Lipophilic Chemicals from Diesel Exhaust Particles Trigger Calcium Response in Human Endothelial Cells via Aryl Hydrocarbon Receptor Non-Genomic Signalling

 , , ,
, , ,
Abstract
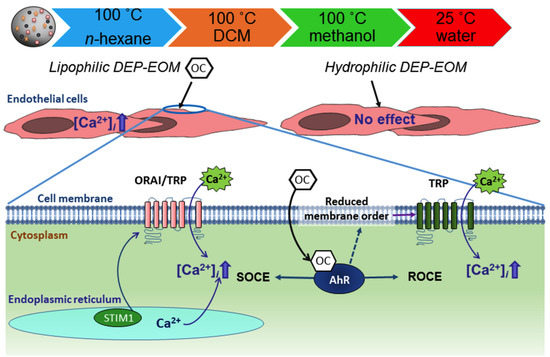
:
1. Introduction
2. Results
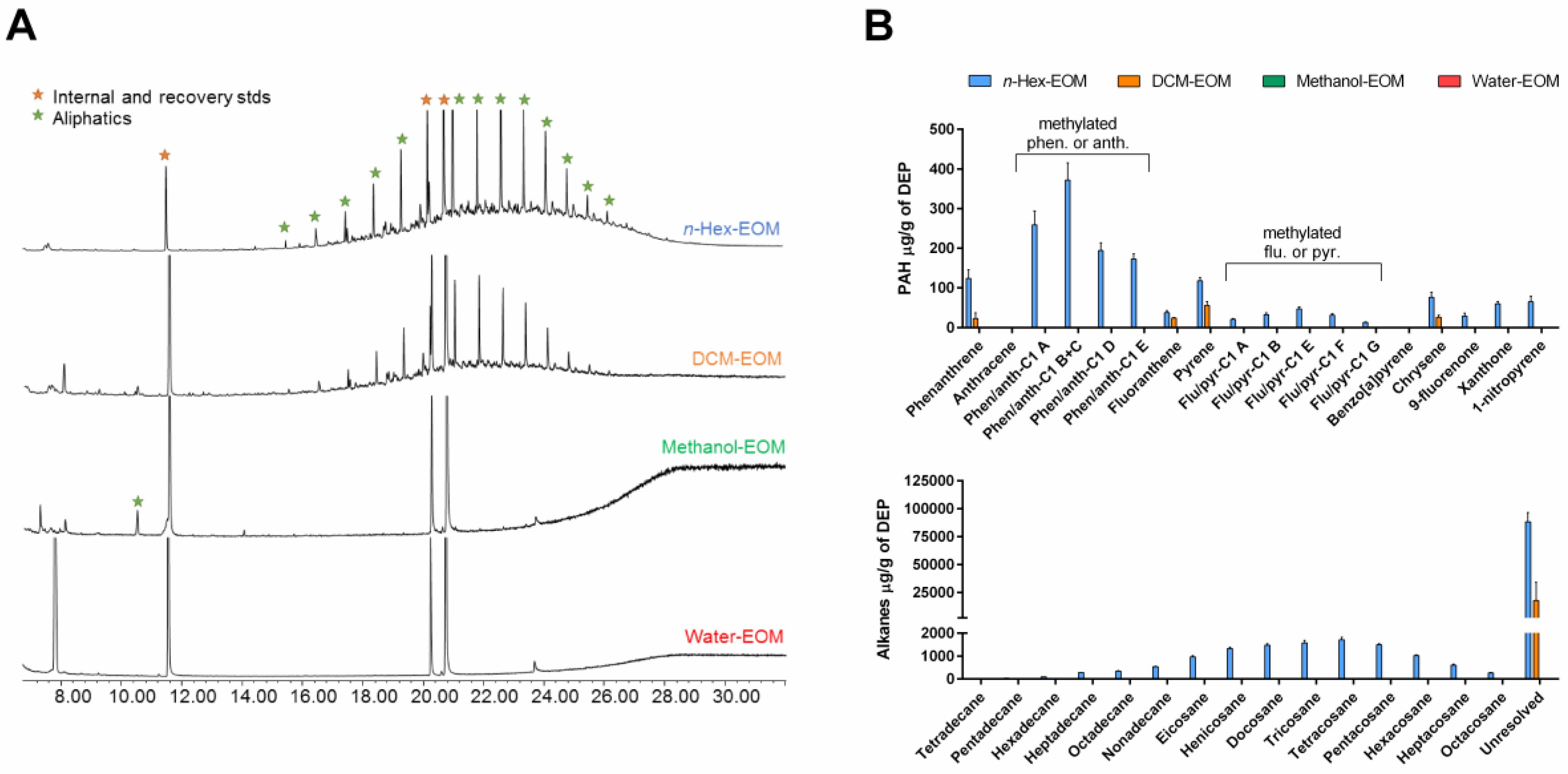
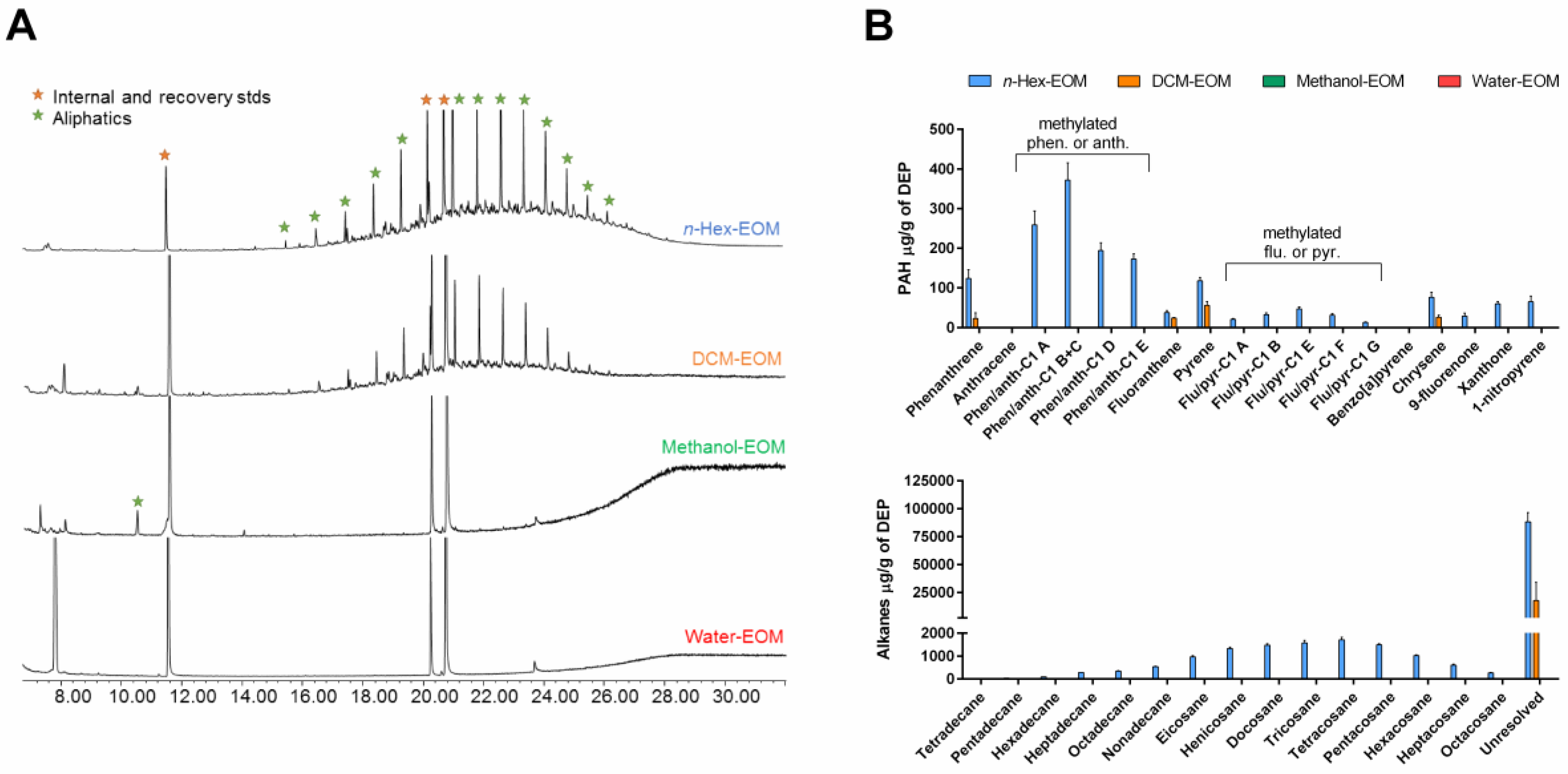
2.1. Characterization of Organic Chemicals Present in DEP-EOM
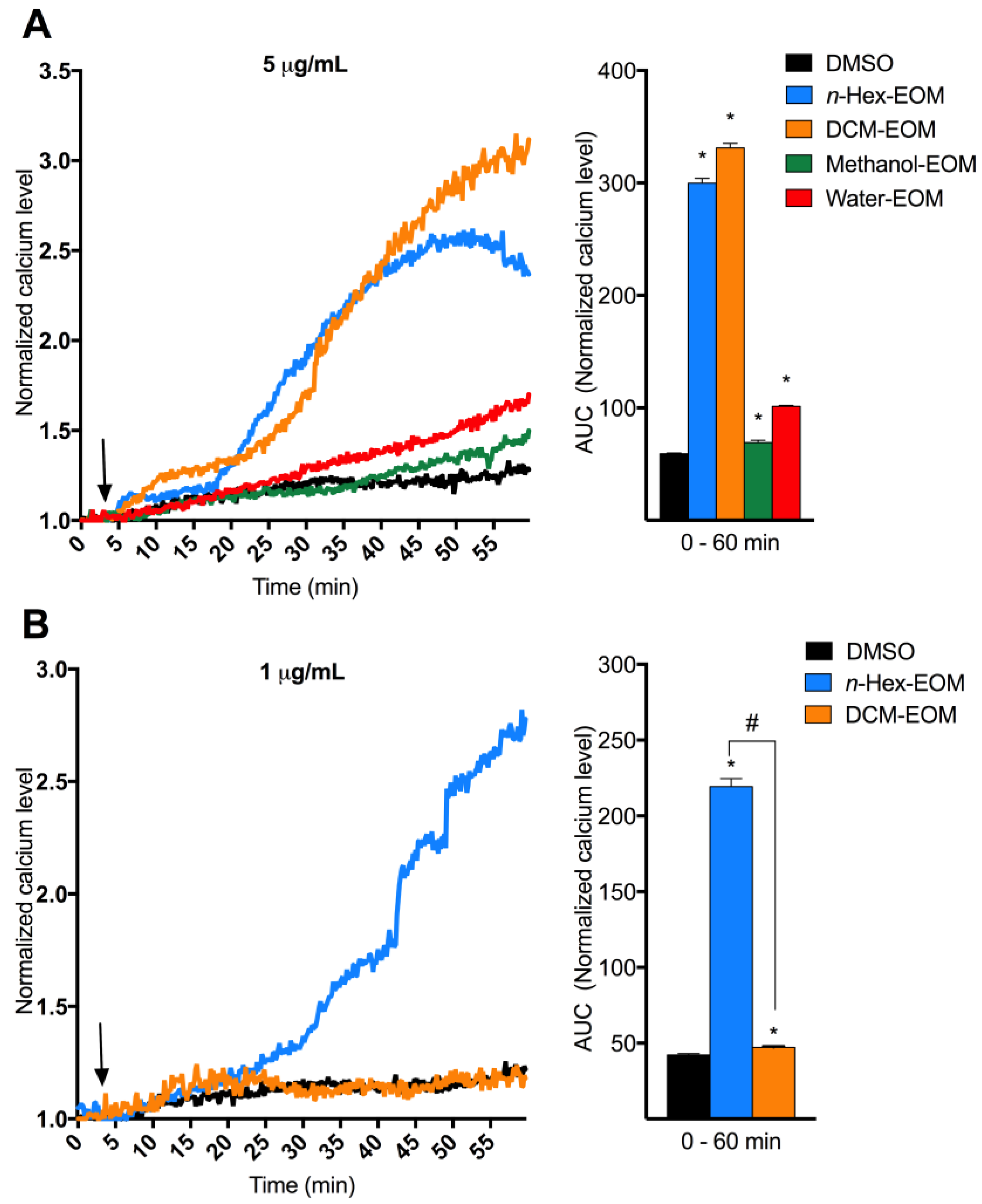
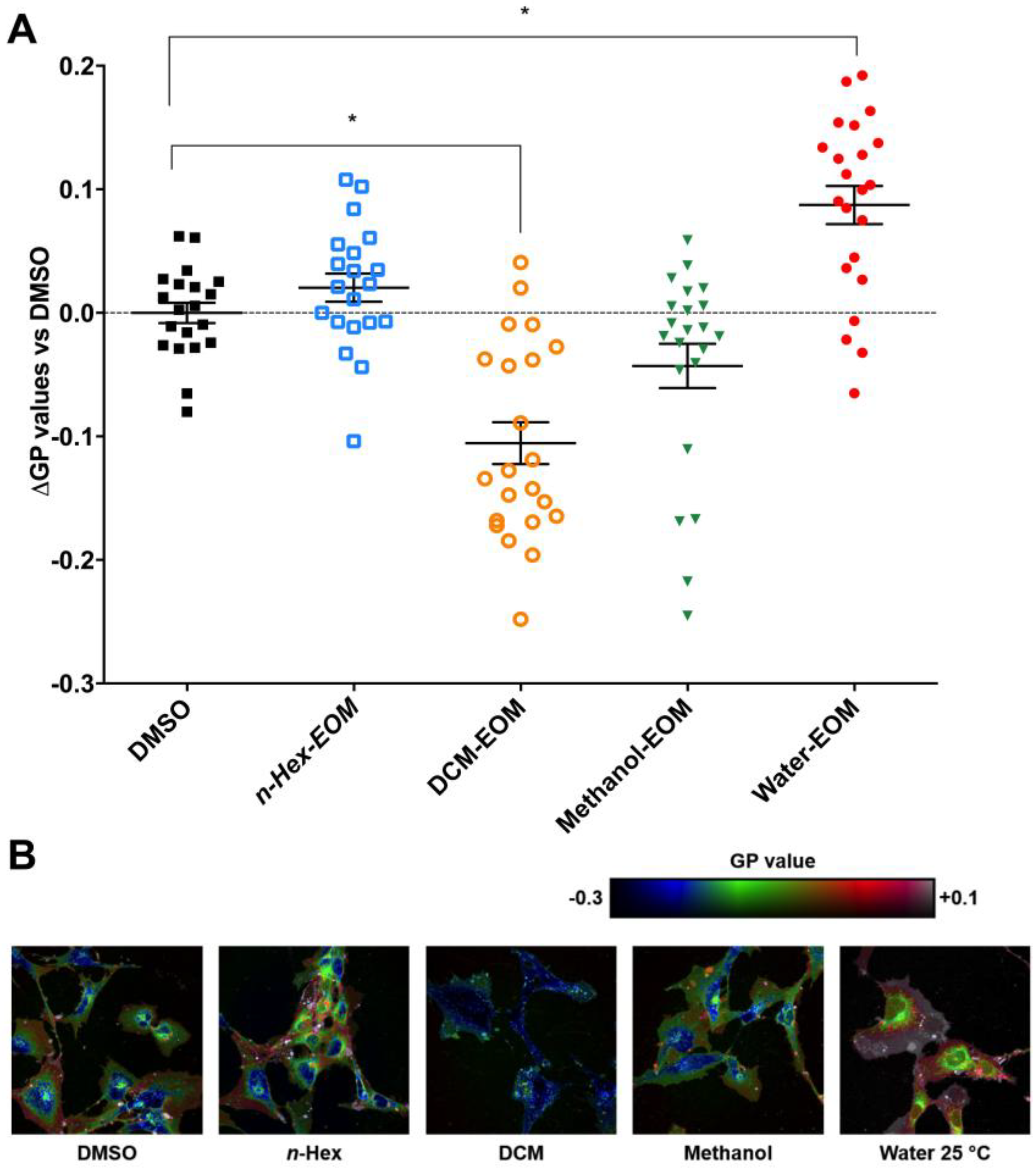
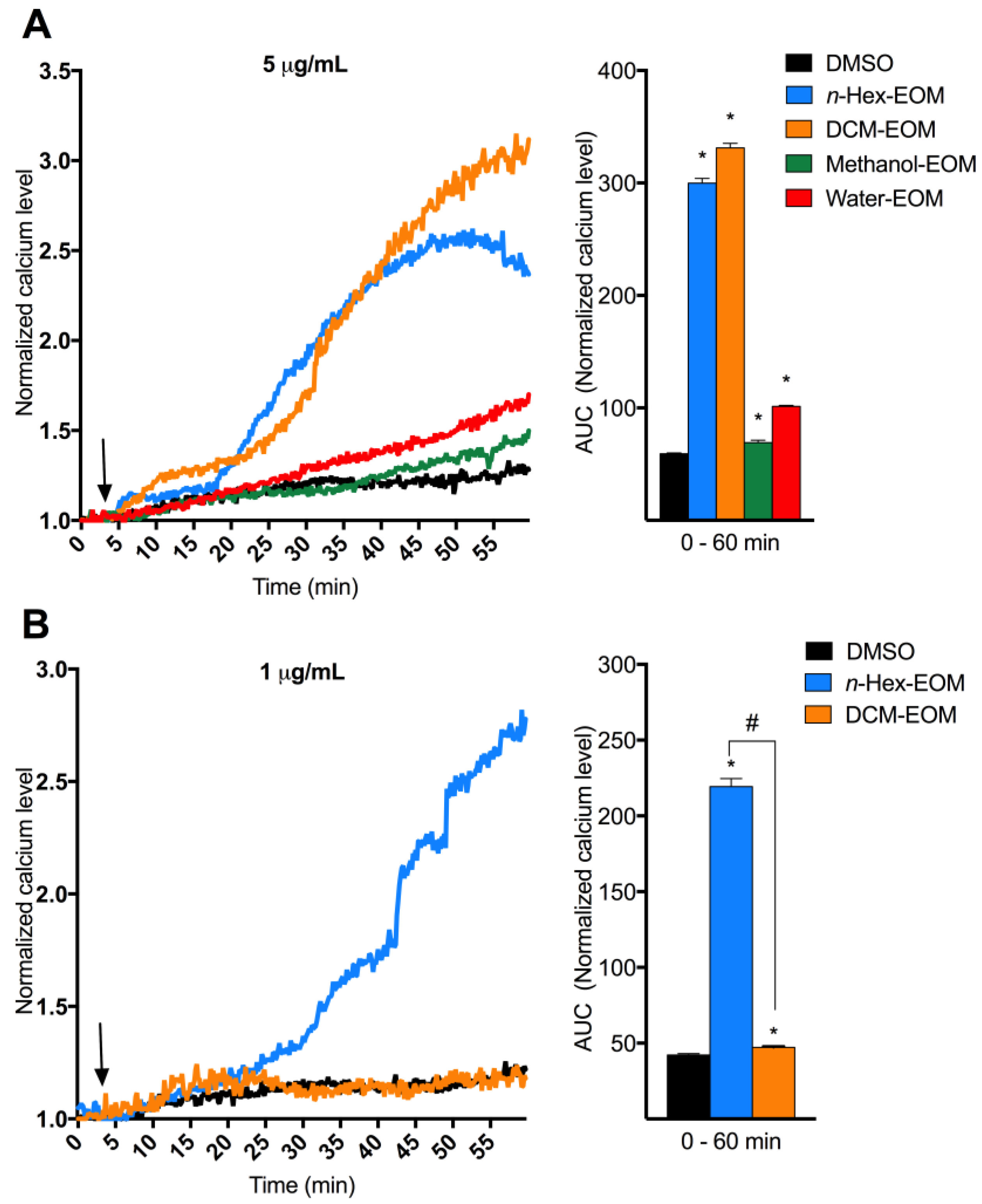
2.2. DEP-EOM Triggered Increase of [Ca2+]i in HMEC-1
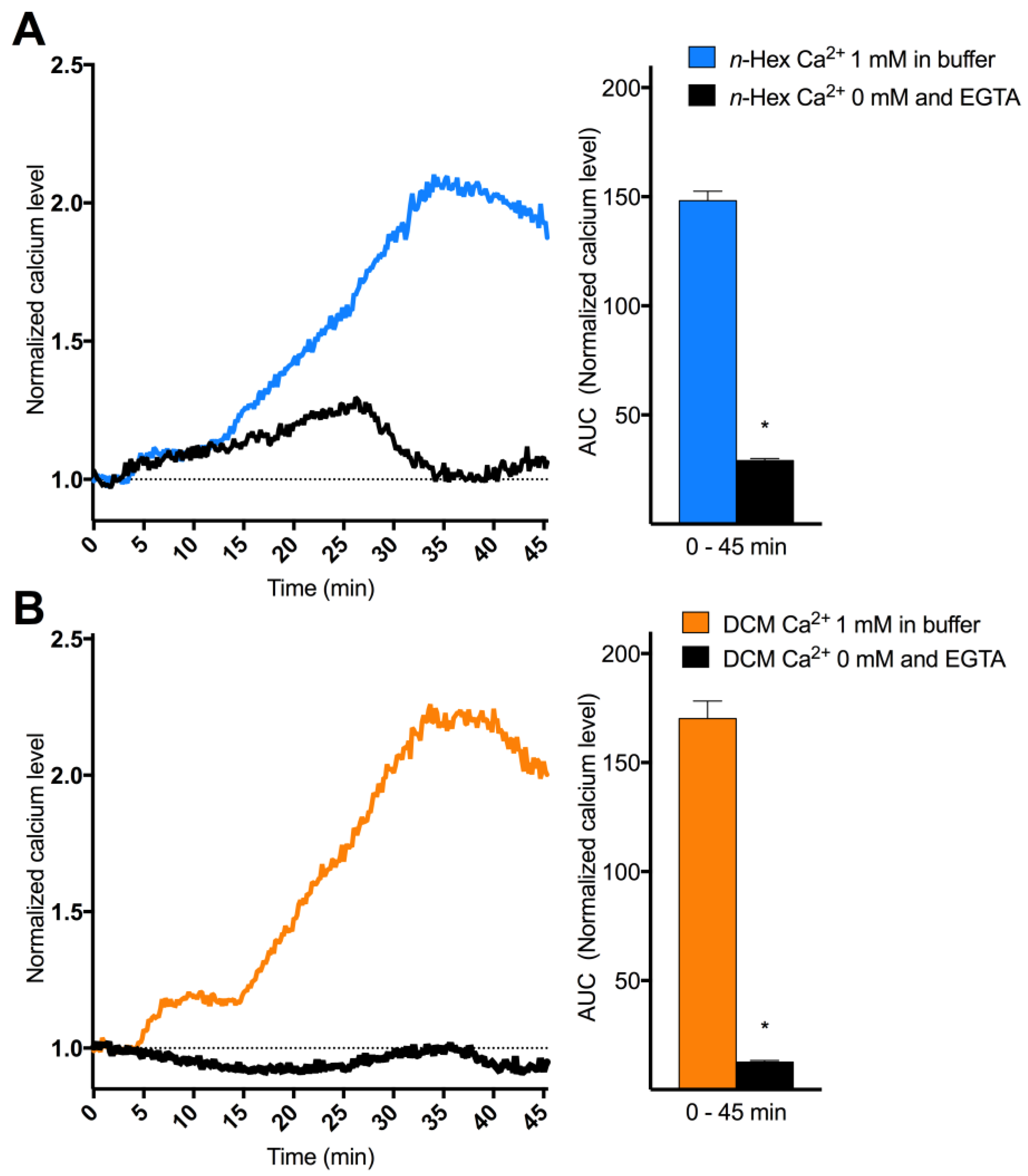
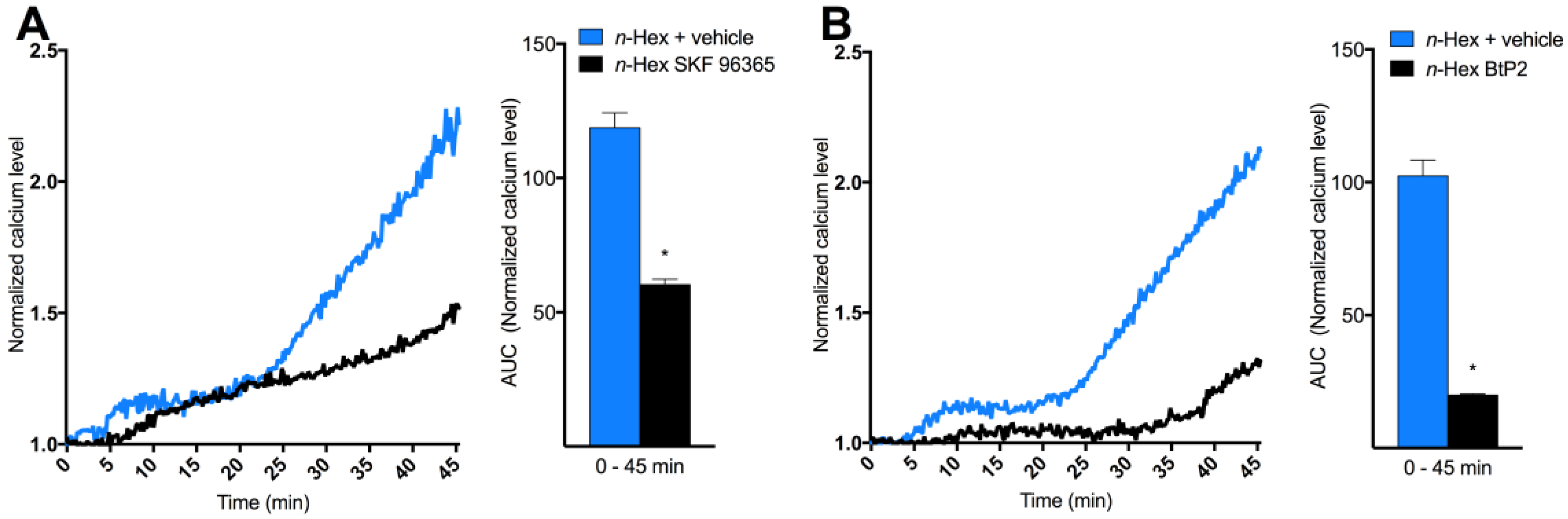
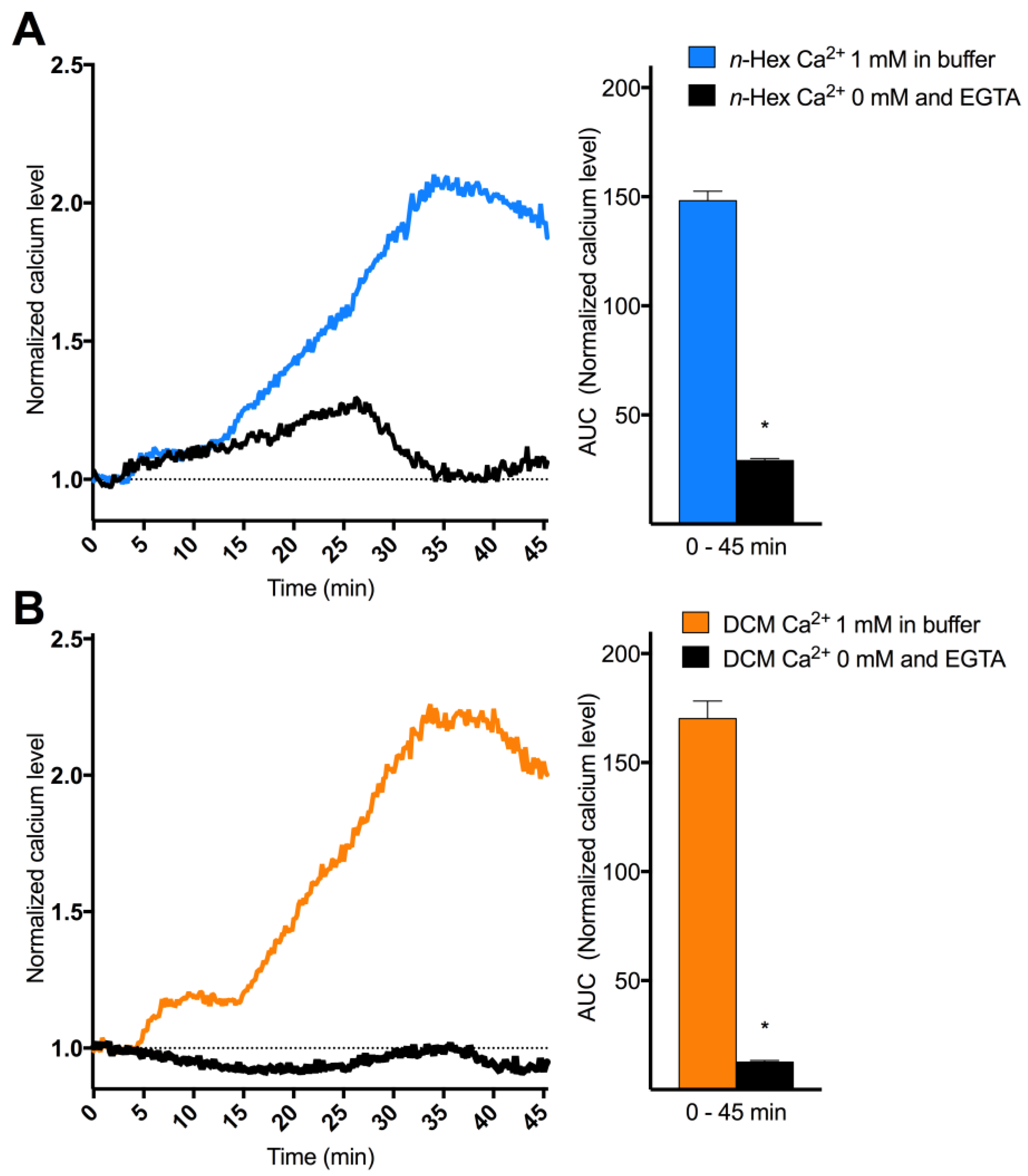
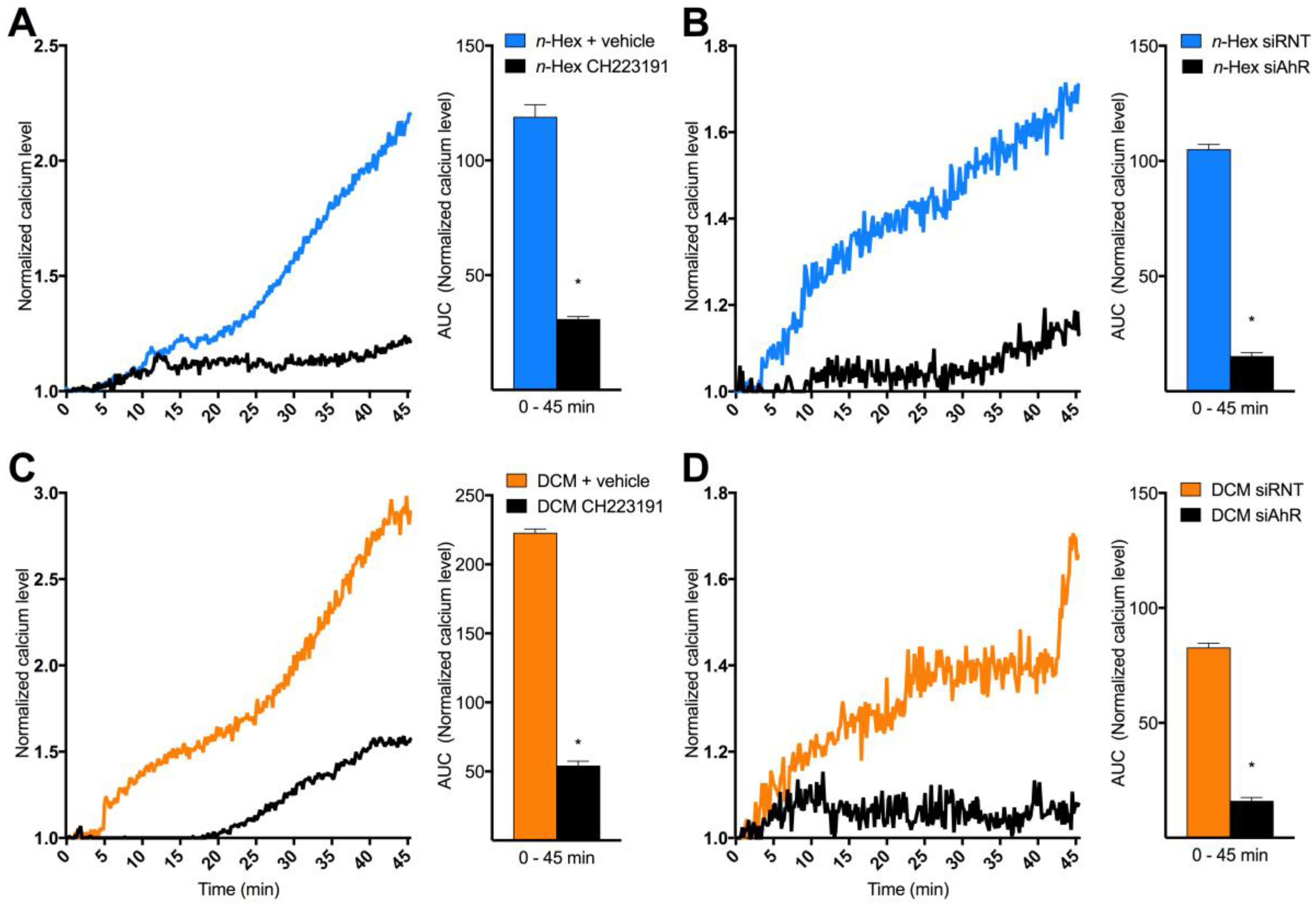
2.3. Origin and Mechanisms Involved in DEP-EOM-Triggered [Ca2+]i Increases
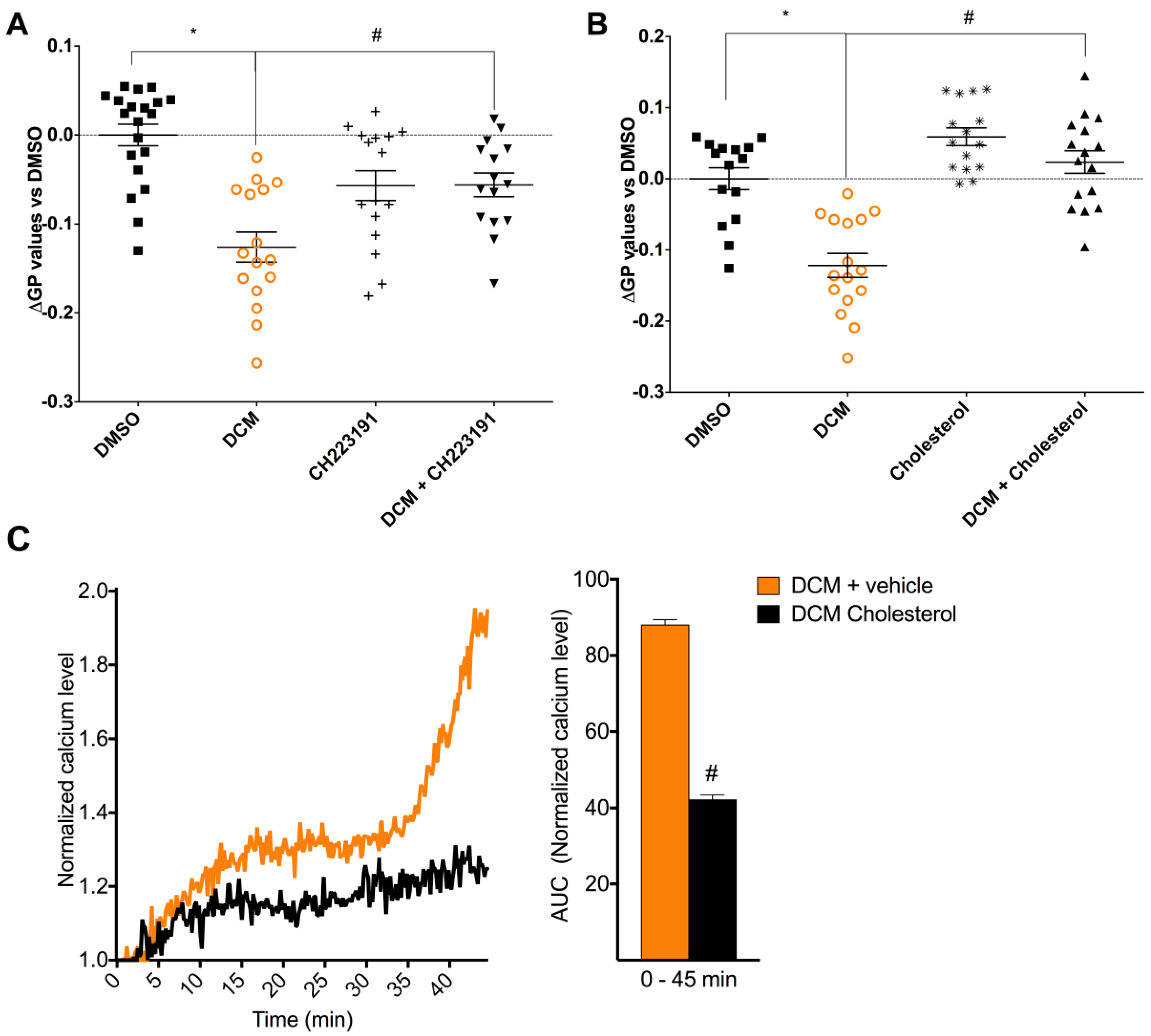
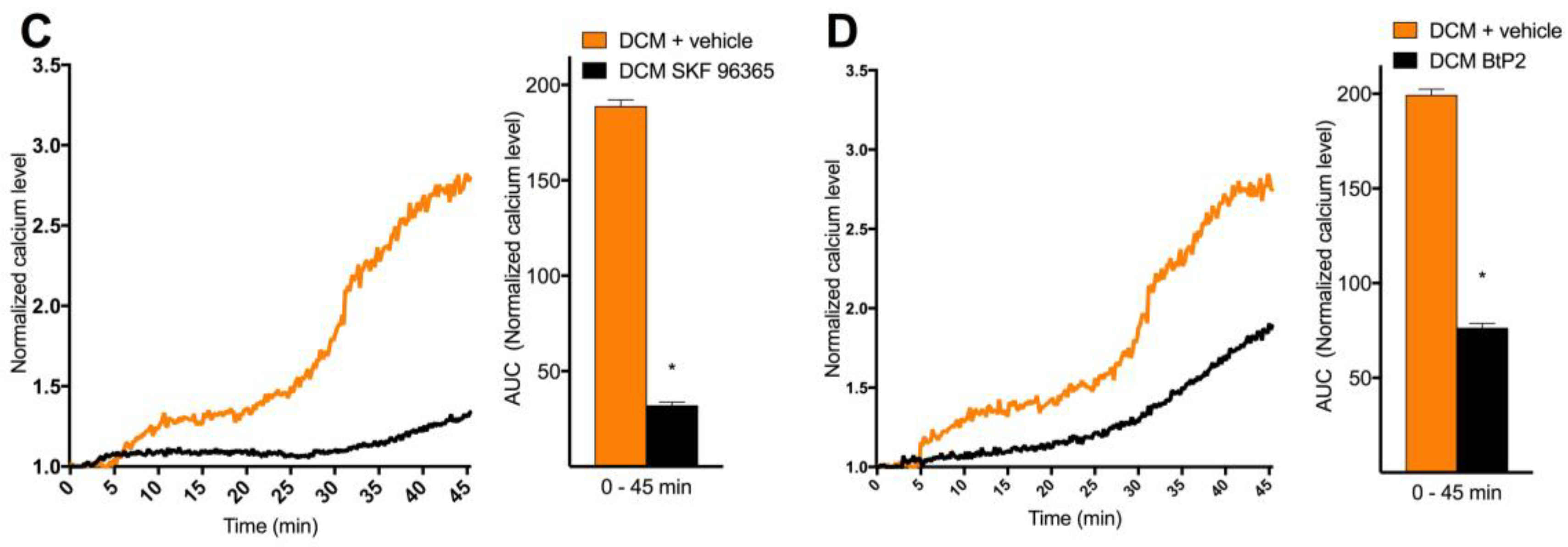
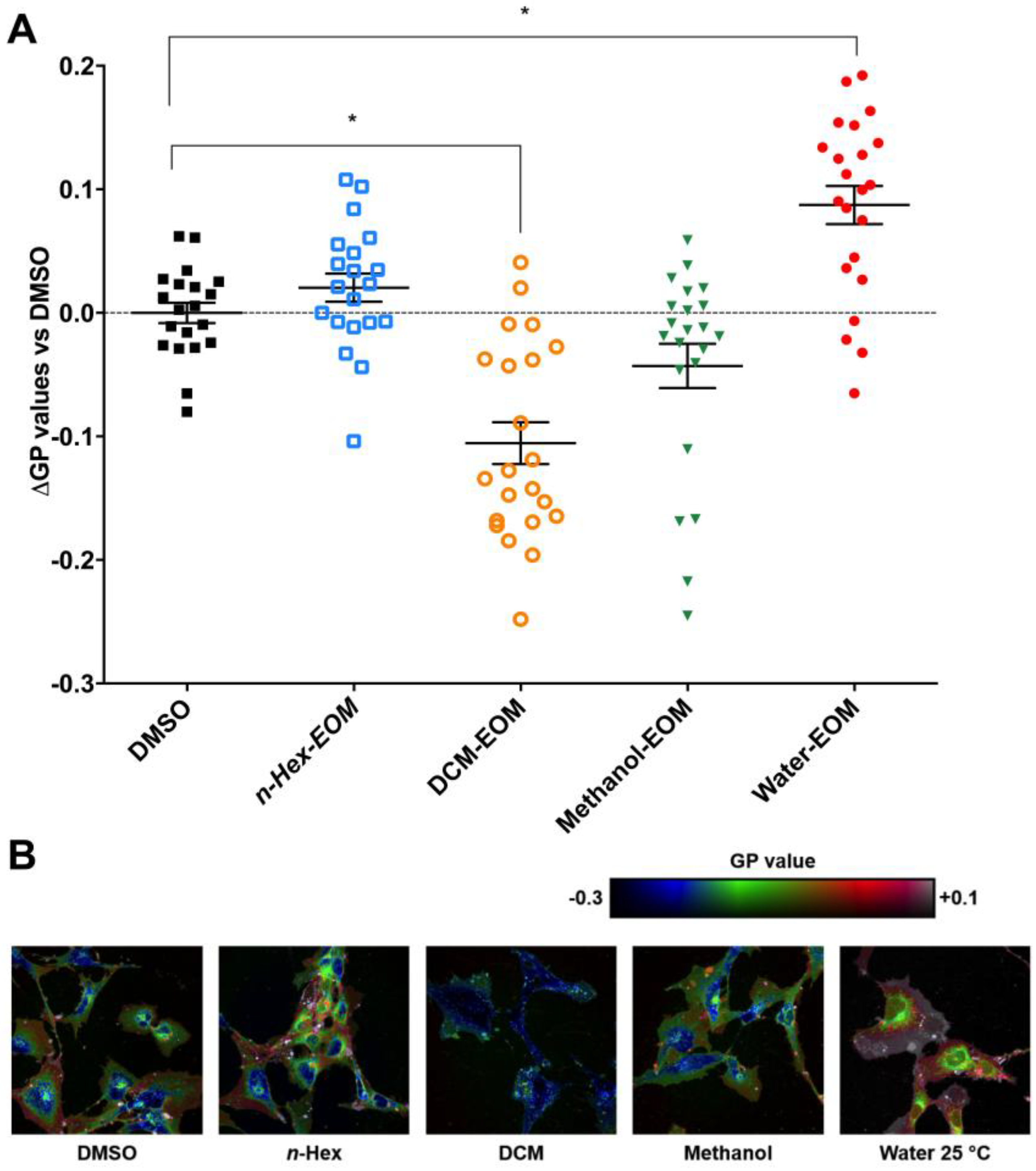
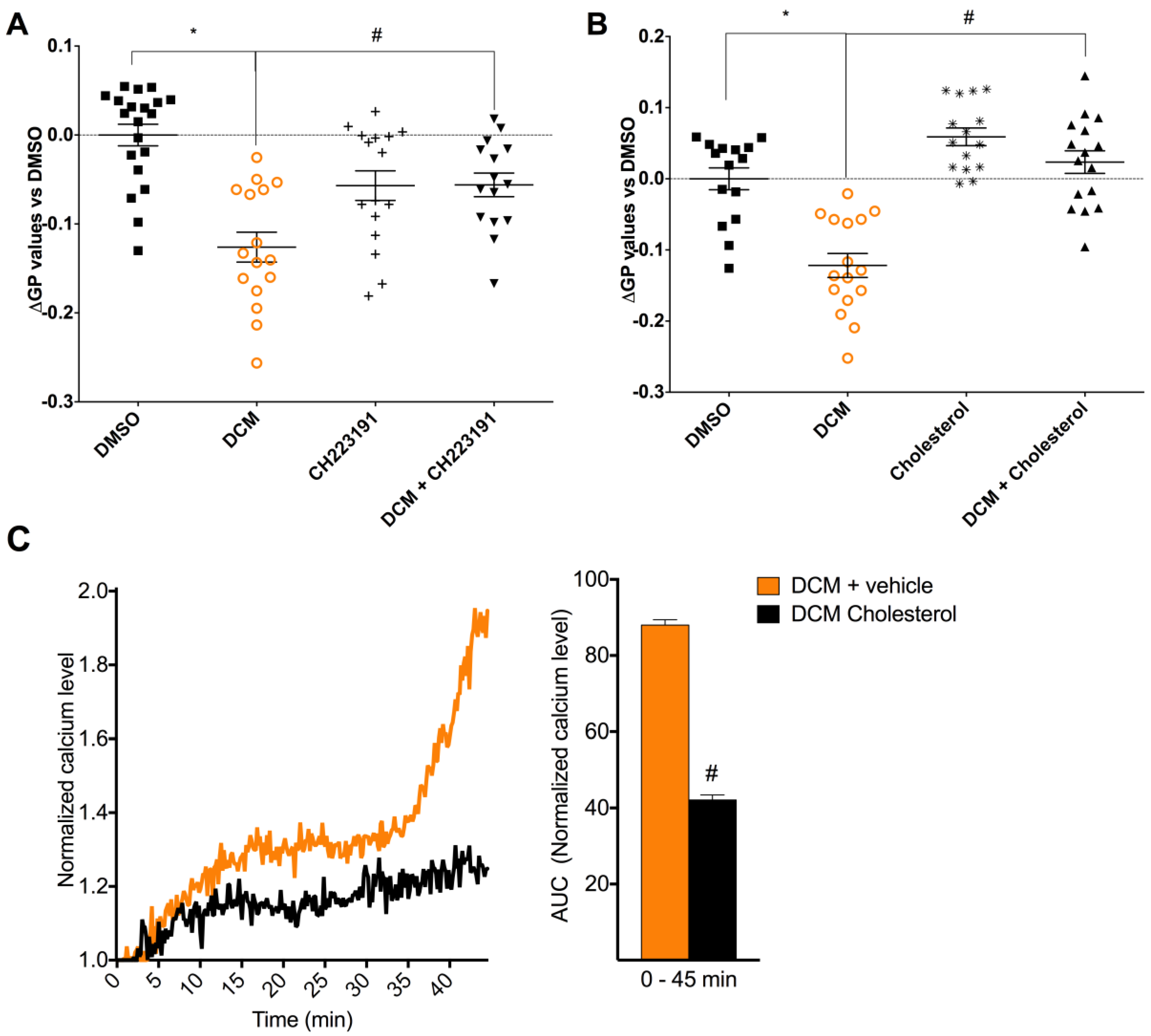
2.4. DCM-EOM Induced Effects on Membrane Order, Mechanisms Involved and Link to [Ca2+]i
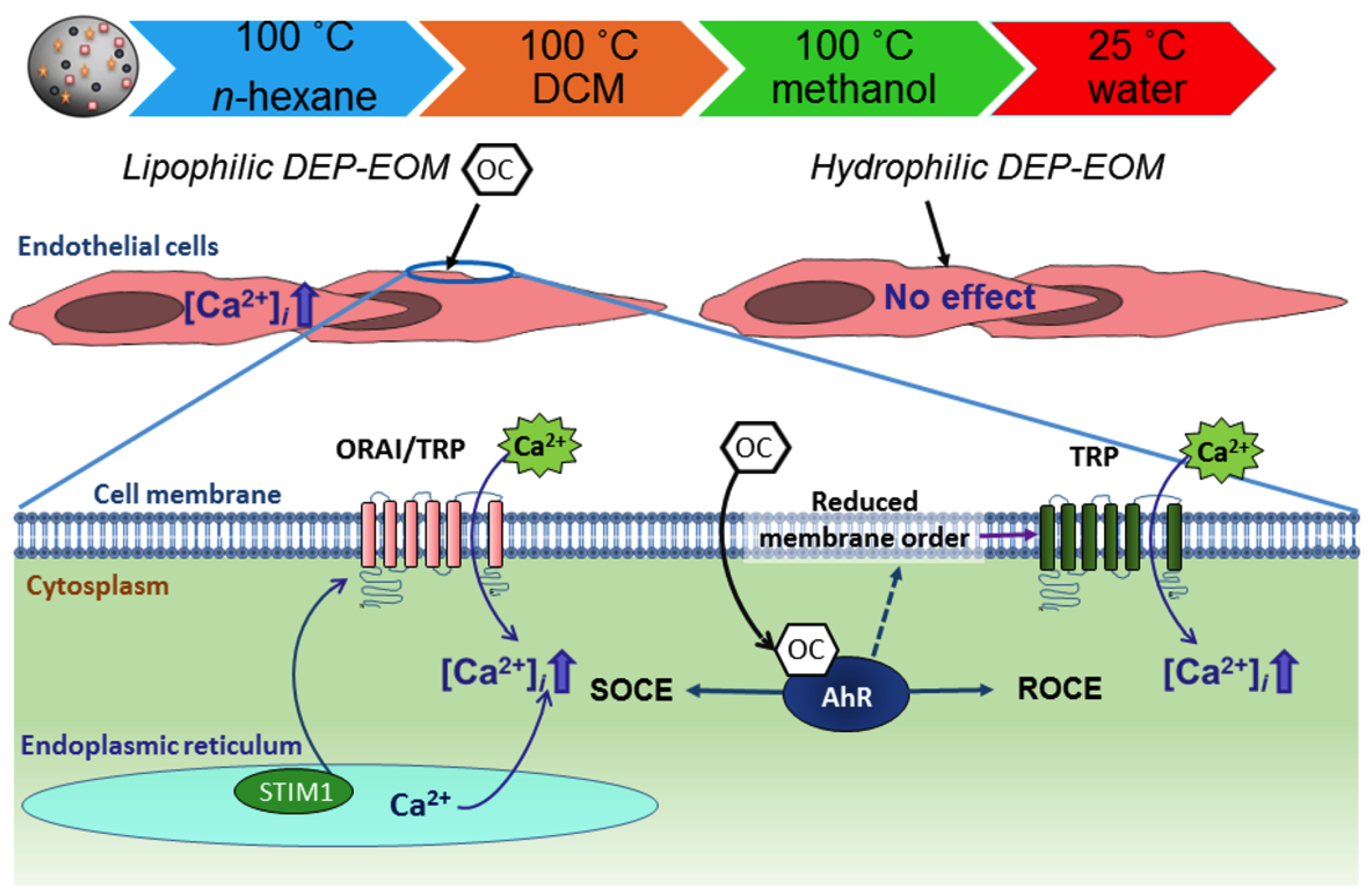
3. Discussion
4. Material and Methods
4.1. Chemicals
4.2. Diesel Exhaust Particles and Chemical Extraction
4.3. Chemical Analysis
4.4. Cell Culture
4.5. Small Interference RNA (SiRNA) Transfection
4.6. Calcium Measurements
4.7. Determination of Structural Perturbation of Plasma Membrane
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AhR | aryl hydrocarbon receptor |
| B[a]P | benzo[a]pyrene |
| β2AR | β2ARadrenergic receptors |
| CVD | cardiovascular disease |
| Cav1 | caveolin-1 |
| CYP | cytochrome P450 |
| Ca2+ | calcium |
| [Ca2+]i | intracytoplasmic calcium concentration |
| OC | organic chemicals |
| DEPs | diesel exhaust particles |
| DEP-EOM | extractable organic matter of DEP |
| DCM-EOM | dichloromethane |
| DMSO | dimethyl sulfoxide |
| HMEC-1 | human microvascular endothelial cells |
| IP3R | inositol trisphosphate receptor |
| GPCRs | G-protein coupled receptors |
| Methanol-EOM | methanol |
| NF-κB | nuclear factor-κB |
| n-Hex-EOM | DEP-EOM extracted by: n-hexane |
| PAHs | polycyclic aromatic hydrocarbons |
| PHECs | primary human endothelial cells |
| PM | particulate matter |
| ROS | reactive oxygen species |
| Water-EOM | water |
| XREs | AhR-response elements |
References
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Rajagopalan, S. Chronic air pollution exposure and endothelial dysfunction: What you can’t see-can harm you. J. Am. Coll. Cardiol. 2012, 60, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Davies, T.S. Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth. Factor Rev. 2015, 26, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Lawal, A.O.; Davids, L.M.; Marnewick, J.L. Diesel exhaust particles and endothelial cells dysfunction: An update. Toxicol. In Vitro 2016, 32, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Urch, B.; Brook, J.R.; Wasserstein, D.; Brook, R.D.; Rajagopalan, S.; Corey, P.; Silverman, F. Relative contributions of PM2.5 chemical constituents to acute arterial vasoconstriction in humans. Inhal. Toxicol. 2004, 16, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Christophersen, D.V.; Jacobsen, N.R.; Skovmand, A.; Gouveia, A.C.; Andersen, M.H.; Kermanizadeh, A.; Jensen, D.M.; Danielsen, P.H.; Roursgaard, M.; et al. Atherosclerosis and vasomotor dysfunction in arteries of animals after exposure to combustion-derived particulate matter or nanomaterials. Crit. Rev. Toxicol. 2016, 46, 437–476. [Google Scholar] [CrossRef] [PubMed]
