion pair amphiphile with double-chained cationic surfactant. Colloid Polym. Sci. 2014, 292, 589–597. [Google Scholar] [CrossRef]
- Walker, S.A.; Zasadzinski, J.A. Electrostatic Control of Spontaneous Vesicle Aggregation. Langmuir 1997, 13, 5076–5081. [Google Scholar] [CrossRef]
- Kuo, A.T.; Chang, C.H.; Shinoda, W. Molecular Dynamics Study of Catanionic Bilayers Composed of Ion Pair Amphiphile with Double-Tailed Cationic Surfactant. Langmuir 2012, 28, 8156–8164. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.S.; Wen, C.F.; Yang, Y.M. Development of ethosome-like catanionic vesicles for dermal drug delivery. J. Taiwan Inst. Chem. Eng. 2012, 43, 830–838. [Google Scholar] [CrossRef]
- Chiu, C.W.; Chang, C.H.; Yang, Y.M. Gelation of ethosome-like catanionic vesicles by water-soluble polymers: Ethanol and cholesterol effects. Soft Matter 2013, 9, 7628–7636. [Google Scholar] [CrossRef]
- Lee, C.H.; Yang, Y.M.; Chang, C.H. Enhancing physical stability of positively charged catanionic vesicles in the presence of calcium chloride via cholesterol-induced fluidic bilayer characteristic. Colloid Polym. Sci. 2014, 292, 2519–2527. [Google Scholar] [CrossRef]
- Kuo, A.T.; Chang, C.H. Cholesterol-induced condensing and disordering effects on a rigid catanionic bilayer: A molecular dynamics study. Langmuir 2014, 30, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.-Y.; Chiu, C.-C. Interplay between alkyl chain asymmetry and cholesterol addition in the rigid ion pair amphiphile bilayer systems. J. Chem. Phys. 2017, 146, 35102–35111. [Google Scholar] [CrossRef] [PubMed]
- Soussan, E.; Cassel, S.; Blanzat, M.; Rico-Lattes, I. Drug delivery by soft matter: Matrix and vesicular carriers. Angew. Chem. Int. Ed. Engl. 2009, 48, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Bramer, T.; Dew, N.; Edsman, K. Pharmaceutical applications for catanionic mixtures. J. Pharm. Pharmacol. 2007, 59, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Li, S. Calorimetric and molecular mechanics studies of the thermotropic phase behavior of membrane phospholipids. Biochim. Biophys. Acta 1999, 1422, 273–307. [Google Scholar] [CrossRef]
- Marsh, D. CRC Handbook of Lipid Bilayers, 1st ed.; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Lee, W.H.; Tang, Y.L.; Chiu, T.C.; Yang, Y.M. Synthesis of Ion-Pair Amphiphiles and Calorimetric Study on the Gel to Liquid-Crystalline Phase Transition Behavior of Their Bilayers. J. Chem. Eng. Data 2015, 60, 1119–1125. [Google Scholar] [CrossRef]
- Silvius, J.R. Thermotropic Phase Transitions of Pure Lipids in Model Membranes and Their Modifications by Membrane Proteins; John Wiley & Sons, Inc.: New York, NY, USA, 1982. [Google Scholar]
- Leekumjorn, S.; Sum, A.K. Molecular studies of the gel to liquid-crystalline phase transition for fully hydrated DPPC and DPPE bilayers. Biochim. Biophys. Acta 2007, 1768, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.S.; Yu, Z.W. Structural Characterization on the Gel to Liquid-Crystal Phase Transition of Fully Hydrated DSPC and DSPE Bilayers. J. Phys. Chem. B 2009, 113, 8114–8123. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Tian, C.-A.; Chiu, C.-C. The Effects of Alkyl Chain Combinations on the Structural and Mechanical Properties of Biomimetic Ion Pair Amphiphile Bilayers. Bioengineering 2017, 4, 84. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Earla, D.J.; Deema, M.W. Parallel Tempering: Theory, Applications, and New Perspectives. Phys. Chem. Chem. Phys. 2005, 7, 3910–3916. [Google Scholar] [CrossRef]
