Ion Channels in Pulmonary Hypertension: A Therapeutic Interest?


 , and
, and 
Abstract
1. Introduction
1.1. Ion Channels in Pulmonary Vasculature
- -
- The cation channels, which comprise Na+, Ca2+, and K+ channels
- -
- The anion channels, which include Cl− channels and bicarbonate channels
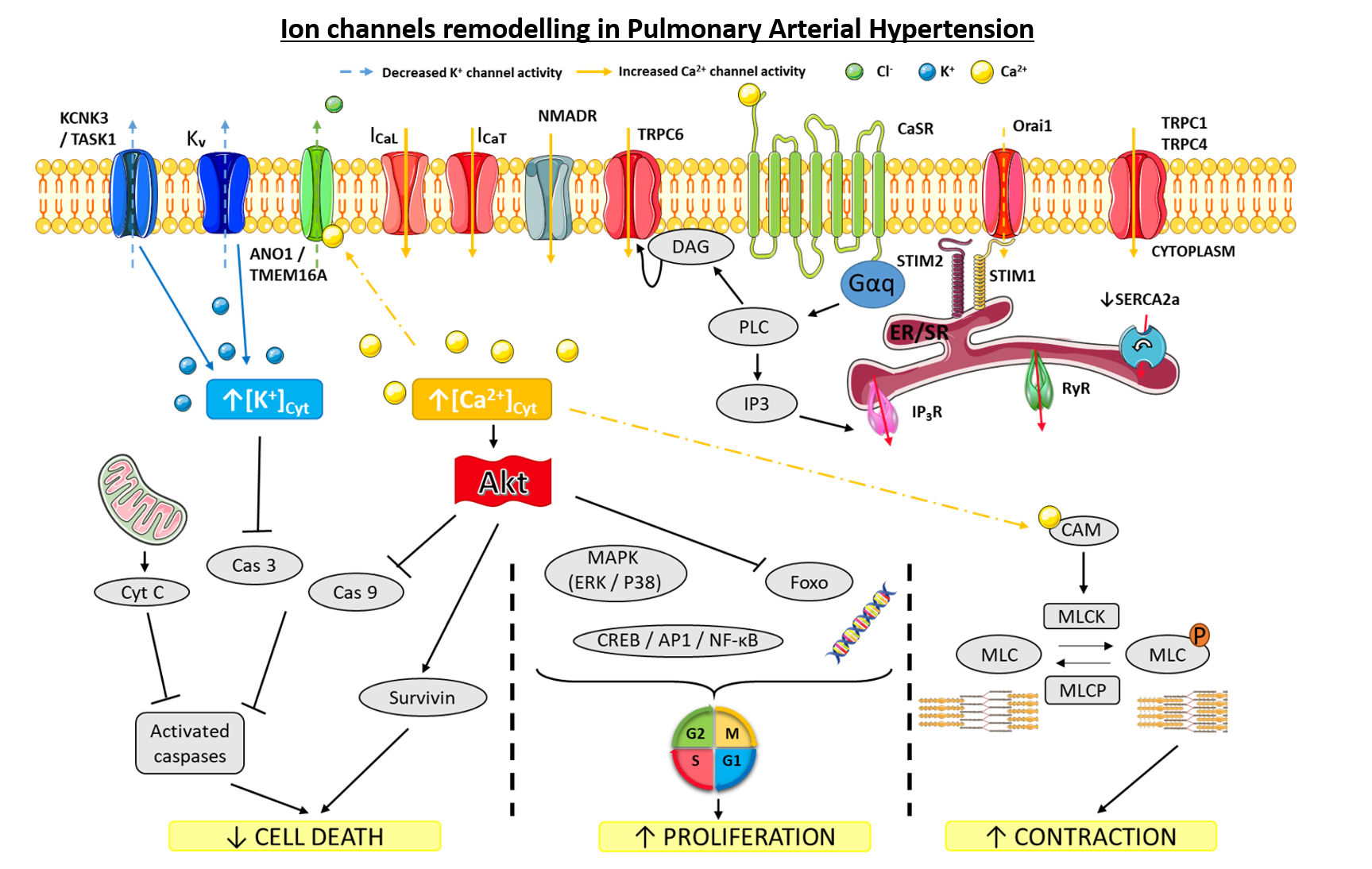
1.2. Role of Ion Channels in Pulmonary Arterial Tone
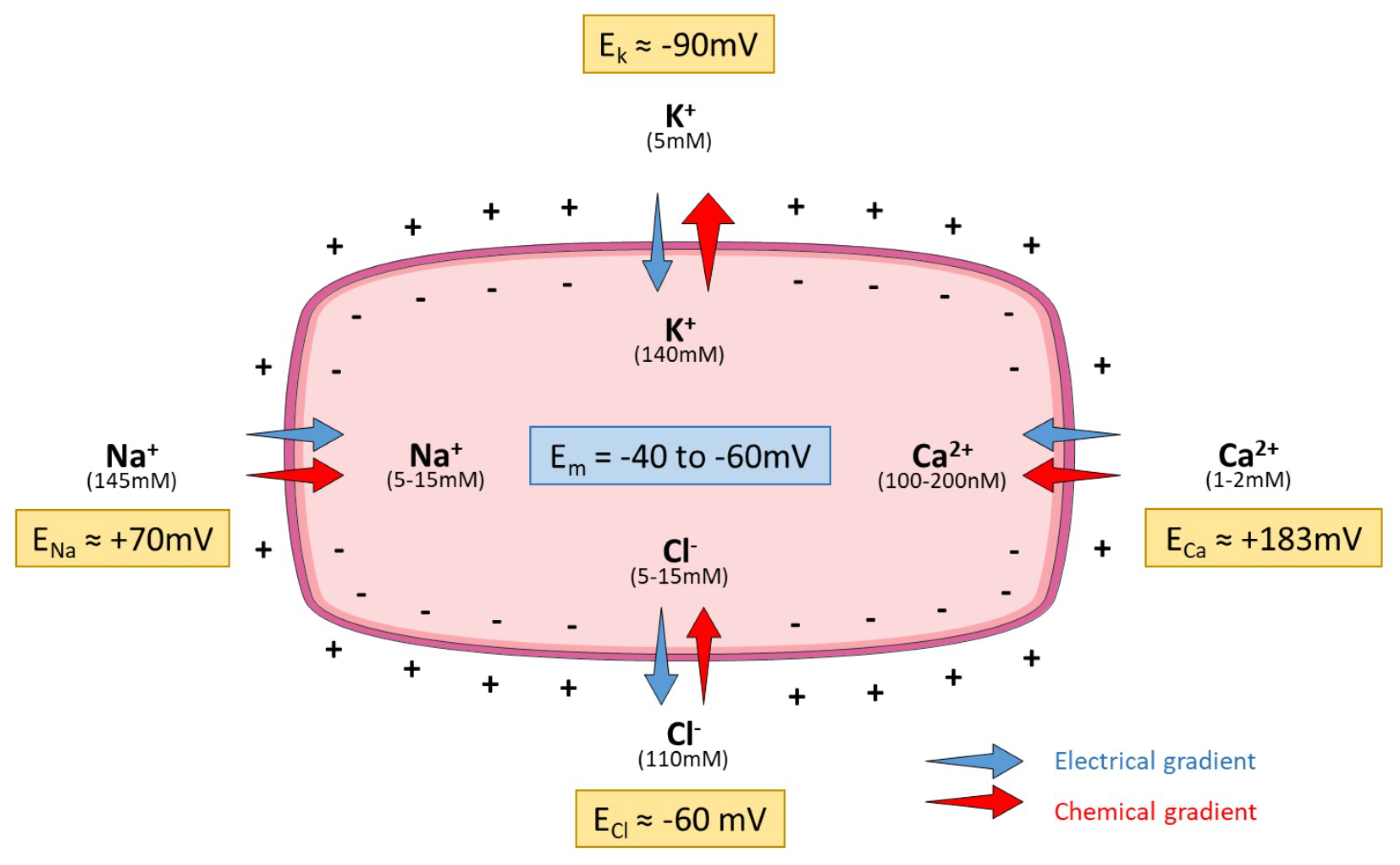
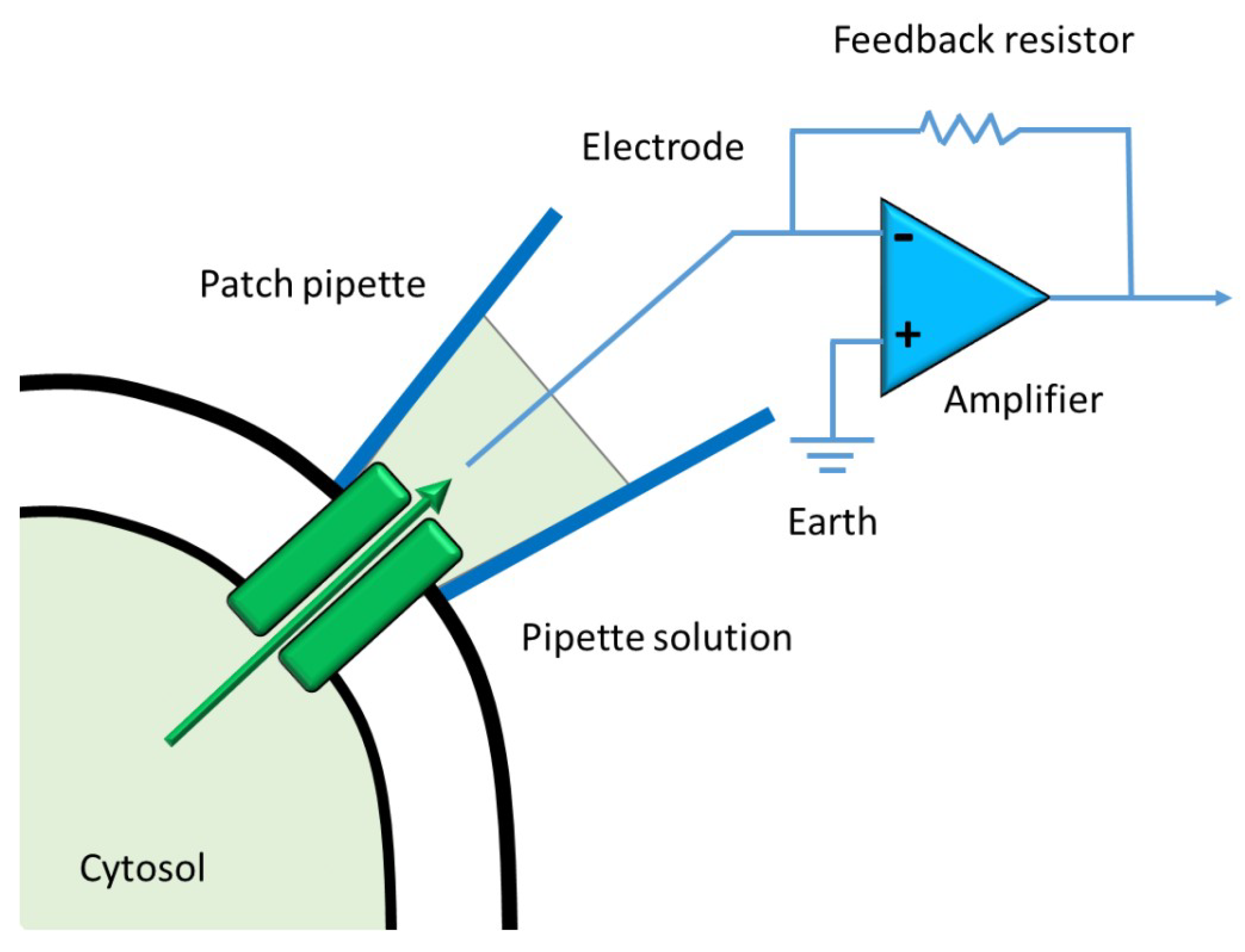
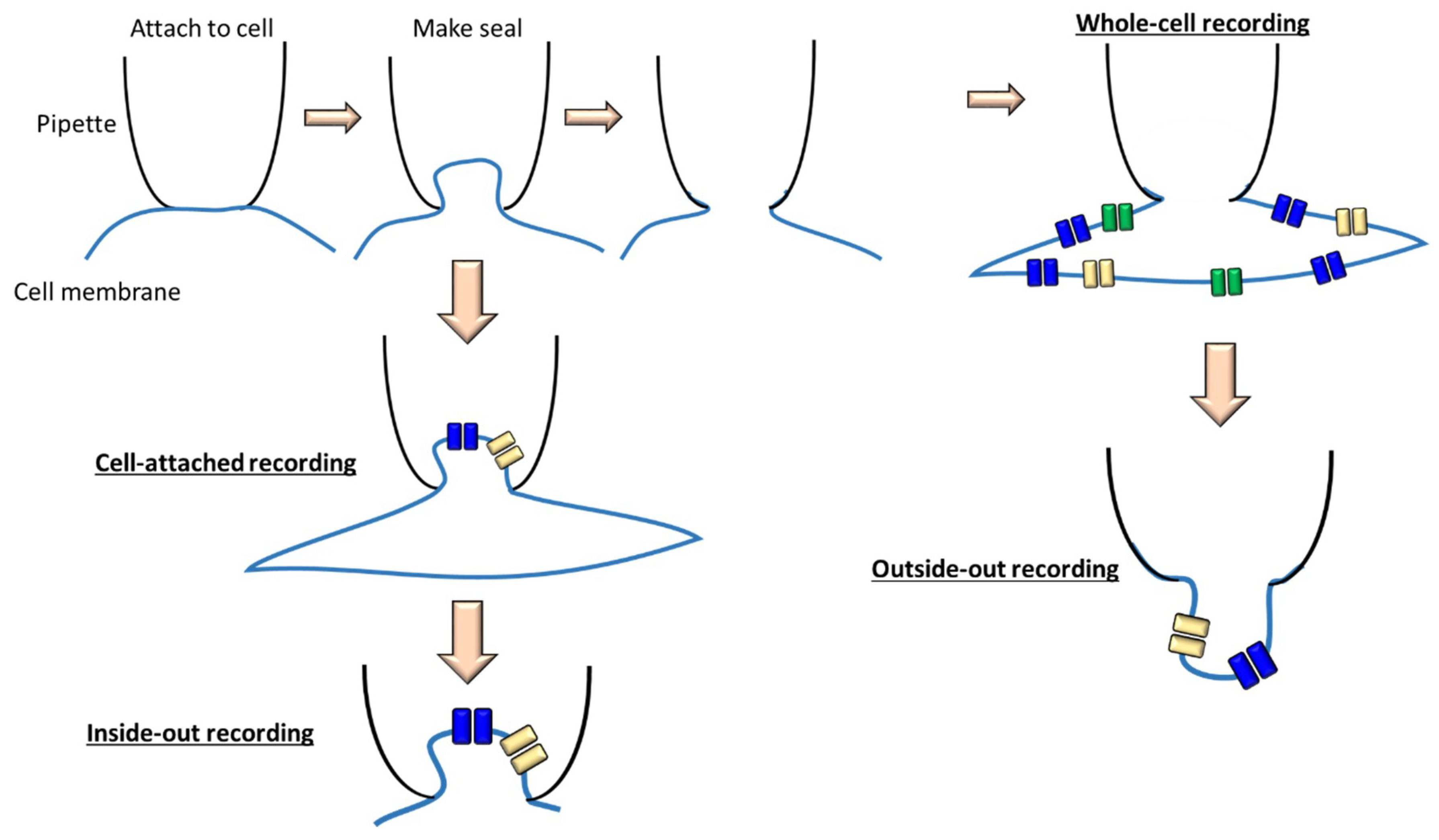
1.3. Current Methods to Measure Ion Channels Activities
1.4. Preclinical Animal Model of PH
2. Functional and Molecular Classification of Ion Channels in the Pulmonary Circulation
2.1. K+ Channels
2.1.1. Inward Rectifier Channel (Kir) Family
2.1.2. ATP-Sensitive K+ Channel (KATP)
Pulmonary Arterial Smooth Muscle Cells
Pulmonary Arterial Endothelial Cells
2.1.3. Voltage-Gated K+ Channels (Kv)
Pulmonary Smooth Muscle Cells
Pulmonary Endothelial Cells
2.1.4. Ca2+-Activated K+ Channels (KCa)
Pulmonary Smooth Muscle Cells
Pulmonary Endothelial Cells
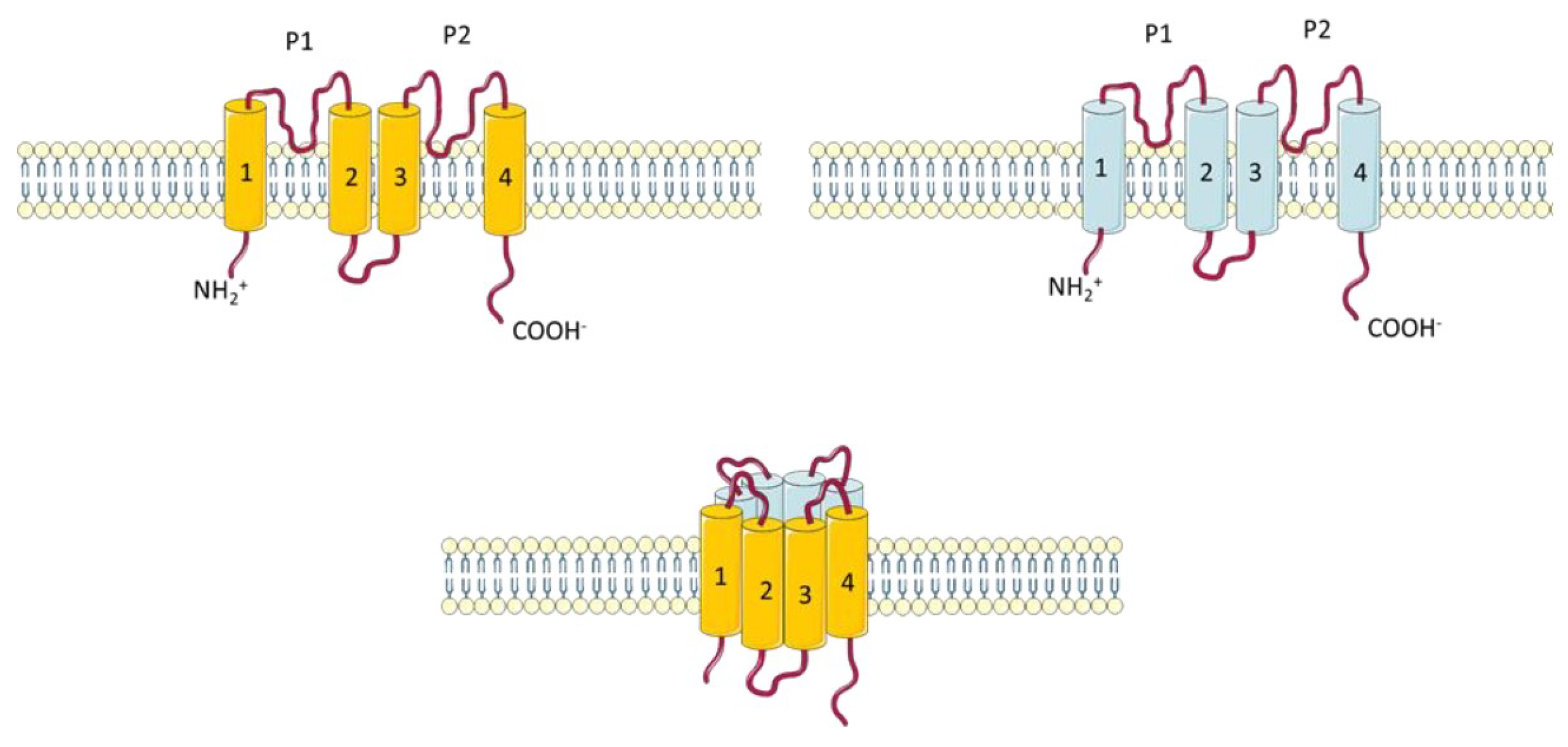
2.1.5. Two Pore Potassium Channels (K2P)
Pulmonary Arterial Smooth Muscle Cells
Pulmonary Arterial Endothelial Cells
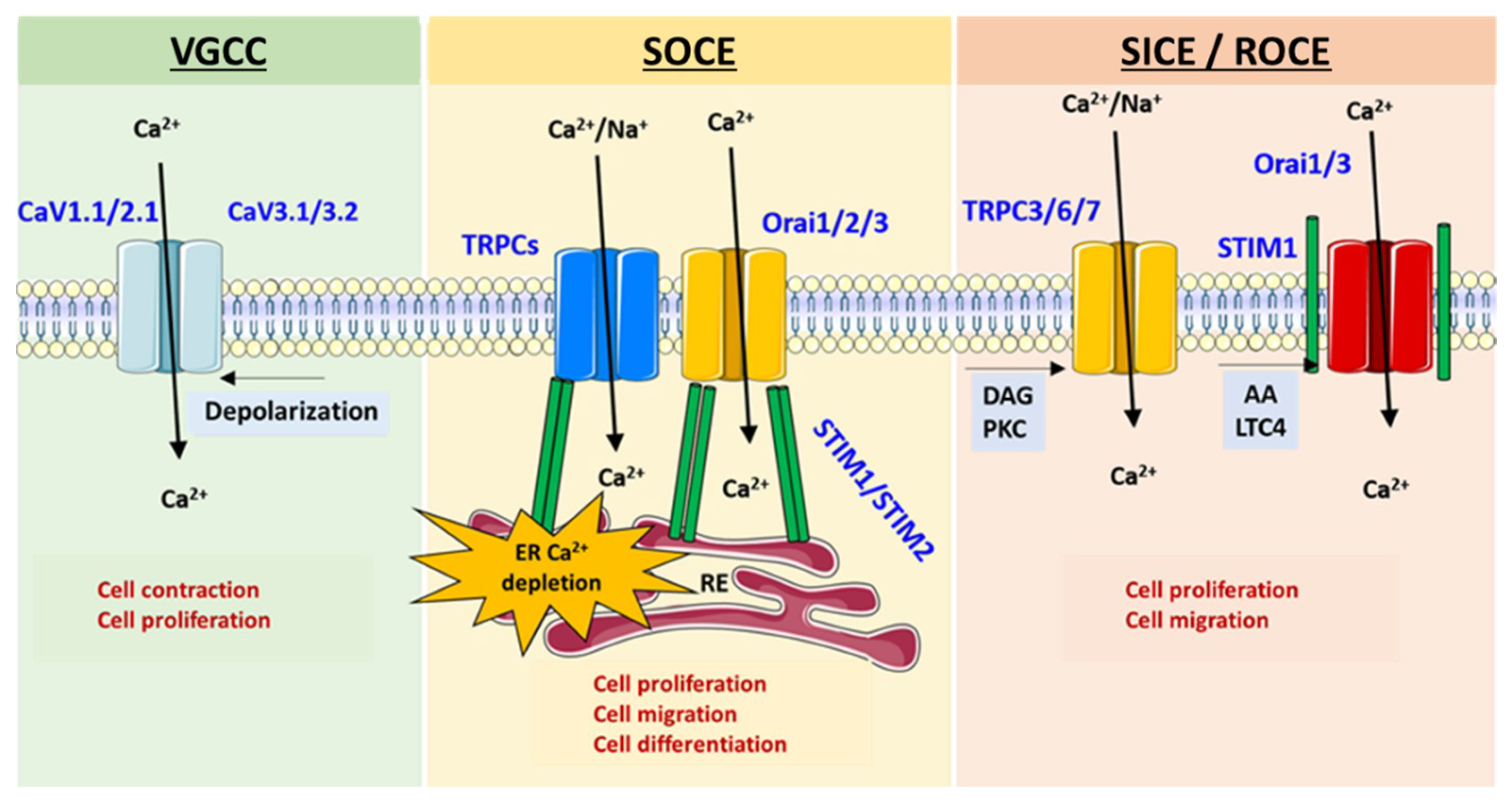
2.2. Ca2+ Channels
- (i)
- Voltage-Gated Ca2+ Channels (VGCC)
- (ii)
- Non-voltage-dependent Store-Operated Ca2+ Entry (SOCE)
- (iii)
- Non-voltage-dependent Store-Independent Ca2+ Entry (SICE) also called Receptor-Operated Ca2+ Entry (ROCE)
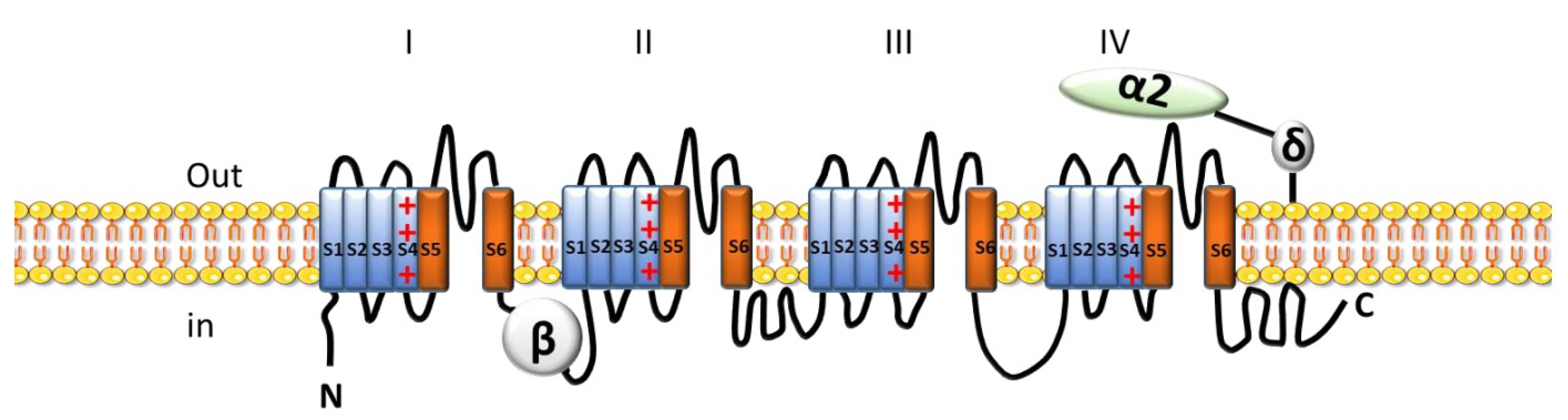
2.2.1. Voltage-Gated Ca2+ Channels (VGCC)
Pulmonary Arterial Smooth Muscle Cells
Pulmonary Arterial Endothelial Cells
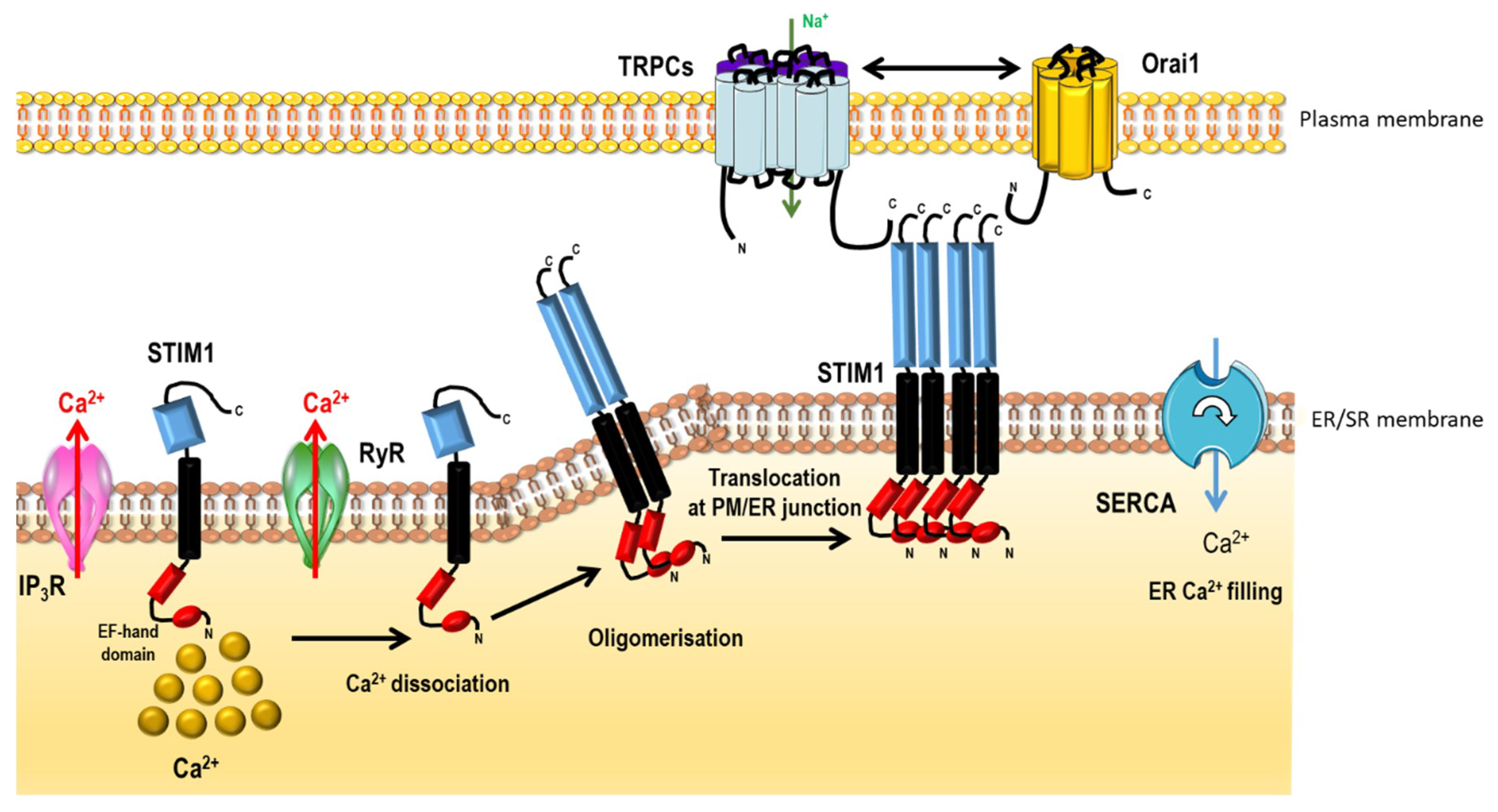
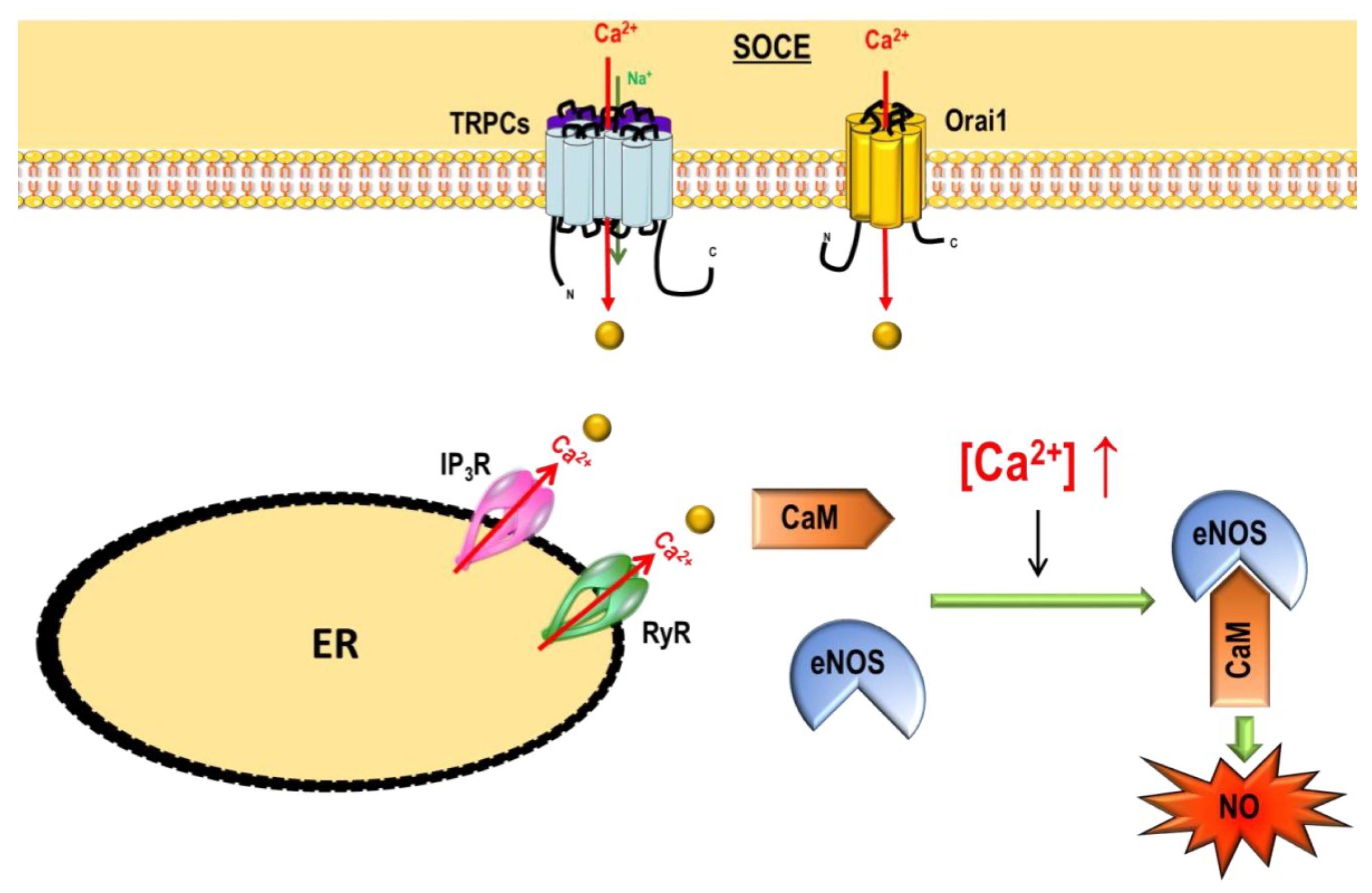
2.2.2. Non-Voltage-Dependent Store-Operated Ca2+ Entry (SOCE)
2.2.3. Non-Voltage-Dependent Store-Independent Ca2+ Entry (SICE)
Pulmonary Arterial Smooth Muscle Cells
Pulmonary Arterial Endothelial Cells
2.2.4. Other Ca2+ or Cationic Channels in Pulmonary Arteries
2.2.5. N-Methyl-d-Aspartate Receptor (NMDAR)
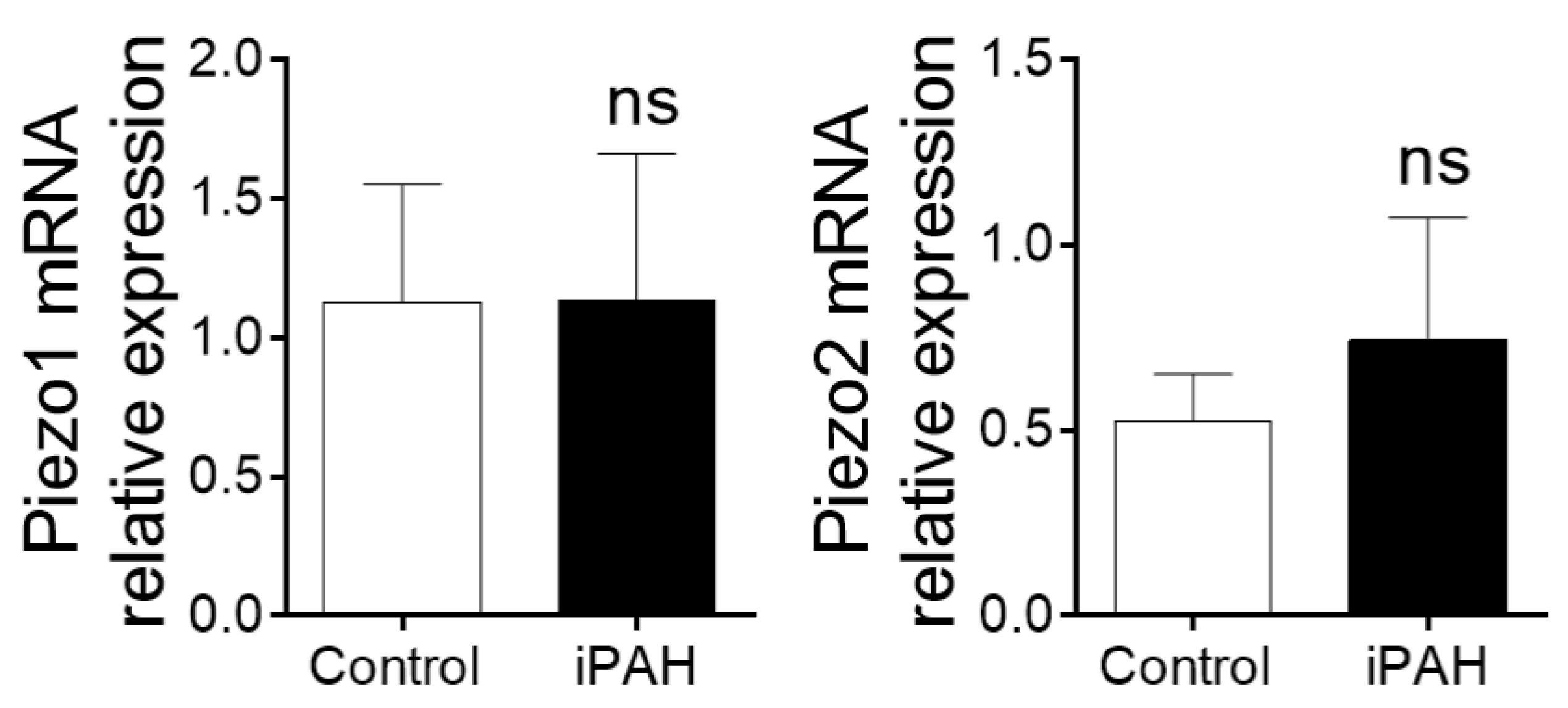
2.2.6. Ca2+ Stretch Channels (Piezo1–2)
2.3. Na+ Channels
2.3.1. Epithelial Na+ channels (ENaC)
2.3.2. Voltage-Gated Na+ Channels
2.4. Cl− channels
2.4.1. TMEM16A Family
2.4.2. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)
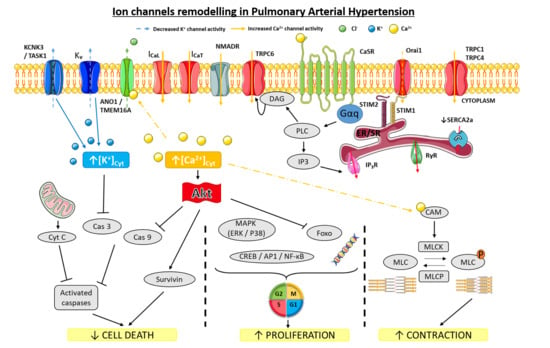
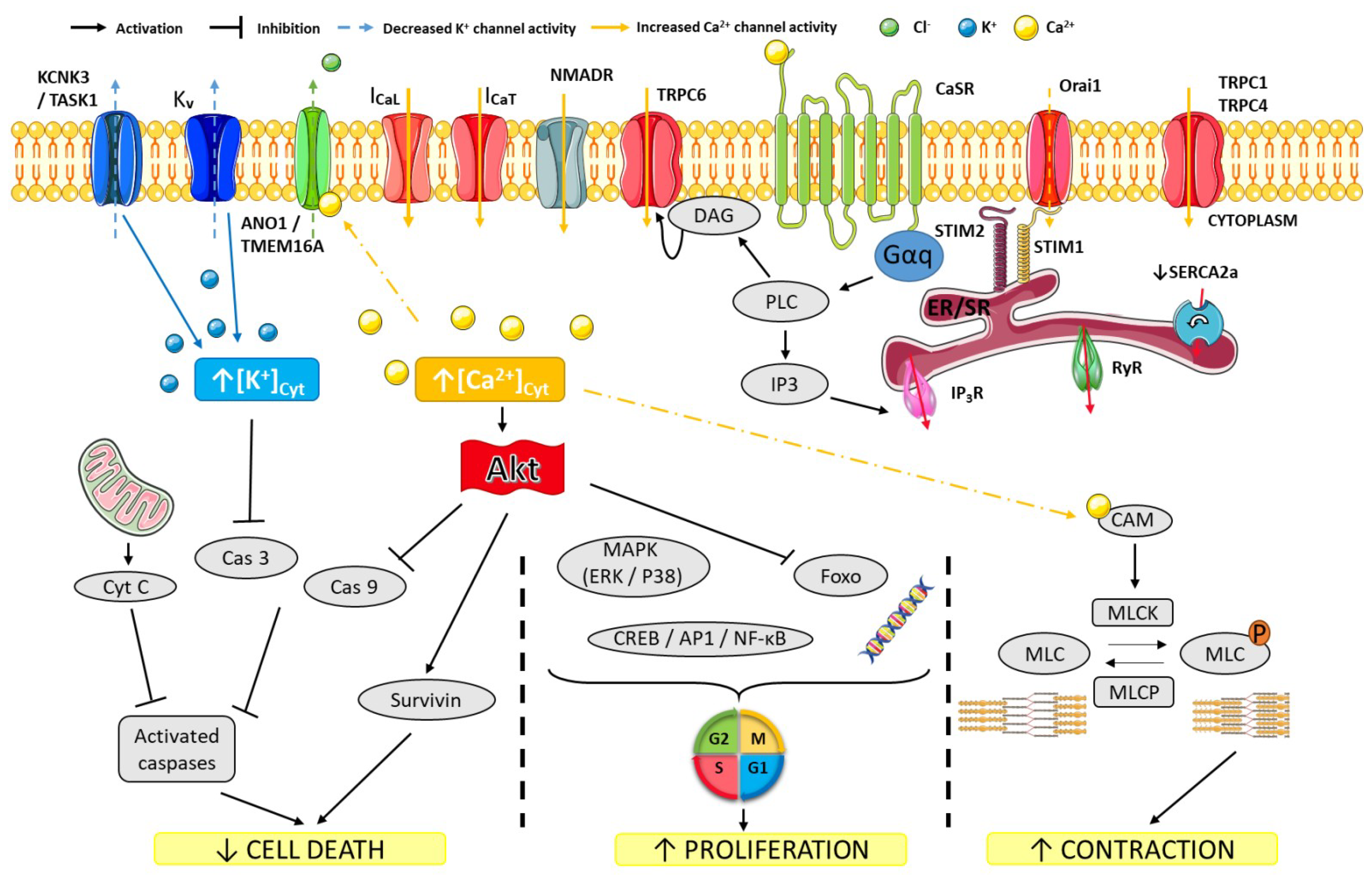
3. Ion Channels in PAH Pathophysiology
3.1. K+ Channels
3.1.1. Voltage-Gated K+ Channels in PAH
3.1.2. Ca2+-Activated K+ Channels in PAH
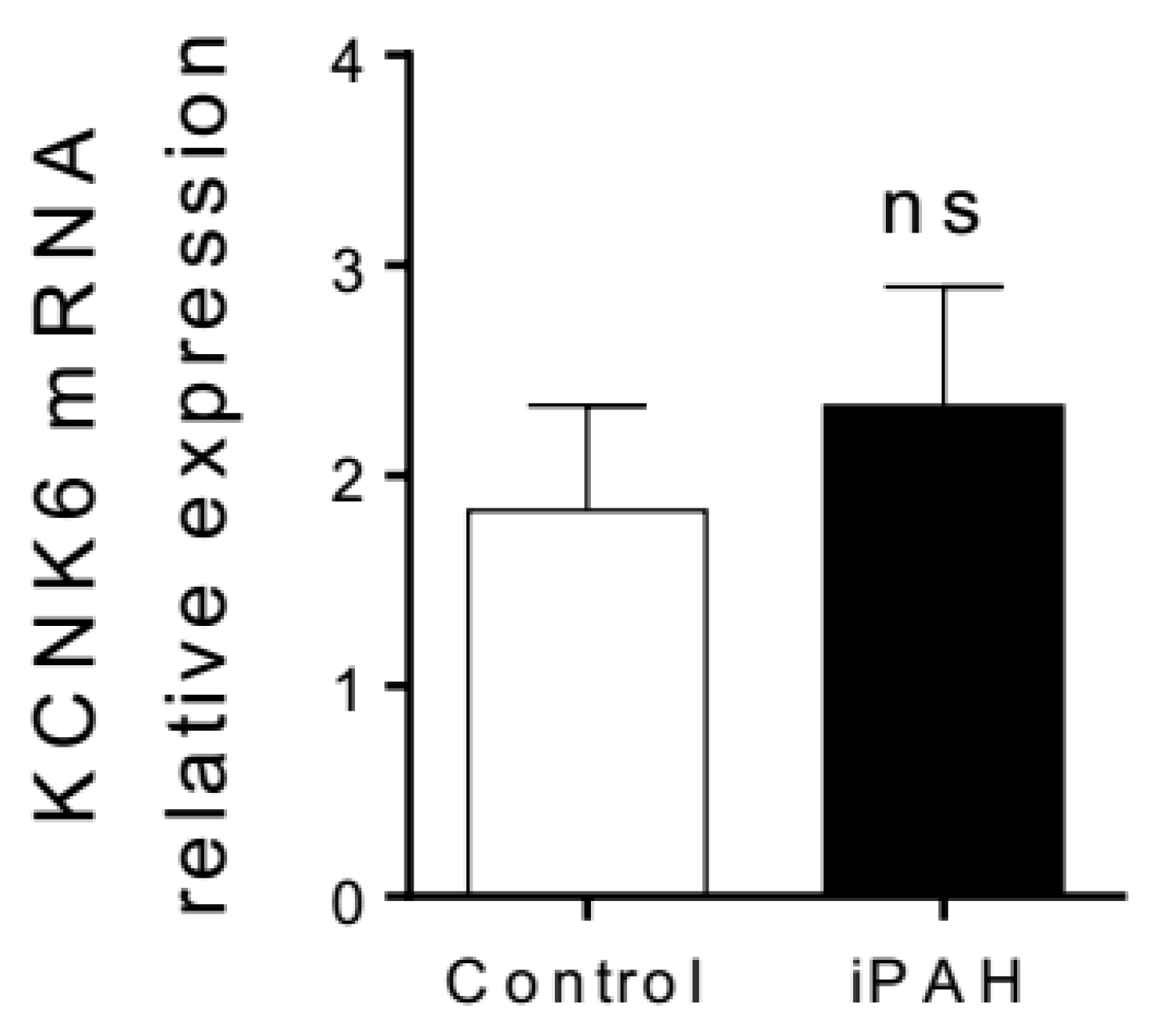
3.1.3. K2P Channels in PAH
3.2. Ca2+ Channels
3.2.1. Voltage-Gated Ca2+ Channels in PAH
3.2.2. SOCE in PAH
3.2.3. TRPC Channels in PAH