- Tornqvist, H.; Mills, N.L.; Gonzalez, M.; Miller, M.R.; Robinson, S.D.; Megson, I.L.; Macnee, W.; Donaldson, K.; Soderberg, S.; Newby, D.E.; et al. Persistent endothelial dysfunction in humans after diesel exhaust inhalation. Am. J. Respir. Crit. Care Med. 2007, 176, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, S.; Takizawa, H.; Takami, K.; Desaki, M.; Okazaki, H.; Kasama, T.; Kobayashi, K.; Yamamoto, K.; Nakahara, K.; Tanaka, M.; et al. Benzene-extracted components are important for the major activity of diesel exhaust particles: Effect on interleukin-8 gene expression in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2001, 24, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Bonvallot, V.; Baeza-Squiban, A.; Baulig, A.; Brulant, S.; Boland, S.; Muzeau, F.; Barouki, R.; Marano, F. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am. J. Respir. Cell Mol. Biol. 2001, 25, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Totlandsdal, A.I.; Herseth, J.I.; Bolling, A.K.; Kubatova, A.; Braun, A.; Cochran, R.E.; Refsnes, M.; Ovrevik, J.; Lag, M. Differential effects of the particle core and organic extract of diesel exhaust particles. Toxicol. Lett. 2012, 208, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Keebaugh, A.J.; Sioutas, C.; Pakbin, P.; Schauer, J.J.; Mendez, L.B.; Kleinman, M.T. Is atherosclerotic disease associated with organic components of ambient fine particles? Sci. Total Environ. 2015, 533, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Alshaarawy, O.; Elbaz, H.A.; Andrew, M.E. The association of urinary polycyclic aromatic hydrocarbon biomarkers and cardiovascular disease in the US population. Environ. Int. 2016, 89–90, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Trasande, L.; Urbina, E.M.; Khoder, M.; Alghamdi, M.; Shabaj, I.; Alam, M.S.; Harrison, R.M.; Shamy, M. Polycyclic aromatic hydrocarbons, brachial artery distensibility and blood pressure among children residing near an oil refinery. Environ. Res. 2015, 136, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, C.E.; Gerde, P.; Hanberg, A.; Jernstrom, B.; Johansson, C.; Kyrklund, T.; Rannug, A.; Tornqvist, M.; Victorin, K.; Westerholm, R. Cancer risk assessment, indicators, and guidelines for polycyclic aromatic hydrocarbons in the ambient air. Environ. Health Perspect. 2002, 110, 451–488. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Jiang, X.; Cheng, S.; Chen, C.; Cao, X.; Tu, B. Effects of coke oven emissions and benzo[a]pyrene on blood pressure and electrocardiogram in coke oven workers. J. Occup. Health 2017, 59, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.; Murphy, G.; Barker, S.; Henk, W.; Penn, L. Combustion-derived ultrafine particles transport organic toxicants to target respiratory cells. Environ. Health Perspect. 