Inhibition of HIF1α-Dependent Upregulation of Phospho-l-Plastin Resensitizes Multiple Myeloma Cells to Frontline Therapy

Abstract
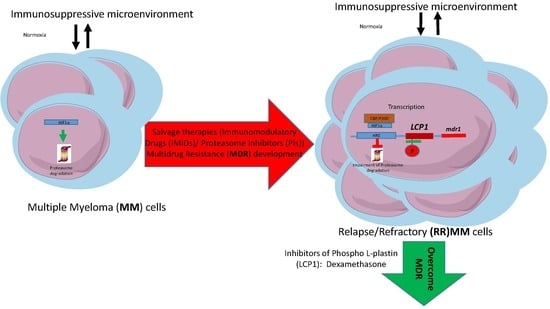
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
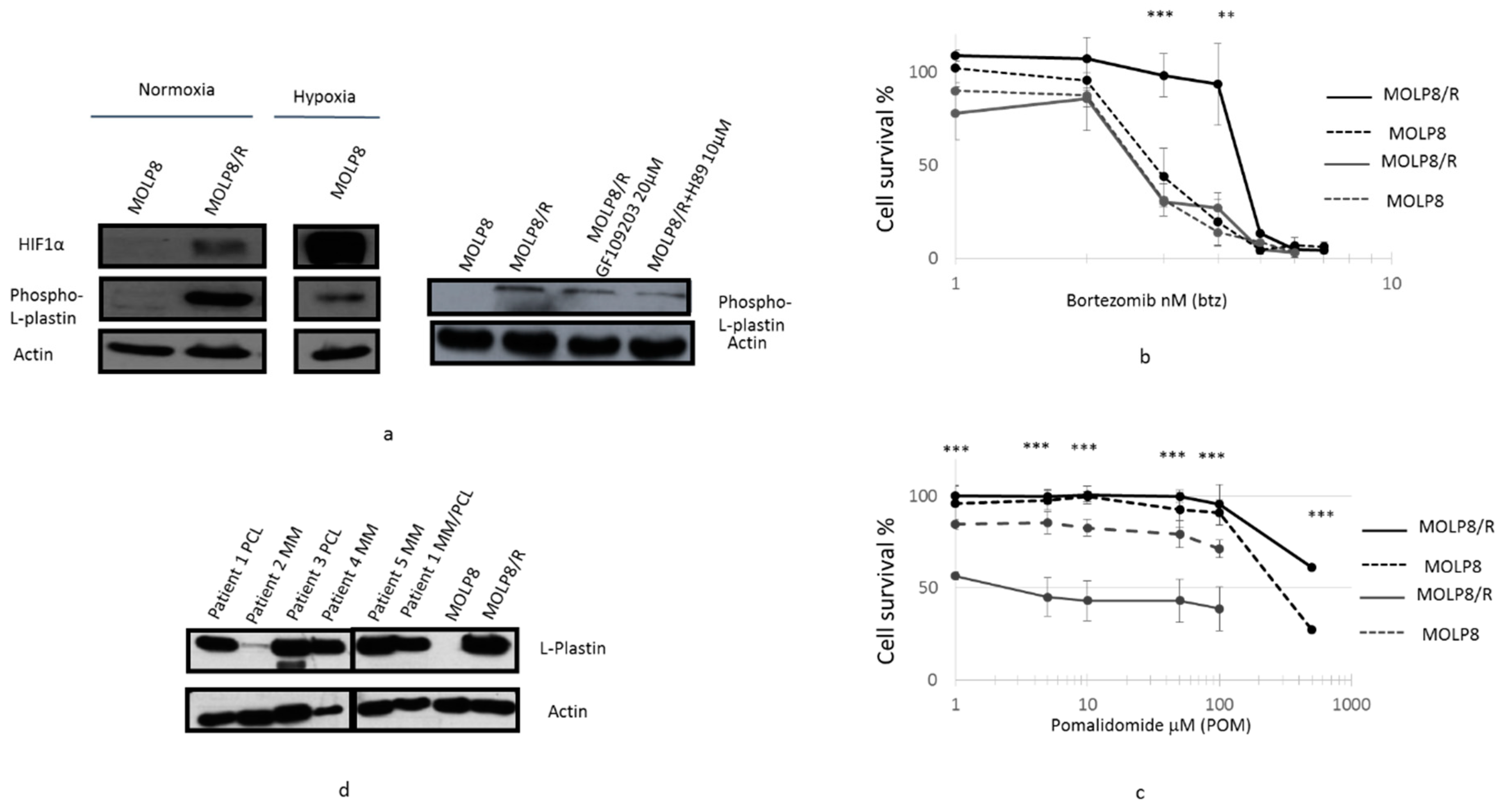
2.1. Drug Resistance Study to New Drugs Used in MM Treatment
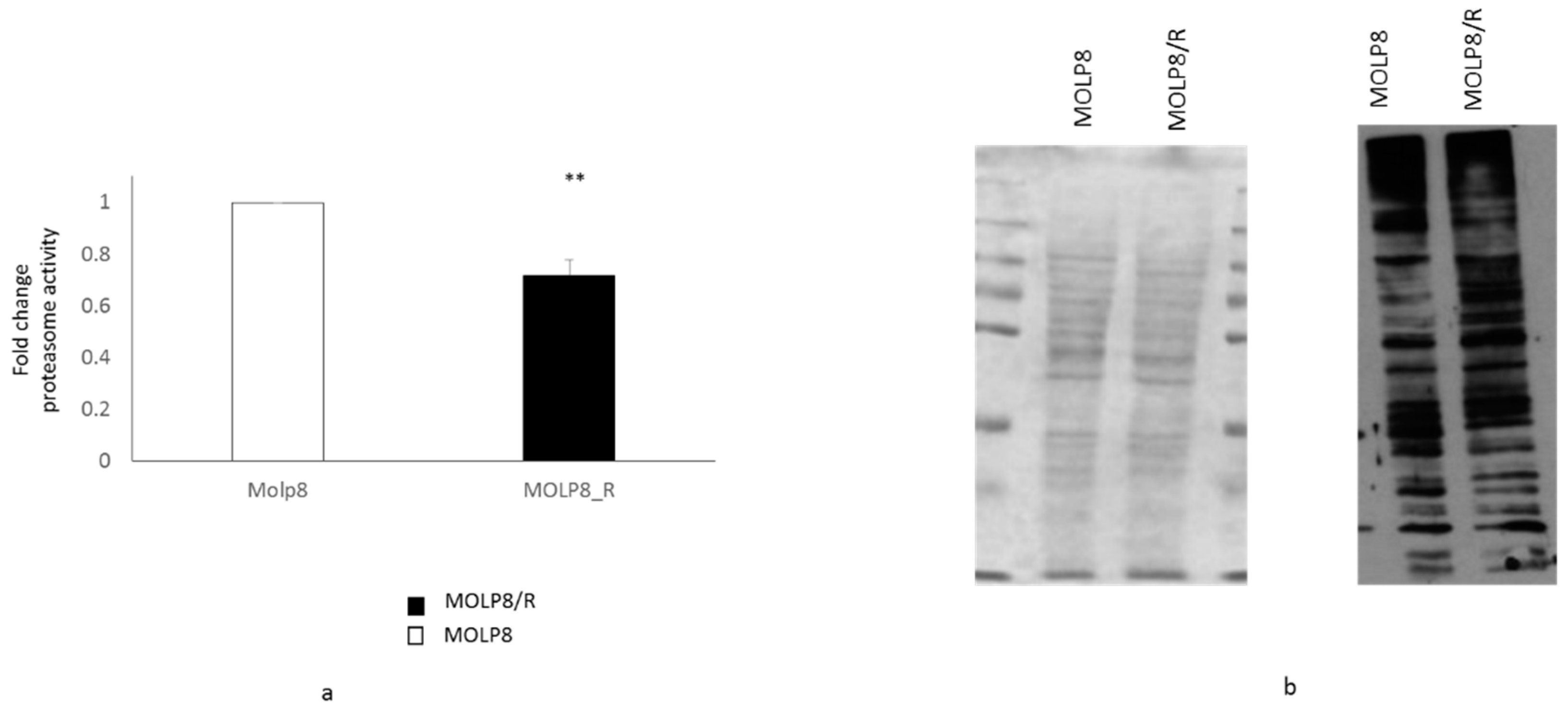
2.2. Measure of Proteasome Activity
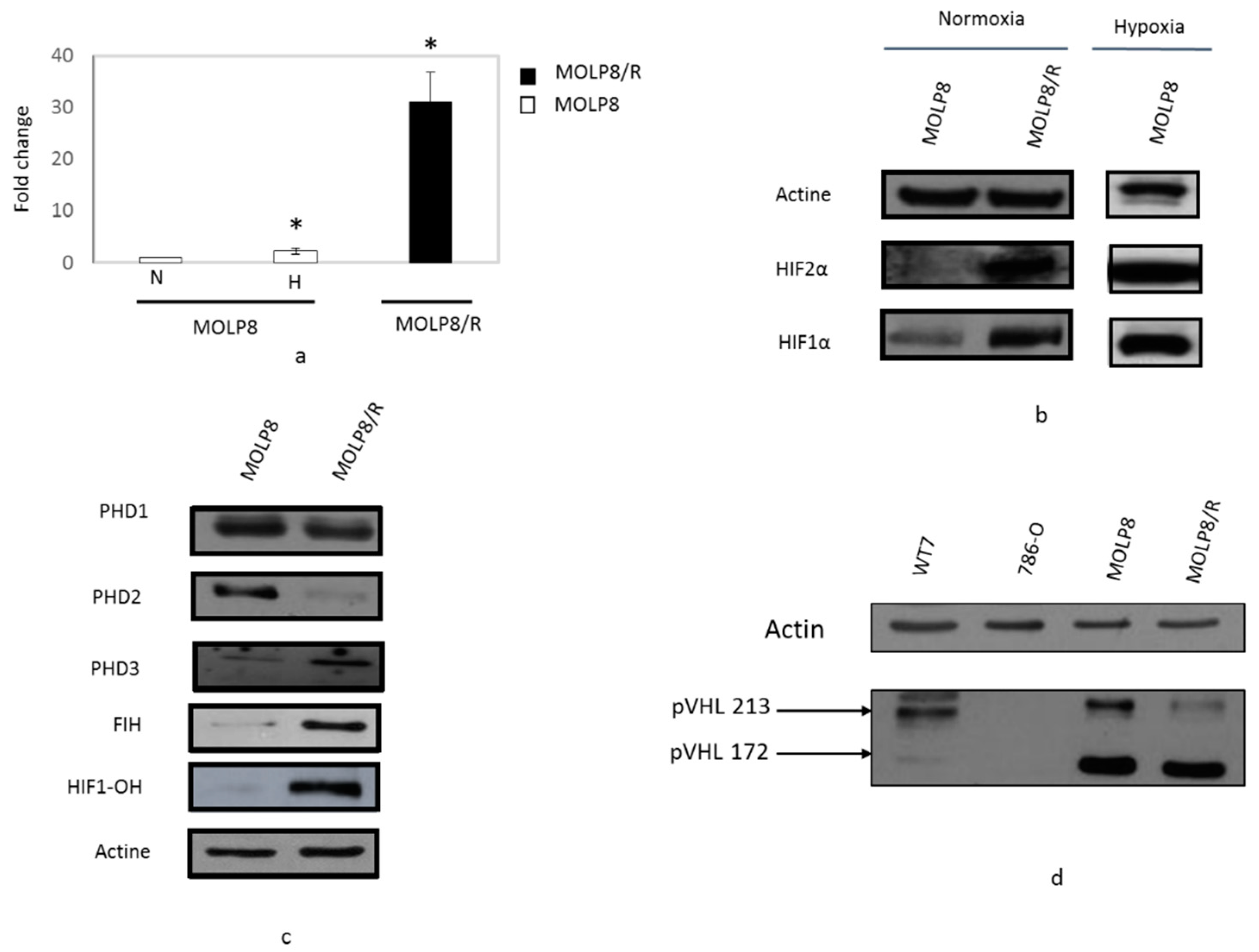
2.3. Overexpression of HIF1α, HIF2α, and HIF-OH in MOLP8/R Cell Line
2.4. Study of Degradation Pathway of HIF1α in Normoxia Conditions
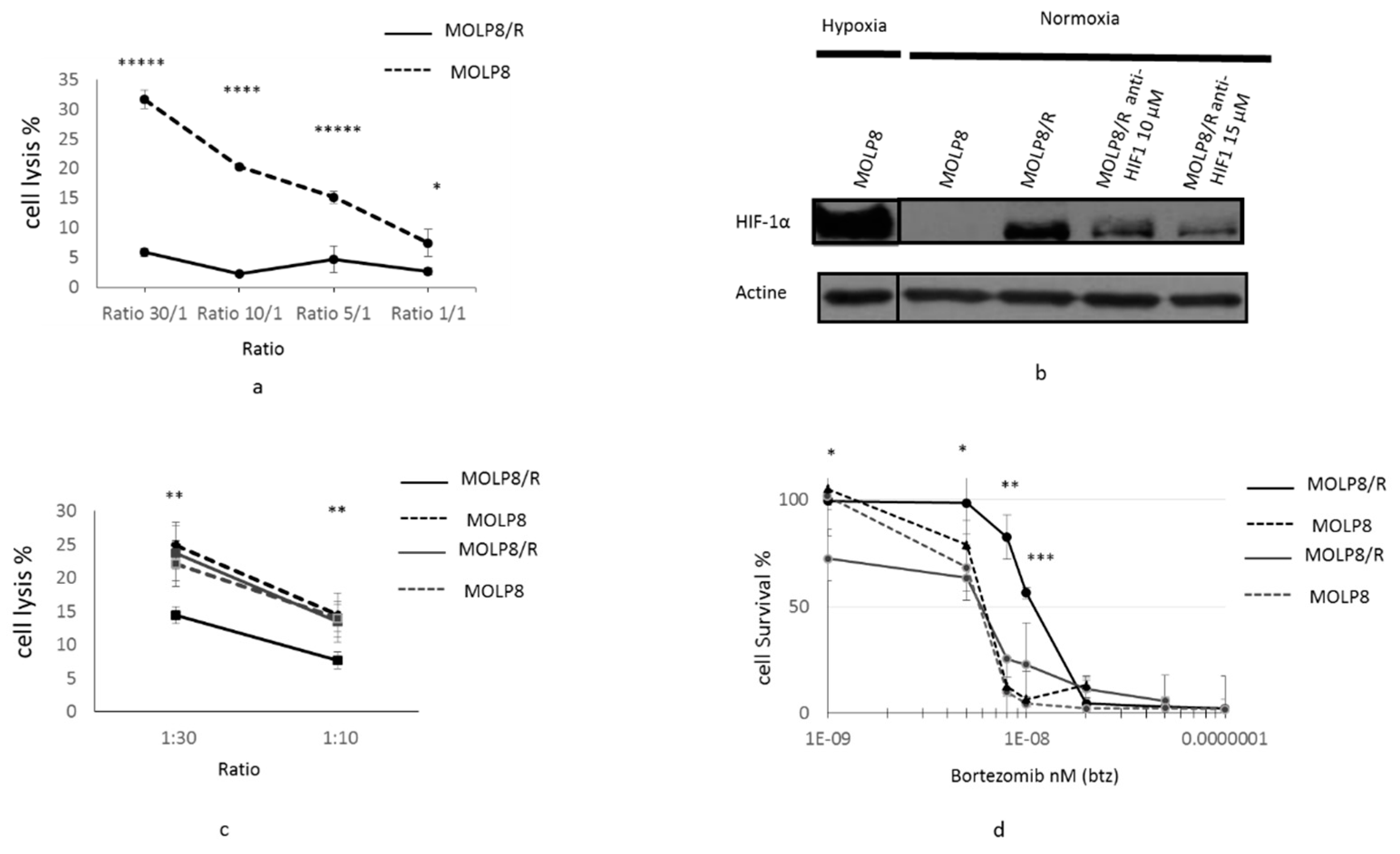
2.5. Resistance to NK Cell Lysis
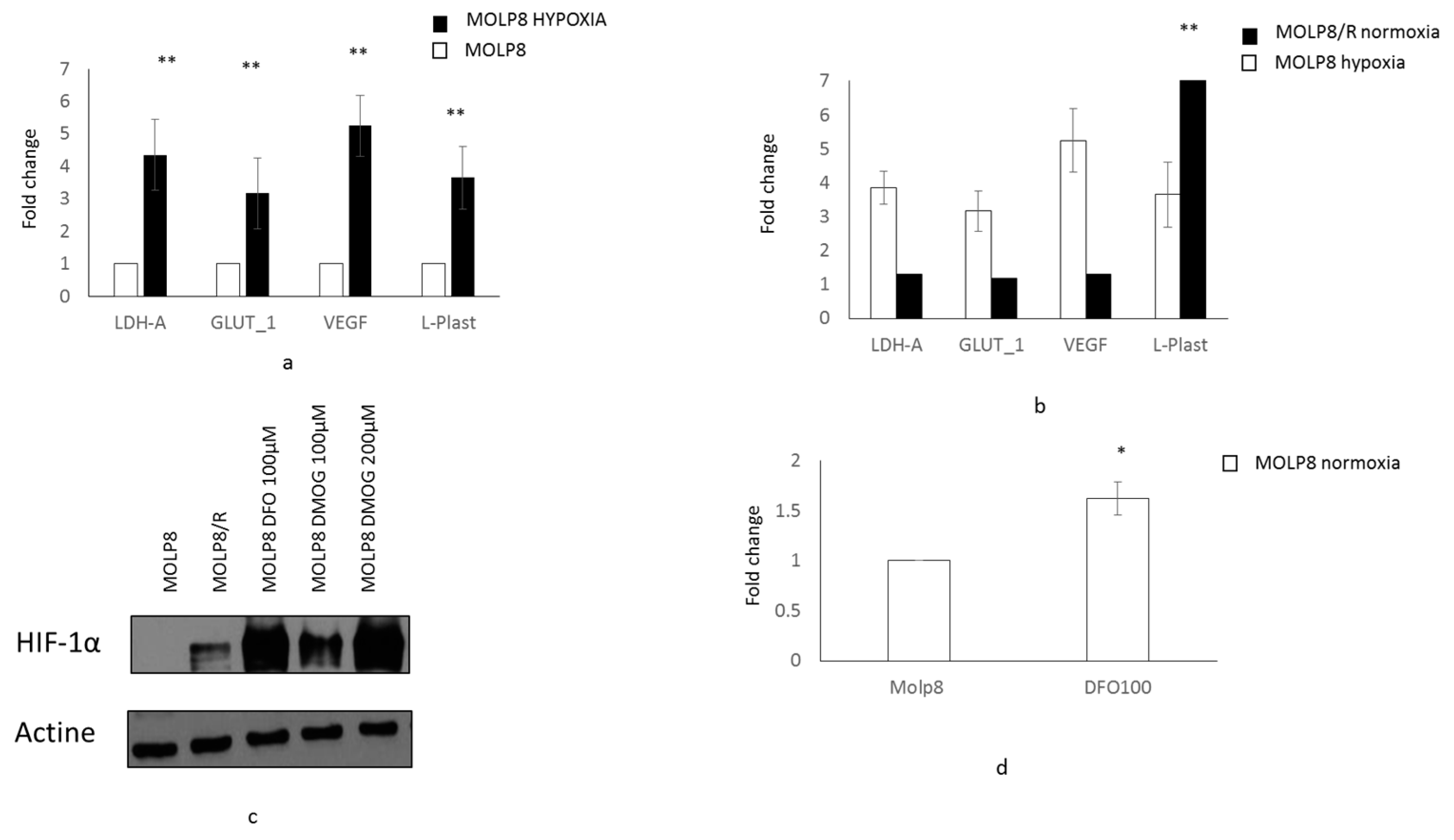
2.6. Main Reference Genes Targeted by HIF1α Transcription Factor
2.7. l-Plastin and Phospho l-Plastin Expression in Normoxia and Hypoxia
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Viability (XTT) Assay
4.3. Protein Extraction
4.4. Antibodies
4.5. Measure of the Proteasome Activity
4.6. Resistance Assay to NK92 Cytotoxicity
4.7. RNA Extraction
4.8. q-PCR Protocol
4.9. Data Are Calculated as a Fold Change Compared to Reference Sample
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MM | multiple myeloma |
| IMiDs | immunomodulatory drugs |
| PIs | proteasome inhibitors |
| HIF1α | hypoxia-inducible factor 1-alpha |
| ABC | transporter ATP-binding cassette transporter |
| Pgp | P-glycoprotein |
| MRP1 | multidrug resistance-associated protein 1 |
| BCRP | breast cancer resistance protein |
| MDR | multidrug resistance |
| HRE | hypoxia-response element |
| FDA | Food and Drug Administration |
| PHD | prolyl hydroxylase |
| FIH | factor-inhibiting HIF1α |
| NK | natural killer |
| VEGF | vascular endothelial growth factor |
| GLUT1 | glucose transporter 1 |
| LDH-A | lactate dehydrogenase A |
| PKA | protein kinase A |
References