3.2.4. Other TRP Channels in PAH Pathophysiology
3.2.5. TRPM
3.2.6. Acid-Sensing Ion Channels (ASIC)
3.2.7. Ca2+-Sensing Receptor (CaSR)
3.2.8. Other Non-Voltage Gated Ca2+ Channels
3.2.9. NMDAR in PAH
3.3. Na+ Channels
3.4. Cl− Channels
3.4.1. CFTR
3.4.2. Ca2+ Activated Cl− Channels
4. Targeting Ion Channels and Transporters in PAH
4.1. Direct Pharmacological Action on Ion Channels in PAH
4.1.1. Voltage Gated Ca2+ Channels
4.1.2. Kv1.5 Gene Therapy
4.1.3. KATP Channels
4.2. Indirect Pharmacological Action on Ion Channels in PAH
4.2.1. KCNK3/TASK-1
4.2.2. KATP Channels
4.2.3. Ca2+-Activated K Channels (KCa)
4.2.4. TRPC1 and TRPC6
4.3. Untested Pharmacological Compounds in PAH
4.3.1. Kv Channels
4.3.2. TRPCs and Orais Channels
4.3.3. TRPV Channels
4.3.4. NMDAR Channels
4.3.5. ASIC Channels
4.3.6. TMEM16A Channel
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [PubMed]
- Humbert, M.; Lau, E.M.T.; Montani, D.; Jaïs, X.; Sitbon, O.; Simonneau, G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 2014, 130, 2189–2208. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, M.; Ruffenach, G.; Meloche, J.; Bonnet, S. Adaptation and remodelling of the pulmonary circulation in pulmonary hypertension. Can. J. Cardiol. 2015, 31, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R; Humbert, M. Pathobiology of pulmonary arterial hypertension: understanding the roads less travelled. Eur. Respir. Review 2017, 146, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, M.-R.; Guignabert, C.; Montani, D.; Girerd, B.; Jaïs, X.; Savale, L.; Hervé, P.; Montpréville, V.T. de; Mercier, O.; Sitbon, O.; et al. BMPR2 mutation status influences bronchial vascular changes in pulmonary arterial hypertension. Eur. Respir. 2016, 48, 1668–1681. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Bailly, S.; Humbert, M. Restoring BMPRII functions in pulmonary arterial hypertension: opportunities, challenges and limitations. Expert Opin. Ther. Targets 2017, 21, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Eyries, M.; Szezepanski, I.; Ladouceur, M.; Nadaud, S.; Bonnet, D.; Soubrier, F. Genetic analyses in a cohort of children with pulmonary hypertension. Eur. Respir. J. 2016, 48, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.H.F.; Roofthooft, M.T.R.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R. M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Trégouët, D.-A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, M.S.; Roman-Campos, D.; Terrenoire, C.; Jnani, J.; Sampson, K.J.; Chung, W.K.; Kass, R.S. The Impact of Heterozygous KCNK3 Mutations Associated With Pulmonary Arterial Hypertension on Channel Function and Pharmacological Recovery. J. Am. Heart Assoc. 2017, 6, e006465. [Google Scholar] [CrossRef] [PubMed]