2005, 113, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Gerde, P.; Muggenburg, B.A.; Lundborg, M.; Dahl, A.R. The rapid alveolar absorption of diesel soot-adsorbed benzo[a]pyrene: Bioavailability, metabolism and dosimetry of an inhaled particle-borne carcinogen. Carcinogenesis 2001, 22, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Brinchmann, B.C.; Skuland, T.; Rambøl, M.H.; Szoke, K.; Brinchmann, J.E.; Gutleb, A.C.; Moschini, E.; Kubátová, A.; Kukowski, K.; Ferrec, E.L.; et al. Lipophilic components of diesel exhaust particles induce pro-inflammatory responses in human endothelial cells through AhR dependent pathway(s). Part. Fibre Toxicol. 2018, in press. [Google Scholar]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.P.; Bradfield, C.A. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008, 21, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Savouret, J.-E.; Berdeaux, A.; Casper, R.E. The aryl hydrocarbon receptor and its xenobiotic ligands-triggers CVD. Nutr. Metab. Cardiovasc. Dis. 2003, 13, 104–113. [Google Scholar] [CrossRef]
- Kopf, P.G.; Huwe, J.K.; Walker, M.K. Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are associated with increased superoxide. Cardiovasc. Toxicol. 2008, 8, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Guyot, E.; Chevallier, A.; Barouki, R.; Coumoul, X. The AhR twist: Ligand-dependent AhR signaling and pharmaco-toxicological implications. Drug. Discov. Today 2013, 18, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Rabson, A.B.; Gallo, M.A. Ah receptor and NF-kappaB interactions: Mechanisms and physiological implications. Chem. Biol. Interact. 2002, 141, 97–115. [Google Scholar] [CrossRef]
- Matsumura, F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem. Pharmacol. 2009, 77, 608–626. [Google Scholar] [CrossRef] [PubMed]
- Tomkiewicz, C.; Herry, L.; Bui, L.C.; Metayer, C.; Bourdeloux, M.; Barouki, R.; Coumoul, X. The aryl hydrocarbon receptor regulates focal adhesion sites through a non-genomic FAK/Src pathway. Oncogene 2013, 32, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Senadheera, S.; Grayson, T.H.; Welsh, D.G.; Murphy, T.V. Calcium and endothelium-mediated vasodilator signaling. Adv. Exp. Med. Biol. 2012, 740, 811–831. [Google Scholar] [PubMed]
- Moller, P.; Mikkelsen, L.; Vesterdal, L.K.; Folkmann, J.K.; Forchhammer, L.; Roursgaard, M.; Danielsen, P.H.; Loft, S. Hazard identification of particulate matter on vasomotor dysfunction and progression of atherosclerosis. Crit. Rev. Toxicol. 2011, 41, 339–368. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Haddock, R.E.; Hill, C.E.; Chadha, P.S.; Kerr, P.M.; Welsh, D.G.; Plane, F. What’s where and why at a vascular myoendothelial microdomain signalling complex. Clin. Exp. Pharmacol. Physiol. 2009, 36, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Garland, C.J. Recent developments in vascular endothelial cell transient receptor potential channels. Circ. Res. 2005, 97, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Majkova, Z.; Toborek, M.; Hennig, B. The role of caveolae in endothelial cell dysfunction with a focus on nutrition and environmental toxicants. J. Cell Mol. Med. 2010, 14, 2359–2370. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Hasan, G. IP3R, store-operated Ca2+ entry and neuronal Ca2+ homoeostasis in Drosophila. Biochem. Soc. Trans. 2012, 40, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Erxleben, C.; Abramowitz, J.; Flockerzi, V.; Zhu, M.X.; Armstrong, D.L.; Birnbaumer, L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc. Natl. Acad. Sci. USA 2008, 105, 2895–2900. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium. 2017, 63, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W.; Tomita, T. Phospholipase C signaling and calcium influx. Adv. Biol. Regul. 2012, 52, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Fariss, M.W.; Gilmour, M.I.; Reilly, C.A.; Liedtke, W.; Ghio, A.J. Emerging mechanistic targets in lung injury induced by combustion-generated particles. Toxicol. Sci. 2013, 132, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kanju, P.; Patterson, M.; Chew, W.L.; Cho, S.H.; Gilmour, I.; Oliver, T.; Yasuda, R.; Ghio, A.; Simon, S.A.; et al. TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ. Health Perspect. 2011, 119, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Smedlund, K.; Bah, M.; Vazquez, G. On the role of endothelial TRPC3 channels in endothelial dysfunction and cardiovascular disease. Cardiovasc. Hematol. Agents Med. Chem. 2012, 10, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.N.; Earley, S. TRP channel Ca2+ sparklets: Fundamental signals underlying endothelium-dependent hyperpolarization. Am. J. Physiol. Cell Physiol. 2013, 305, C999–C1008. [Google Scholar] [CrossRef] [PubMed]
- Earley, S. TRPA1 channels in the vasculature. Br. J. Pharmacol. 2012, 167, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Barath, S.; Mills, N.L.; Lundback, M.; Tornqvist, H.; Lucking, A.J.; Langrish, J.P.; Soderberg, S.; Boman, C.; Westerholm, R.; Londahl, J.; et al. Impaired vascular function after exposure to diesel exhaust generated at urban transient running conditions. Part. Fibre Toxicol. 2010, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; Le Ferrec, E.; Lagadic-Gossmann, D.; Fardel, O. Aryl hydrocarbon receptor-independent up-regulation of intracellular calcium concentration by environmental polycyclic aromatic hydrocarbons in human endothelial HMEC-1 cells. Environ. Toxicol. 2012, 27, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; Le Ferrec, E.; Holme, J.A.; Fardel, O.; Lagadic-Gossmann, D.; Ovrevik, J. Calcium signaling and beta2-adrenergic receptors regulate 1-nitropyrene induced CXCL8 responses in BEAS-2B cells. Toxicol. Vitro 2014, 28, 1153–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deering-Rice, C.E.; Romero, E.G.; Shapiro, D.; Hughen, R.W.; Light, A.R.; Yost, G.S.; Veranth, J.M.; Reilly, C.A. Electrophilic components of diesel exhaust particles (DEP) activate transient receptor potential ankyrin-1 (TRPA1): a probable mechanism of acute pulmonary toxicity for DEP. Chem. Res. Toxicol. 2011, 24, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z.; Zeng, F.; Boulay, G.; Grimm, C.; Harteneck, C.; Beech, D.J. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: A differential, extracellular and voltage-dependent effect. Br. J. Pharmacol. 2005, 145, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Z.; Gu, Q.; Wang, C.; Colton, C.K.; Tang, J.; Kinoshita-Kawada, M.; Lee, L.Y.; Wood, J.D.; Zhu, M.X. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 2004, 279, 35741–35748. [Google Scholar] [CrossRef] [PubMed]