- Rajkumar, S.V.; Kumar, S. Multiple myeloma: Diagnosis and treatment. Mayo Clin. Proc. 2016, 91, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Myeloma today: Disease definitions and treatment advances. Am. J. Hematol. 2016, 91, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Lub, S.; Maes, K.; Menu, E.; de Bruyne, E.; Vanderkerken, K.; van Valckenborgh, E. Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget 2016, 7, 6521–6537. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Lin, S.F. Mechanisms of drug resistance in relapse and refractory multiple myeloma. BioMed Res. Int. 2015, 2015, 341430. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.; D’Agnano, I.; Vitelli, G.; Vona, R.; Marino, M.; Mottolese, M.; Zuppi, C.; Capoluongo, E.; Ameglio, F. c-MYC deregulation is involved in melphalan resistance of multiple myeloma: Role of PDGF-BB. Int. J. Immunopathol. Pharmacol. 2006, 19, 67–79. [Google Scholar] [PubMed]
- Chang-Yew Leow, C.; Gerondakis, S.; Spencer, A. MEK inhibitors as a chemotherapeutic intervention in multiple myeloma. Blood Cancer J. 2013, 3, e105. [Google Scholar] [CrossRef] [PubMed]
- Karadimitris, A.; Chaidos, A.; Caputo, V.; Goudevenou, K.; Ponnusamy, K.; Xiao, X. Myeloma propagating cells, drug resistance and relapse. Stem Cells 2015, 33, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Di Luca, A.; Mleczko, J.; Meleady, P.; Henry, M.; Pesic, M.; Cabrera, D.; van Liempd, S.; Lima, R.T.; O’Connor, R.; et al. Identification of the metabolic alterations associated with the multidrug resistant phenotype in cancer and their intercellular transfer mediated by extracellular vesicles. Sci. Rep. 2017, 7, 44541. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.; Xiao, H.Q.; Oba, B.K. P-glycoprotein expression in plasma-cell myeloma is associated with resistance to VAD. Blood 1989, 74, 913–917. [Google Scholar] [PubMed]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of myeloma. Annu. Rev. Pathol. 