- Navas Tejedor, P.; Tenorio Castaño, J.; Palomino Doza, J.; Arias Lajara, P.; Gordo Trujillo, G.; López Meseguer, M.; Román Broto, A.; Lapunzina Abadía, P.; Escribano Subía, P. An homozygous mutation in KCNK3 is associated with an aggressive form of hereditary pulmonary arterial hypertension. Clin. Genet. 2017, 91, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Best, D.H.; Sumner, K.L.; Smith, B.P.; Damjanovich-Colmenares, K.; Nakayama, I.; Brown, L.M.; Ha, Y.; Paul, E.; Morris, A.; Jama, M.A.; et al. EIF2AK4 Mutations in Patients Diagnosed With Pulmonary Arterial Hypertension. Chest 2017, 151, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Higasa, K.; Ogawa, A.; Terao, C.; Shimizu, M.; Kosugi, S.; Yamada, R.; Date, H.; Matsubara, H.; Matsuda, F. A burden of rare variants in BMPR2 and KCNK3 contributes to a risk of familial pulmonary arterial hypertension. BMC Pulm. Med. 2017, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef] [PubMed]
- Wipff, J.; Dieudé, P.; Guedj, M.; Ruiz, B.; Riemekasten, G.; Cracowski, J.L.; Matucci-Cerinic, M.; Melchers, I.; Humbert, M.; Hachulla, E.; et al. Association of a KCNA5 gene polymorphism with systemic sclerosis-associated pulmonary arterial hypertension in the European Caucasian population. Arthritis Rheum. 2010, 62, 3093–3100. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.-Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Remillard, C.V.; Fantozzi, I.; Mandegar, M.; Sison, T.T.; Zhang, S.; Burg, E.; Yuan, J.X.-J. Diversity of voltage-dependent K+ channels in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L226–L238. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Chabot, S.; Antigny, F.; Perros, F.; Provencher, S.; Bonnet, S. Potassium channels in pulmonary arterial hypertension. Eur. Respir. J. 2015, 46, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Firth, A.L.; Yuan, J.X.-J. Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr. Physiol. 2011, 1, 1555–1602. [Google Scholar] [PubMed]
- Xiong, P.Y.; Potus, F.; Chan, W.; Archer, S.L. Models and Molecular Mechanisms of World Health Organization Group 2 to 4 Pulmonary Hypertension. Hypertension 2018, 71, 34–55. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.; Bloch, K.; Archer, S.L. Rodent models of pulmonary hypertension: harmonisation with the world health organisation’s categorisation of human PH. Int. J. Clin. Pract. Suppl. 