- Ogata, K.; Uchida, W.; Nakamura, S. Understanding Thermal Phases in Atomic Detail by All-Atom Molecular-Dynamics Simulation of a Phospholipid Bilayer. J. Phys. Chem. B 2014, 118, 14353–14365. [Google Scholar] [CrossRef] [PubMed]
- Ogata, K.; Nakamura, S. Improvement of Parameters of the AMBER Potential Force Field for Phospholipids for Description of Thermal Phase Transitions. J. Phys. Chem. B 2015, 119, 9726–9739. [Google Scholar] [CrossRef] [PubMed]
- Jambeck, J.P.M.; Lyubartsev, A.P. Derivation and Systematic Validation of a Refined All-Atom Force Field for Phosphatidylcholine Lipids. J. Phys. Chem. B 2012, 116, 3164–3179. [Google Scholar] [CrossRef] [PubMed]
- Kucerka, N.; Nieh, M.-P.; Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Bimchim. Biophys. Acta 2011, 1808, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Tristram-Nagle, S.; Kucerka, N.; Nagle, J.F. Temperature Dependence of Structure, Bending Rigidity, and Bilayer Interactions of Dioleoylphosphatidylcholine Bilayers. Biophys. J. 2008, 94, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Chaban, V. Computationally efficient prediction of area per lipid. Chem. Phys. Lett. 2014, 616-617, 25–29. [Google Scholar] [CrossRef]
- Singer, M.A.; Finegold, L. Permeability and morphology of low temperature phases in bilayer of single and mixtures of phosphatidylcholines. Biochim. Biophys. Acta 1985, 816, 303–312. [Google Scholar] [CrossRef]
- Mortensen, K.; Pfeiffer, W.; Sackmann, E.; Knoll, W. Structural properties of a phosphatidylcholine-cholesterol system as studied by small-angle neutron scattering: Ripple structure and phase diagram. Biochim. Biophys. Acta 1988, 945, 221–245. [Google Scholar] [CrossRef]
- Tristram-Nagle, S.; Zhang, R.; Suter, R.M.; Worthington, C.R.; Sun, W.; Nagle, J.F. Measurement of chain tilt order angle in fully hydrated bilyers of gel phase lecithins. Biophys. J. 1994, 64, 1097–1109. [Google Scholar] [CrossRef]
- Tristram-Nagle, S.; Liu, Y.; Legleiter, J.; Nagle, J.F. Structure of Gel Phase DMPC Determined by X-Ray Diffraction. Biophys. J. 2002, 83, 3324–3335. [Google Scholar] [CrossRef]
- Sun, W.J.; Tristram-Nagle, S.; Suter, R.M.; Nagle, J.F. Structure of gel phase saturated lecithin bilayers: Temperature and chain length dependence. Biophys. J. 1996, 71, 885–891. [Google Scholar] [CrossRef]
- Schindler, H.; Seelig, J. Deuterium Order Parameters in Relation to Thermodynamic Properties of a Phospholipid Bilayer. A Statistical Mechanical Interpretation. Biochemistry 1975, 14, 2283–2287. [Google Scholar] [CrossRef] [PubMed]
- Heller, H.; Schaefer, M.; Schulten, K. Molecular Dynamics Simulation of a Bilayer of 200 Lipids in the Gel and in the Liquid-Crystal Phases. J. Phys. Chem. B 1993, 97, 8343–8360. [Google Scholar] [CrossRef]
- Feller, S.E.; Pastor, R.W. Constant surface tension simulations of lipid bilayers: The sensitivity of surface areas and compressibilities. J. Chem. Phys. 1999, 111, 1281–1287. [Google Scholar] [CrossRef]