2011, 6, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M. Myeloma as a model for the process of metastasis: Implications for therapy. Blood 2012, 120, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Munshi, N.C. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015, 125, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.C.; Wenning, L.A.; Miller, W.M.; Papoutsakis, E.T. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I. Krogh’s model. Biophys. J. 2001, 81, 675–684. [Google Scholar] [CrossRef]
- Cicione, C.; Muinos-Lopez, E.; Hermida-Gomez, T.; Fuentes-Boquete, I.; Diaz-Prado, S.; Blanco, F.J. Effects of severe hypoxia on bone marrow mesenchymal stem cells differentiation potential. Stem Cells Int. 2013, 2013, 232896. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, A.; Tiwary, B.N. Hypoxia inducible factor-1α and multiple myeloma. Int. J. Adv. Res. 2016, 4, 706–715. [Google Scholar]
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene 2014, 33, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metast. Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P.; et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. Mechanisms of motility in metastasizing cells. Mol. Cancer Res. 2010, 8, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Lommel, M.J.; Trairatphisan, P.; Gabler, K.; Laurini, C.; Muller, A.; Kaoma, T.; Vallar, L.; Sauter, T.; Schaffner-Reckinger, E. l-Plastin Ser5 phosphorylation in breast cancer cells and in vitro is mediated by RSK downstream of the ERK/MAPK pathway. FASEB J. 2016, 30, 1218–1233. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Giganti, A.; De Corte, V.; Catillon, M.; Bruyneel, E.; Lentz, D.; Plastino, J.; Gettemans, J.; Friederich, E. Phosphorylation on Ser5 increases the F-actin-binding activity of l-Plastin and promotes its targeting to sites of actin assembly in cells. J. Cell Sci. 2006, 119, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Chun, Y.S.; Lee, D.S.; Huang, L.E.; Park, J.W. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 2008, 111, 3131–3136. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Payvandi, F.; Wu, L.; Zhang, L.H.; Hariri, R.J.; Man, H.W.; Chen, R.S.; Muller, G.W.; Hughes, C.C.; Stirling, D.I.; et al. The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc. Res. 2009, 77, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Borsi, E.; Perrone, G.; Terragna, C.; Martello, M.; Zamagni, E.; Tacchetti, P.; Pantani, L.; Brioli, A.; Dico, A.F.; Zannetti, B.A.; et al. HIF-1alpha inhibition blocks the cross talk between multiple myeloma plasma cells and tumor microenvironment. Exp. Cell Res. 2014, 328, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Messai, Y.; Noman, M.Z.; Hasmim, M.; Janji, B.; Tittarelli, A.; Boutet, M.; Baud, V.; Viry, E.; Billot, K.; Nanbakhsh, A.; et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. 2014, 74, 6820–6832. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Brown, E.J. Immune complex-induced integrin activation and l-Plastin phosphorylation require protein kinase A. J. Biol. Chem. 1999, 274, 24349–24356. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Vallar, L.; Al Tanoury, Z.; Bernardin, F.; Vetter, G.; Schaffner-Reckinger, E.; Berchem, G.; Friederich, E.; Chouaib, S. The actin filament cross-linker l-Plastin confers resistance to TNF-alpha in MCF-7 breast cancer cells in a phosphorylation-dependent manner. J. Cell. Mol. Med. 2010, 14, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Lochner, A.; Moolman, J.A. The many faces of H89: A review. Cardiovasc. Drug Rev. 2006, 24, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Mitsiades, C.S.; Orlowski, R.Z.; Anderson, K.C. Future agents and treatment directions in multiple myeloma. Expert Rev. Hematol. 2014, 7, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Chanan-Khan, A.A.; Swaika, A.; Paulus, A.; Kumar, S.K.; Mikhael, J.R.