2011, 65, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Toba, M.; Alzoubi, A.; Ito, M.; Fagan, K.A.; Cool, C.D.; Voelkel, N.F.; McMurtry, I.F.; Oka, M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010, 121, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L. Potassium channel diversity in the pulmonary arteries and pulmonary veins: implications for regulation of the pulmonary vasculature in health and during pulmonary hypertension. Pharmacol. Ther. 2007, 115, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Quayle, J.M.; McCarron, J.G.; Brayden, J.E.; Nelson, M.T. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am. J. Physiol. Cell Physiol. 1993, 265, C1363–C1370. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Droogmans, G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef] [PubMed]
- Wellman, G.C.; Bevan, J.A. Barium inhibits the endothelium-dependent component of flow but not acetylcholine-induced relaxation in isolated rabbit cerebral arteries. J. Pharmacol. Exp. Ther. 1995, 274, 47–53. [Google Scholar] [PubMed]
- Clement, J.P.; Kunjilwar, K.; Gonzalez, G.; Schwanstecher, M.; Panten, U.; Aguilar-Bryan, L.; Bryan, J. Association and stoichiometry of KATP channel subunits. Neuron 1997, 18, 827–838. [Google Scholar] [CrossRef]
- Brayden, J.E. Functional roles of KATP channels in vascular smooth muscle. Clin. Exp. Pharmacol. Physiol. 2002, 29, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zong, F.; Wang, H.; Wang, Q.; Xie, W.; Wang, H. Iptakalim, a novel ATP-sensitive potassium channel opener, inhibits pulmonary arterial smooth muscle cell proliferation by downregulation of PKC-α. J. Biomed. Res. 2011, 25, 392–401. [Google Scholar] [CrossRef]
- Murphy, M.E.; Brayden, J.E. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J. Physiol. 1995, 486, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Al-Mehdi, A.-B.; Levitan, I.; Stevens, T.; Fisher, A.B. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2003, 285, C959–C967. [Google Scholar] [CrossRef] [PubMed]
- Korovkina, V.P.; England, S.K. Detection and implications of potassium channel alterations. Vascul. Pharmacol. 2002, 38, 3–12. [Google Scholar] [CrossRef]
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Li-Smerin, Y.; Hackos, D.H.; Swartz, K.J. alpha-helical structural elements within the voltage-sensing domains of a K+ channel. J. Gen. Physiol. 2000, 115, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.A.; Han, J.; Jung, I.D.; Park, W.S. Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res. 2008, 44, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Mecham, R.P. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu. Rev. Physiol. 1997, 59, 89–144. [Google Scholar] [CrossRef] [PubMed]