- Pani, B.; Singh, B.B. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium. 2009, 45, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, P.; Phillips, R. Membrane-protein interactions in mechanosensitive channels. Biophys. J. 2005, 88, 880–902. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.A. Influence of increased membrane cholesterol on membrane fluidity and cell function in human red blood cells. J. Supramol. Struct. 1978, 8, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Lawal, A.O.; Zhang, M.; Dittmar, M.; Lulla, A.; Araujo, J.A. Heme oxygenase-1 protects endothelial cells from the toxicity of air pollutant chemicals. Toxicol. Appl. Pharmacol. 2015, 284, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.W.; Zhao, W.; Li, N.; Barajas, B.; Kleinman, M.; Sioutas, C.; Horvath, S.; Lusis, A.J.; Nel, A.; Araujo, J.A. Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol. 2007, 8, R149. [Google Scholar] [CrossRef] [PubMed]
- DeMarini, D.M.; Brooks, L.R.; Warren, S.H.; Kobayashi, T.; Gilmour, M.I.; Singh, P. Bioassay-directed fractionation and salmonella mutagenicity of automobile and forklift diesel exhaust particles. Environ. Health Perspect. 2004, 112, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Zeng, W.; Yuan, J.P.; Shin, D.M.; Worley, P.F.; Muallem, S. Native Store-operated Ca2+ Influx Requires the Channel Function of Orai1 and TRPC1. J. Biol. Chem. 2009, 284, 9733–9741. [Google Scholar] [CrossRef] [PubMed]
- Ahmmed, G.U.; Mehta, D.; Vogel, S.; Holinstat, M.; Paria, B.C.; Tiruppathi, C.; Malik, A.B. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J. Biol. Chem. 2004, 279, 20941–20949. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; Horn, N.A.; Huynh, T.; Kelava, L.; Lansman, J.B. Evidence TRPV4 contributes to mechanosensitive ion channels in mouse skeletal muscle fibers. Channels 2012, 6, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Hildebrand, M.E.; Garcia, E.; Snutch, T.P. The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharmacol. 2010, 160, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Hartmannsgruber, V.; Heyken, W.T.; Kacik, M.; Kaistha, A.; Grgic, I.; Harteneck, C.; Liedtke, W.; Hoyer, J.; Kohler, R. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS ONE 2007, 2, e827. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Plummer, N.W.; George, M.D.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. USA 2009, 106, 3202–3206. [Google Scholar] [CrossRef] [PubMed]