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q. Pomalidomide: The new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. 2013, 3, e143. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Kumar, S.K.; Orlowski, R.Z.; Cook, G.; Richardson, P.G.; Gertz, M.A.; Giralt, S.; Mateos, M.V.; Leleu, X.; Anderson, K.C. Management of relapsed and refractory multiple myeloma: Novel agents, antibodies, immunotherapies and beyond. Leukemia 2018, 32, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Harousseau, J.L.; Durie, B.; Anderson, K.C.; Dimopoulos, M.; Kyle, R.; Blade, J.; Richardson, P.; Orlowski, R.; Siegel, D.; et al. Consensus recommendations for the uniform reporting of clinical trials: Report of the International Myeloma Workshop Consensus Panel 1. Blood 2011, 117, 4691–4695. [Google Scholar] [CrossRef] [PubMed]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Naymagon, L.; Abdul-Hay, M. Novel agents in the treatment of multiple myeloma: A review about the future. J. Hematol. Oncol. 2016, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Zamagni, E.; Tacchetti, P.; Pantani, L.; Cavo, M. Anti-CD38 and anti-SLAMF7: The future of myeloma immunotherapy. Expert Rev. Hematol. 2018, 11, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Gorgun, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sanchez, L.; Siegel, D.S.; Wang, M.L. Elotuzumab for the treatment of multiple myeloma. J. Hematol. Oncol. 2016, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.; Wang, Y.; Siegel, D.S.; Wang, M.L. Daratumumab: A first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J. Hematol. Oncol. 2016, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, J.S.; Touzeau, C.; Moreau, P. The role of SLAMF7 in multiple myeloma: Impact on therapy. Expert Rev. Clin. Immunol. 2017, 13, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Karakashev, S.V.; Reginato, M.J. Progress toward overcoming hypoxia-induced resistance to solid tumor therapy. Cancer Manag. Res. 2015, 7, 253–264. [Google Scholar] [PubMed]