- Hayabuchi, Y. The Action of Smooth Muscle Cell Potassium Channels in the Pathology of Pulmonary Arterial Hypertension. Pediatr. Cardiol. 2017, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kuhr, F.K.; Smith, K.A.; Song, M.Y.; Levitan, I.; Yuan, J.X.-J. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1546–H1562. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Souil, E.; Dinh-Xuan, A.T.; Schremmer, B.; Mercier, J.C.; El Yaagoubi, A.; Nguyen-Huu, L.; Reeve, H.L.; Hampl, V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J. Clin. Investig. 1998, 101, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J.; Wang, J.; Juhaszova, M.; Gaine, S.P.; Rubin, L.J. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet Lond. Engl. 1998, 351, 726–727. [Google Scholar] [CrossRef]
- Coppock, E.A.; Martens, J.R.; Tamkun, M.M. Molecular basis of hypoxia-induced pulmonary vasoconstriction: role of voltage-gated K+ channels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L1–L12. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.R.; Kozlowski, R.Z. Kv channel subunit expression in rat pulmonary arteries. Lung 2001, 179, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Firth, A.L.; Han, J.; Ko, E.A. Patho-, physiological roles of voltage-dependent K+ channels in pulmonary arterial smooth muscle cells. J. Smooth Muscle Res. 2010, 46, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Standen, N.B.; Quayle, J.M. K+ channel modulation in arterial smooth muscle. Acta Physiol. Scand. 1998, 164, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, M.T.; López-López, J.R. Are Kv channels the essence of O2 sensing? Circ. Res. 2000, 86, 490–491. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.N.; Platoshyn, O.; Li, L.; Guo, X.; Golovina, V.A.; Yuan, J.X.-J.; Wang, J.-Y. Activation of K+ channels and increased migration of differentiated intestinal epithelial cells after wounding. Am. J. Physiol. Cell Physiol. 2002, 282, C885–C898. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Golovina, V.A.; Bailey, C.L.; Limsuwan, A.; Krick, S.; Juhaszova, M.; Seiden, J.E.; Rubin, L.J.; Yuan, J.X. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am. J. Physiol. Cell Physiol. 2000, 279, C1540–C1549. [Google Scholar] [CrossRef] [PubMed]
- Tajsic, T.; Morrell, N.W. Smooth muscle cell hypertrophy, proliferation, migration and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 295–317. [Google Scholar] [PubMed]
- Joshi, S.; Sedivy, V.; Hodyc, D.; Herget, J.; Gurney, A.M. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J. Pharmacol. Exp. Ther. 2009, 329, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Hogg, D.S.; McMurray, G.; Kozlowski, R.Z. Endothelial cells freshly isolated from small pulmonary arteries of the rat possess multiple distinct K+ current profiles. Lung 2002, 180, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995, 268, C799–C822. [Google Scholar] [CrossRef] [PubMed]