- Marlowe, J.L.; Puga, A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J. Cell. Biochem. 2005, 96, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Matsumura, F. Roles of cytosolic phospholipase A2 and Src kinase in the early action of 2,3,7,8-tetrachlorodibenzo-p-dioxin through a nongenomic pathway in MCF10A cells. Mol. Pharmacol. 2008, 74, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.; Hoffer, A.; Zhou, S.; Bohm, J.M.; Leikauf, G.D.; Shertzer, H.G. Sustained Increase in Intracellular Free Calcium and Activation of Cyclooxygenase-2 Expression in Mouse Hepatoma Cells Treated with Dioxin. Biochem. Pharmacol. 1997, 54, 1287–1296. [Google Scholar] [CrossRef]
- Rey-Barroso, J. The Dioxin receptor modulates Caveolin-1 mobilization during directional migration: Role of cholesterol. Cell Commun. Signal. 2014, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Iwamuro, Y.; Miwa, S.; Minowa, T.; Enoki, T.; Zhang, X.F.; Ishikawa, M.; Hashimoto, N.; Masaki, T. Activation of two types of Ca2+-permeable nonselective cation channel by endothelin-1 in A7r5 cells. Br. J. Pharmacol. 1998, 124, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Amaro, M.; Reina, F.; Hof, M.; Eggeling, C.; Sezgin, E. Laurdan and Di-4-ANEPPDHQ probe different properties of the membrane. J. Phys. D Appl. Phys. 2017, 50, 134004. [Google Scholar] [CrossRef] [PubMed]
- Totlandsdal, A.I.; Cassee, F.R.; Schwarze, P.; Refsnes, M.; Lag, M. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells. Part. Fibre Toxicol. 2010, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Andrysik, Z.; Vondracek, J.; Marvanova, S.; Ciganek, M.; Neca, J.; Pencikova, K.; Mahadevan, B.; Topinka, J.; Baird, W.M.; Kozubik, A.; et al. Activation of the aryl hydrocarbon receptor is the major toxic mode of action of an organic extract of a reference urban dust particulate matter mixture: The role of polycyclic aromatic hydrocarbons. Mutat. Res. 2011, 714, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Y.; Scholze, A.; Kreutz, R.; Wehland-von-Trebra, M.; Zidek, W.; Zhu, Z.M.; Tepel, M. Monocytes from spontaneously hypertensive rats show increased store-operated and second messenger-operated calcium influx mediated by transient receptor potential canonical Type 3 channels. Am. J. Hypertens. 2007, 20, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Y.; Thilo, F.; Scholze, A.; Wittstock, A.; Zhao, Z.G.; Harteneck, C.; Zidek, W.; Zhu, Z.M.; Tepel, M. Increased store-operated and 1-oleoyl-2-acetyl-sn-glycerol-induced calcium influx in monocytes is mediated by transient receptor potential canonical channels in human essential hypertension. J. Hypertens. 2007, 25, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.M.; Adar, S.D.; Szpiro, A.A.; Jorgensen, N.W.; Van Hee, V.C.; Barr, R.G.; O’Neill, M.S.; Herrington, D.M.; Polak, J.F.; Kaufman, J.D. Vascular responses to long- and short-term exposure to fine particulate matter: MESA Air (Multi-Ethnic Study of Atherosclerosis and Air Pollution). J. Am. Coll. Cardiol. 2012, 60, 2158–2166. [Google Scholar] [CrossRef] [PubMed]