- Mutlu, P.; Kiraz, Y.; Gunduz, U.; Baran, Y. An update on molecular biology and drug resistance mechanisms of multiple myeloma. Crit. Rev. Oncol. Hematol. 2015, 96, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.R.; Jaiswal, R.; Brown, R.D.; Luk, F.; Bebawy, M. Multiple myeloma and persistence of drug resistance in the age of novel drugs (Review). Int. J. Oncol. 2016, 49, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Begicevic, R.R.; Falasca, M. ABC Transporters in cancer stem cells: Beyond chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; van de Donk, N.; Zweegman, S.; Lokhorst, H.M. Current and new therapeutic strategies for relapsed and refractory multiple myeloma: An update. Drugs 2018, 78, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Spektor, T.M.; Stampleman, L.; Bessudo, A.; Rosen, P.J.; Klein, L.M.; Woliver, T.; Flam, M.; Eshaghian, S.; Nassir, Y.; et al. Safety and efficacy of pomalidomide, dexamethasone and pegylated liposomal doxorubicin for patients with relapsed or refractory multiple myeloma. Br. J. Haematol. 2018, 180, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Merz, M.; Jauch, A.; Hielscher, T.; Bochtler, T.; Schonland, S.O.; Seckinger, A.; Hose, D.; Bertsch, U.; Neben, K.; Raab, M.S.; et al. Prognostic significance of cytogenetic heterogeneity in patients with newly diagnosed multiple myeloma. Blood Adv. 2018, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besse, A.; Stolze, S.C.; Rasche, L.; Weinhold, N.; Morgan, G.J.; Kraus, M.; Bader, J.; Overkleeft, H.S.; Besse, L.; Driessen, C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised international staging system for multiple myeloma: A report from international myeloma working group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.M.; Rajkumar, S.V. Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J. 2015, 5, e365. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Kastritis, E.; Symeonidis, A.S.; Giannakoulas, N.; Katodritou, E.; Delimpasi, S.; Repousis, P.; Terpos, E.; Dimopoulos, M.A. Immunoglobulin D myeloma: Clinical features and outcome in the era of novel agents. Eur. J. Haematol. 2014, 92, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Eble, J.M.; Moon, E.; Yuan, H.; Weitzel, D.H.; Landon, C.D.; Nien, C.Y.; Hanna, G.; Rich, J.N.; Provenzale, J.M.; et al. Tumor cells upregulate normoxic HIF-1alpha in response to doxorubicin. Cancer Res. 2013, 73, 6230–6242. [Google Scholar] [CrossRef] [PubMed]
- Roncuzzi, L.; Pancotti, F.; Baldini, N. Involvement of HIF-1alpha activation in the doxorubicin resistance of human osteosarcoma cells. Oncol. Rep. 2014, 32, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, M.; Vacchelli, E.; Eggermont, A.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Lenalidomide-based immunochemotherapy. Oncoimmunology 2013, 2, e26494. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Cavadas, M.A.; Nguyen, L.K.; Cheong, A. Hypoxia-inducible factor (HIF) network: Insights from mathematical models. Cell Commun. Signal. 2013, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.K.; Cavadas, M.A.; Scholz, C.C.; Fitzpatrick, S.F.; Bruning, U.; Cummins, E.P.; Tambuwala, M.M.; Manresa, M.C.; Kholodenko, B.N.; Taylor, C.T.; et al. A dynamic model of the hypoxia-inducible factor 1alpha (HIF-1α) network. J. Cell Sci. 2013, 126, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A review in the theme: Cellular responses to hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Germeraad, W.T.; Rouschop, K.M.; Steeghs, E.M.; van Gelder, M.; Bos, G.M.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Wabnitz, G.H.; Michalke, F.; Stober, C.; Kirchgessner, H.; Jahraus, B.; van den Boomen, D.J.; Samstag, Y. l-Plastin phosphorylation: A novel target for the immunosuppressive drug dexamethasone in primary human T cells. Eur. J. Immunol. 