- Guéguinou, M.; Chantôme, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca2+ channels: the complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.M.; Silvia, C.; Hirschberg, B.; Bond, C.T.; Adelman, J.P.; Maylie, J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA 1997, 94, 11651–11656. [Google Scholar] [CrossRef] [PubMed]
- Joiner, W.J.; Wang, L.Y.; Tang, M.D.; Kaczmarek, L.K. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. USA 1997, 94, 11013–11018. [Google Scholar] [CrossRef] [PubMed]
- Stocker, M. Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurosci. 2004, 5, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Soh, H.; Park, C.-S. Localization of divalent cation-binding site in the pore of a small conductance Ca2+-activated K+ channel and its role in determining current-voltage relationship. Biophys. J. 2002, 83, 2528–2538. [Google Scholar] [CrossRef]
- Bolotina, V.M.; Najibi, S.; Palacino, J.J.; Pagano, P.J.; Cohen, R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Fisslthaler, B.; Popp, R.; Kiss, L.; Potente, M.; Harder, D.R.; Fleming, I.; Busse, R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999, 401, 493–497. [Google Scholar] [PubMed]
- Vang, A.; Mazer, J.; Casserly, B.; Choudhary, G. Activation of endothelial BKCa channels causes pulmonary vasodilation. Vascul. Pharmacol. 2010, 53, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Kroigaard, C.; Dalsgaard, T.; Nielsen, G.; Laursen, B.E.; Pilegaard, H.; Köhler, R.; Simonsen, U. Activation of endothelial and epithelial K(Ca) 2.3 calcium-activated potassium channels by NS309 relaxes human small pulmonary arteries and bronchioles. Br. J. Pharmacol. 2012, 167, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Connolly, M.; Nagaraj, C.; Tang, B.; Bálint, Z.; Popper, H.; Smolle-Juettner, F.M.; Lindenmann, J.; Kwapiszewska, G.; Aaronson, P.I.; et al. Peroxisome Proliferator–Activated Receptor–β/δ, the Acute Signaling Factor in Prostacyclin-Induced Pulmonary Vasodilation. Am. J. Respir. Cell Mol. Biol. 2012, 46, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Lesage, F.; Lazdunski, M. Molecular and functional properties of two-pore-domain potassium channels. Am. J. Physiol. Renal Physiol. 2000, 279, F793–F801. [Google Scholar] [CrossRef] [PubMed]
- Duprat, F.; Lesage, F.; Fink, M.; Reyes, R.; Heurteaux, C.; Lazdunski, M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 1997, 16, 5464–5471. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Li, Y.; Tang, B.; Hanze, J.; Eul, B.; Bohle, R.M.; Wilhelm, J.; Morty, R.E.; Brau, M.E.; Weir, E.K.; et al. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ. Res. 2006, 98, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Honoré, E. Molecular physiology of oxygen-sensitive potassium channels. Eur. Respir. J. 2001, 18, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Gnatenco, C. TASK-5, a new member of the tandem-pore K(+) channel family. Biochem. Biophys. Res. Commun. 2001, 284, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Gardener, M.J.; Johnson, I.T.; Burnham, M.P.; Edwards, G.; Heagerty, A.M.; Weston, A.H. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br. J. Pharmacol. 2004, 142, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Wanstall, J.C. The pulmonary vasodilator properties of potassium channel opening drugs. Gen. Pharmacol. 1996, 27, 599–605. [Google Scholar] [CrossRef]
- Gurney, A.M.; Osipenko, O.N.; MacMillan, D.; McFarlane, K.M.; Tate, R.J.; Kempsill, F.E.J. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ. Res. 2003, 93, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Manoury, B.; Etheridge, S.L.; Reid, J.; Gurney, A.M. Organ culture mimics the effects of hypoxia on membrane potential, K+ channels and vessel tone in pulmonary artery. Br. J. Pharmacol. 2009, 158, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Manoury, B.; Lamalle, C.; Oliveira, R.; Reid, J.; Gurney, A.M. Contractile and electrophysiological properties of pulmonary artery smooth muscle are not altered in TASK-1 knockout mice. J. Physiol. 2011, 589, 3231–3246. [Google Scholar] [CrossRef] [PubMed]
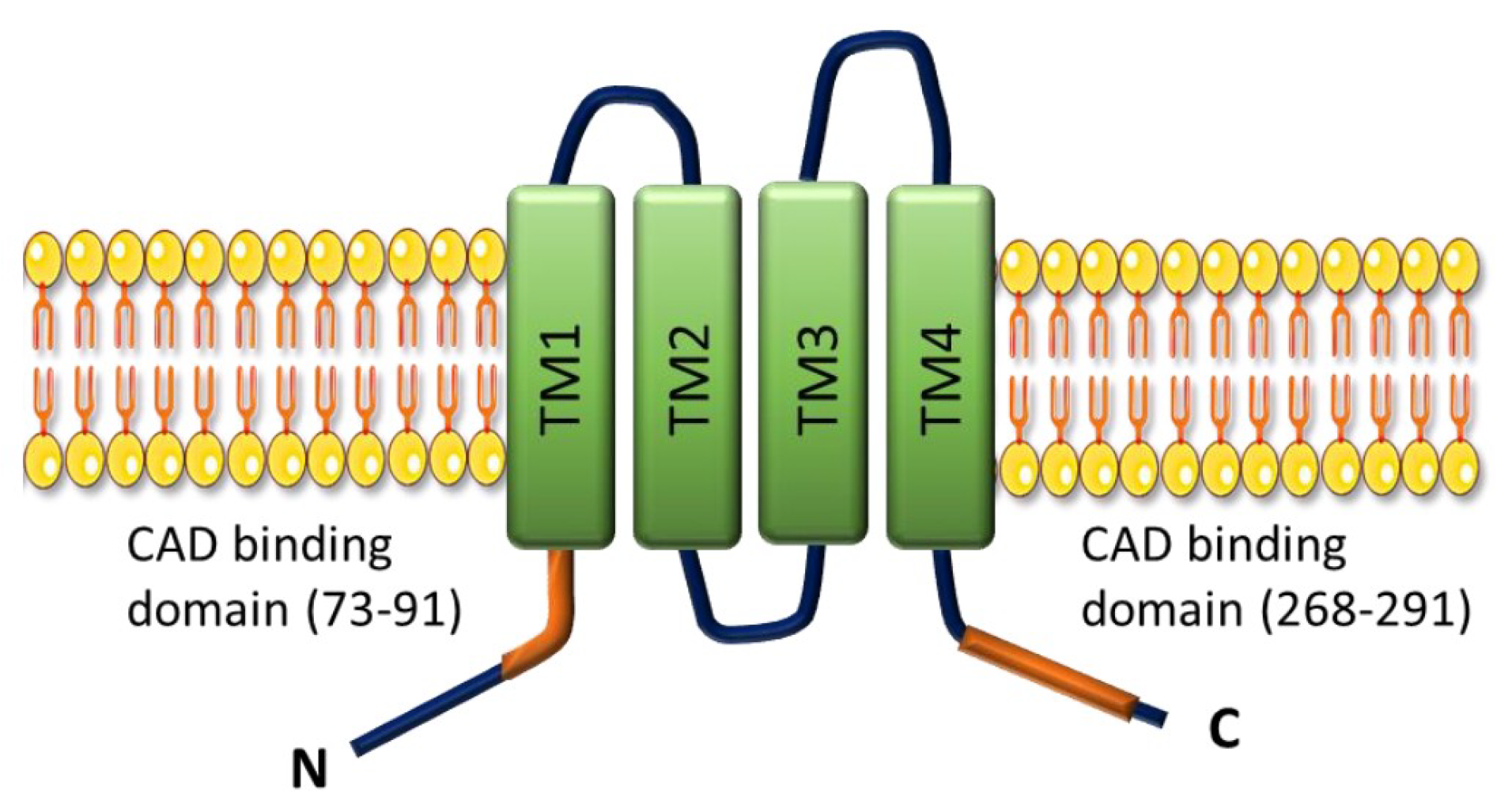
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Péchoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.W.; Ellinor, P.T.; Horne, W.A. Molecular diversity of voltage-dependent Ca2+ channels. Trends Pharmacol. Sci. 1991, 12, 349–354. [Google Scholar] [CrossRef]
- Bers, D. Cardiac excitation-contraction coupling. Available online: https://www-ncbi-nlm-nih-gov.gate2.inist.fr/pubmed/11805843 (accessed on 11 April 2018).
- Ferron, L.; Ruchon, Y.; Renaud, J.-F.; Capuano, V. T-type Ca2+ signalling regulates aldosterone-induced CREB activation and cell death through PP2A activation in neonatal cardiomyocytes. Cardiovasc. Res. 2011, 90, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Rodman, D.M.; Reese, K.; Harral, J.; Fouty, B.; Wu, S.; West, J.; Hoedt-Miller, M.; Tada, Y.; Li, K.-X.; Cool, C.; et al. Low-Voltage-Activated (T-Type) Calcium Channels Control Proliferation of Human Pulmonary Artery Myocytes. Circ. Res. 2005, 96, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Sankhe, S.; Manousakidi, S.; Antigny, F.; Arthur Ataam, J.; Bentebbal, S.; Ruchon, Y.; Lecerf, F.; Sabourin, J.; Price, L.; Fadel, E.; et al. T-type Ca2+ channels elicit pro-proliferative and anti-apoptotic responses through impaired PP2A/Akt1 signaling in PASMCs from patients with pulmonary arterial hypertension. Biochim. Biophys. Acta 2017, 1864, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Hockerman, G.H.; Johnson, B.D.; Abbott, M.R.; Scheuer, T.; Catterall, W.A. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels in transmembrane segment IIIS6 and the pore region of the alpha1 subunit. J. Biol. Chem. 1997, 272, 18759–18765. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed]