- Lucking, A.J.; Lundback, M.; Barath, S.L.; Mills, N.L.; Sidhu, M.K.; Langrish, J.P.; Boon, N.A.; Pourazar, J.; Badimon, J.J.; Gerlofs-Nijland, M.E.; et al. Particle traps prevent adverse vascular and prothrombotic effects of diesel engine exhaust inhalation in men. Circulation 2011, 123, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, M.; le Ferrec, E.; Lagadic-Gossmann, D.; Corre, S.; Gilot, D.; Lecureur, V.; Monteiro, P.; Rauch, C.; Galibert, M.D.; Fardel, O. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J. Biol. Chem. 2006, 281, 19906–19915. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Gilot, D.; le Ferrec, E.; Rauch, C.; Lagadic-Gossmann, D.; Fardel, O. Dioxin-mediated up-regulation of aryl hydrocarbon receptor target genes is dependent on the calcium/calmodulin/CaMKIalpha pathway. Mol. Pharmacol. 2008, 73, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Dharmashankar, K.; Widlansky, M.E. Vascular endothelial function and hypertension: Insights and directions. Curr. Hypertens. Rep. 2010, 12, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Brinchmann, B.C.; Ferrec, E.L.; Podechard, N.; Lagadic-Gossmann, D.; Holme, J.A.; Øvrevik, J. Organic chemicals from diesel exhaust particles affects intracellular calcium, inflammation and β-adrenoceptors in endothelial cells. Toxicol. Lett. 2018. under review. [Google Scholar]
- Araujo, J.A.; Nel, A.E. Particulate matter and atherosclerosis: Role of particle size, composition and oxidative stress. Part. Fibre Toxicol. 2009, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.G.; Cambier, S.; Hennen, J.; Legay, S.; Serchi, T.; Nelissen, I.; Chary, A.; Moschini, E.; Krein, A.; Blomeke, B.; et al. Endothelial responses of the alveolar barrier in vitro in a dose-controlled exposure to diesel exhaust particulate matter. Part. Fibre Toxicol. 2017, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Raftis, J.B.; Langrish, J.P.; McLean, S.G.; Samutrtai, P.; Connell, S.P.; Wilson, S.; Vesey, A.T.; Fokkens, P.H.B.; Boere, A.J.F.; et al. Inhaled Nanoparticles Accumulate at Sites of Vascular Disease. ACS Nano 2017, 11, 4542–4552. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Raftis, J.B.; Langrish, J.P.; McLean, S.G.; Samutrtai, P.; Connell, S.P.; Wilson, S.; Vesey, A.T.; Fokkens, P.H.B.; Boere, A.J.F.; et al. Correction to “Inhaled Nanoparticles Accumulate at Sites of Vascular Disease”. ACS Nano 2017, 11, 10623–10624. [Google Scholar] [CrossRef] [PubMed]
- Totlandsdal, A.I.; Ovrevik, J.; Cochran, R.E.; Herseth, J.I.; Bolling, A.K.; Lag, M.; Schwarze, P.; Lilleaas, E.; Holme, J.A.; Kubatova, A. The occurrence of polycyclic aromatic hydrocarbons and their derivatives and the proinflammatory potential of fractionated extracts of diesel exhaust and wood smoke particles. J. Environ. Sci. Health A Tox. Hazard. Subst. Environ. Eng. 2014, 49, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Kubátová, A.; Lahren, T.J.; Beránek, J.; Smoliakova, I.P.; Braun, A.; Huggins, F.E. Extractable Organic Carbon and its Differentiation by Polarity in Diesel Exhaust, Wood Smoke, and Urban Particulate Matter. Aerosol. Sci. Technol. 2009, 43, 714–729. [Google Scholar] [CrossRef]
- Stevens, T.; Cho, S.H.; Linak, W.P.; Gilmour, M.I. Differential potentiation of allergic lung disease in mice exposed to chemically distinct diesel samples. Toxicol. Sci. 2009, 107, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.A.; Barajas, B.; Kleinman, M.; Wang, X.; Bennett, B.J.; Gong, K.W.; Navab, M.; Harkema, J.; Sioutas, C.; Lusis, A.J.; et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ. Res. 2008, 102, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Cochran, R.E.; Kubátová, A. Pressurised fluid extraction of polycyclic aromatic hydrocarbons and their polar oxidation products from atmospheric particles. Int. J. Environ. Anal. Chem. 2015, 95, 434–452. [Google Scholar] [CrossRef]