2011, 41, 3157–3169. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Jensen, K.V.; Woodman, A.G.; Hyndman, M.E.; Vogel, H.J. The calcium-dependent switch helix of l-Plastin regulates actin bundling. Sci. Rep. 2017, 7, 40662. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Drexler, H.G.; Harashima, A.; Okochi, A.; Hasegawa, A.; Kojima, K.; Orita, K. Induction of CD28 on the new myeloma cell line MOLP-8 with t(11;14)(q13;q32) expressing delta/lambda type immunoglobulin. Leuk. Res. 2004, 28, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, O.; Kibel, A.; Gray, S.; Kaelin, W.G., Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat. Med. 1995, 1, 822–826. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosseler, M.; Marani, V.; Broukou, A.; Lequeux, A.; Kaoma, T.; Schlesser, V.; François, J.-H.; Palissot, V.; Berchem, G.J.; Aouali, N.; et al. Inhibition of HIF1α-Dependent Upregulation of Phospho-l-Plastin Resensitizes Multiple Myeloma Cells to Frontline Therapy. Int. J. Mol. Sci. 2018, 19, 1551. https://doi.org/10.3390/ijms19061551
Bosseler M, Marani V, Broukou A, Lequeux A, Kaoma T, Schlesser V, François J-H, Palissot V, Berchem GJ, Aouali N, et al. Inhibition of HIF1α-Dependent Upregulation of Phospho-l-Plastin Resensitizes Multiple Myeloma Cells to Frontline Therapy. International Journal of Molecular Sciences. 2018; 19(6):1551. https://doi.org/10.3390/ijms19061551
Chicago/Turabian StyleBosseler, Manon, Vanessa Marani, Angelina Broukou, Amandine Lequeux, Tony Kaoma, Vincent Schlesser, Jean-Hugues François, Valérie Palissot, Guy J. Berchem, Nasséra Aouali, and et al. 2018. "Inhibition of HIF1α-Dependent Upregulation of Phospho-l-Plastin Resensitizes Multiple Myeloma Cells to Frontline Therapy" International Journal of Molecular Sciences 19, no. 6: 1551. https://doi.org/10.3390/ijms19061551