- Shinoda, W.; Shinoda, K.; Baba, T.; Mikami, M. Molecular dynamics study of bipolar tetraether lipid membranes. Biophys. J. 2005, 89, 3195–3202. [Google Scholar] [CrossRef] [PubMed]
- Khelashvili, G.; Pabst, G.; Harries, D. Cholesterol Orientation and Tilt Modulus in DMPC Bilayers. J. Phys. Chem. B 2010, 114, 7524–7534. [Google Scholar] [CrossRef] [PubMed]
- Khelashvili, G.; Harries, D. How cholesterol tilt modulates the mechanical properties of saturated and unsaturated lipid membranes. J. Phys. Chem. B 2013, 117, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Khelashvili, G.; Kollmitzer, B.; Heftberger, P.; Pabst, G.; Harries, D. Calculating the Bending Modulus for Multicomponent Lipid Membranes in Different Thermodynamic Phases. J. Chem. Theory Comput. 2013, 9, 3866–3871. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, W.; Shimizu, M.; Okazaki, S. Molecular Dynamics Study on Electrostatic Properties of a Lipid Bilayer: Polarization, Electrostatic Potential, and the Effects on Structure and Dynamics of Water near the Interface. J. Phys. Chem. B 1998, 102, 6647–6654. [Google Scholar] [CrossRef]
- Zhou, F.; Schulten, K. Molecular Dynamics Study of a Membrane-Water Interface. J. Phys. Chem. 1995, 99, 2194–2207. [Google Scholar] [CrossRef]
- Tiwari, S.P.; Rai, N.; Maginn, E.J. Dynamics of actinyl ions in water: A molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2014, 16, 8060–8069. [Google Scholar] [CrossRef] [PubMed]
- Impey, R.W.; Madden, P.A.; McDonald, I.R. Hydration and Mobility of Ions in Solution. J. Phys. Chem. 1983, 87, 5071–5083. [Google Scholar] [CrossRef]
- Guardia, E.; Padro, J.A. Molecular Dynamics Simulation of Single Ions in Aqueous Solutions: Effects of the Flexibility of the Water Molecules. J. Phys. Chem. 1990, 94, 6049–6055. [Google Scholar] [CrossRef]
- Shinoda, W.; Mikami, M.; Baba, T.; Hato, M. Dynamics of a highly branched lipid bilayer: A molecular dynamics study. Chem. Phys. Letters 2004, 390, 35–40. [Google Scholar] [CrossRef]
- Shinoda, K.; Shinoda, W.; Mikami, M. Molecular dynamics simulation of archaeal lipid bilayer with sodium chloride. Phys. Chem. Chem. Phys. 2007, 9, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Robalo, J.R.; Ramalho, J.P.; Huster, D.; Loura, L.M.S. Influence of the sterol aliphatic side chain on membrane properties: A molecular dynamics study. Phys. Chem. Chem. Phys. 2015, 17, 22736–22748. [Google Scholar] [CrossRef] [PubMed]
- De Canto, A.M.T.M.; Robalo, J.R.; Santos, P.D.; Carvalho, A.J.P.; Ramalho, J.P.P.; Loura, L.M.S. Diphenylhexatriene membrane probes DPH and TMA-DPH: A comparative molecular dynamics simulation study. Bimchim. Biophys. Acta 2016, 1858, 2647–2661. [Google Scholar]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on Six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Nose, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Nose, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8596. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1469–1472. [Google Scholar] [CrossRef]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martinez, J.M. Software News and Update Packmol: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Nagle, J.F.; Tristram-Nagle, S. Structure of lipid bilayers. Biochim. Biophys. Acta 2000, 1469, 159–195. [Google Scholar] [CrossRef]