- Zhao, B.; Degroot, D.E.; Hayashi, A.; He, G.; Denison, M.S. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol. Sci. 2010, 117, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Uchida, K.; Takahashi, N.; Iwata, Y.; Wakabayashi, S.; Goto, T.; Kawada, T.; Tominaga, M. Activation of TRPV2 negatively regulates the differentiation of mouse brown adipocytes. Pflugers Arch. Eur. J. Physiol. 2016, 468, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Morales, J.L.; Mottram, L.F.; Iyer, A.; Peterson, B.R.; August, A. Structural requirements for the inhibition of calcium mobilization and mast cell activation by the pyrazole derivative BTP2. Int. J. Biochem. Cell Biol. 2011, 43, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; Levoin, N.; Paris, H.; N’Diaye, M.; Courtois, A.; Uriac, P.; Lagadic-Gossmann, D.; Fardel, O.; Le Ferrec, E. Induction of intracellular calcium concentration by environmental benzo(a)pyrene involves a β2-adrenergic receptor/adenylyl cyclase/Epac-1/inositol 1,4,5-trisphosphate pathway in endothelial cells. J. Biol. Chem. 2012, 287, 4041–4052. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.M.; Rentero, C.; Magenau, A.; Abu-Siniyeh, A.; Gaus, K. Quantitative imaging of membrane lipid order in cells and organisms. Nat. Protoc. 2011, 7, 24–35. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemicals | n-Hex-EOM | DCM-EOM | Methanol-EOM | Water-EOM |
|---|---|---|---|---|
| OC | 153 ± 29 | 113 ± 23 | 62 ± 15 | ND |
| Aliphatics | 100 ± 8 | 17 ± 16 | ND | ND |
| PAHs | 1.65 ± 0.07 | 0.13 ± 0.02 | ND | ND |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brinchmann, B.C.; Le Ferrec, E.; Podechard, N.; Lagadic-Gossmann, D.; Shoji, K.F.; Penna, A.; Kukowski, K.; Kubátová, A.; Holme, J.A.; Øvrevik, J. Lipophilic Chemicals from Diesel Exhaust Particles Trigger Calcium Response in Human Endothelial Cells via Aryl Hydrocarbon Receptor Non-Genomic Signalling. Int. J. Mol. Sci. 2018, 19, 1429. https://doi.org/10.3390/ijms19051429
Brinchmann BC, Le Ferrec E, Podechard N, Lagadic-Gossmann D, Shoji KF, Penna A, Kukowski K, Kubátová A, Holme JA, Øvrevik J. Lipophilic Chemicals from Diesel Exhaust Particles Trigger Calcium Response in Human Endothelial Cells via Aryl Hydrocarbon Receptor Non-Genomic Signalling. International Journal of Molecular Sciences. 2018; 19(5):1429. https://doi.org/10.3390/ijms19051429
Chicago/Turabian StyleBrinchmann, Bendik C., Eric Le Ferrec, Normand Podechard, Dominique Lagadic-Gossmann, Kenji F. Shoji, Aubin Penna, Klara Kukowski, Alena Kubátová, Jørn A. Holme, and Johan Øvrevik. 2018. "Lipophilic Chemicals from Diesel Exhaust Particles Trigger Calcium Response in Human Endothelial Cells via Aryl Hydrocarbon Receptor Non-Genomic Signalling" International Journal of Molecular Sciences 19, no. 5: 1429. https://doi.org/10.3390/ijms19051429