- Coppock, P.S.; Kindt, J.T. Determination of Phase Transition Temperatures for Atomistic Models of Lipids from Temperature-Dependent Stripe Domain Growth Kinetics. J. Phys. Chem. B 2010, 114, 11468–11473. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | a | b | c | |||
|---|---|---|---|---|---|---|
| DCPC | 0.690 ± 0.017 | 0.659 | 2.36 ± 0.07 | 35.61 ± 19.10 | 6.34 ± 0.17 | |
| PC | DCPC | 0.684 ± 0.017 | 0.657 | 2.64 ± 0.05 | 33.55 ± 18.43 | 6.29 ± 0.17 |
| DCPC | 0.679 ± 0.017 | 0.650 | 3.08 ± 0.07 | 32.44 ± 18.15 | 6.31 ± 0.16 | |
| DCPC | 0.617 ± 0.016 | 0.638 | 3.37 ± 0.08 | 30.89 ± 17.73 | 6.27 ± 0.17 | |
| CTMA-CS | 0.578 ± 0.013 | - | 2.69 ± 0.05 | 24.33 ± 14.35 | 3.41 ± 0.13 | |
| IPA | CTMA-CS | 0.569 ± 0.012 | - | 3.28 ± 0.04 | 22.47 ± 13.56 | 3.37 ± 0.12 |
| CTMA-CS | 0.561 ± 0.012 | - | 3.60 ± 0.03 | 20.81 ± 12.78 | 3.34 ± 0.12 | |
| CTMA-CS | 0.556 ± 0.012 | - | 3.88 ± 0.04 | 19.43 ± 12.32 | 3.31 ± 0.12 | |
| System | a | b | ||||
|---|---|---|---|---|---|---|
| DCPC | 0.606 ± 0.013 | - | 2.40 ± 0.05 | 29.46 ± 17.84 | 7.03 ± 0.16 | |
| PC | DCPC | 0.511 ± 0.008 | 0.472 c | 3.17 ± 0.03 | 29.27 ± 10.14 | 6.20 ± 0.16 |
| DCPC | 0.493 ± 0.005 | 0.473 d | 3.42 ± 0.04 | 35.01 ± 6.05 | 5.77 ± 0.15 | |
| DCPC | 0.501 ± 0.004 | 0.473 d | 3.89 ± 0.02 | 37.13 ± 5.30 | 5.74 ± 0.15 | |
| CTMA-CS | 0.422 ± 0.005 | - | 3.15 ± 0.02 | 13.32 ± 8.00 | 2.54 ± 0.08 | |
| IPA | CTMA-CS | 0.426 ± 0.005 | - | 3.64 ± 0.05 | 21.57 ± 5.83 | 2.48 ± 0.08 |
| CTMA-CS | 0.431 ± 0.004 | - | 4.10 ± 0.06 | 24.25 ± 5.32 | 2.60 ± 0.09 | |
| CTMA-CS | 0.441 ± 0.004 | - | 4.28 ± 0.08 | 28.90 ± 5.24 | 2.62 ± 0.08 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, C.-a.; Chiu, C.-c. Importance of Hydrophilic Groups on Modulating the Structural, Mechanical, and Interfacial Properties of Bilayers: A Comparative Molecular Dynamics Study of Phosphatidylcholine and Ion Pair Amphiphile Membranes. Int. J. Mol. Sci. 2018, 19, 1552. https://doi.org/10.3390/ijms19061552
Tian C-a, Chiu C-c. Importance of Hydrophilic Groups on Modulating the Structural, Mechanical, and Interfacial Properties of Bilayers: A Comparative Molecular Dynamics Study of Phosphatidylcholine and Ion Pair Amphiphile Membranes. International Journal of Molecular Sciences. 2018; 19(6):1552. https://doi.org/10.3390/ijms19061552
Chicago/Turabian StyleTian, Ching-an, and Chi-cheng Chiu. 2018. "Importance of Hydrophilic Groups on Modulating the Structural, Mechanical, and Interfacial Properties of Bilayers: A Comparative Molecular Dynamics Study of Phosphatidylcholine and Ion Pair Amphiphile Membranes" International Journal of Molecular Sciences 19, no. 6: 1552. https://doi.org/10.3390/ijms19061552
APA StyleTian, C.-a., & Chiu, C.-c. (2018). Importance of Hydrophilic Groups on Modulating the Structural, Mechanical, and Interfacial Properties of Bilayers: A Comparative Molecular Dynamics Study of Phosphatidylcholine and Ion Pair Amphiphile Membranes. International Journal of Molecular Sciences, 19(6), 1552. https://doi.org/10.3390/ijms19061552