- Perez-Reyes, E. Molecular Physiology of Low-Voltage-Activated T-type Calcium Channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, L.L. T-type Ca2+ channels in vascular smooth muscle: multiple functions. Cell Calcium 2006, 40, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Haynes, J.; Taylor, J.T.; Obiako, B.O.; Stubbs, J.R.; Li, M.; Stevens, T. Cav3.1 (alpha1G) T-type Ca2+ channels mediate vaso-occlusion of sickled erythrocytes in lung microcirculation. Circ. Res. 2003, 93, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, H.; King, J.A.; Sellak, H.; Kuebler, W.M.; Yin, J.; Townsley, M.I.; Shin, H.-S.; Wu, S. Alpha1G T-type calcium channel selectively regulates P-selectin surface expression in pulmonary capillary endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L86–L97. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, H.; Lu, F.; Sellak, H.; Daigle, J.A.; Alexeyev, M.F.; Xi, Y.; Ju, J.; van Mourik, J.A.; Wu, S. Cav3.1 (alpha1G) controls von Willebrand factor secretion in rat pulmonary microvascular endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L833–L844. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.; Gilbert, G.; Roux, E.; Lory, P.; Marthan, R.; Savineau, J.-P.; Quignard, J.-F. T-type calcium channels are involved in hypoxic pulmonary hypertension. Cardiovasc. Res. 2014, 103, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.; Courtois, A.; Dubois, M.; Cussac, L.-A.; Ducret, T.; Lory, P.; Marthan, R.; Savineau, J.-P.; Quignard, J.-F. T-type voltage gated calcium channels are involved in endothelium-dependent relaxation of mice pulmonary artery. Biochem. Pharmacol. 2017, 138, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. Formation and actions of calcium-mobilizing messenger, inositol 1,4,5-trisphosphate. Am. J. Physiol. 1987, 252, G149–G157. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; Kinet, J.-P. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Montell, C. The history of TRP channels, a commentary and reflection. Pflugers. Arch. 2011, 461, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. STIM1 regulates Ca 2+ entry via arachidonate-regulated Ca 2+ -selective (ARC) channels without store depletion or translocation to the plasma membrane: Plasma membrane STIM1 regulates the ARC channels. J. Physiol. 2007, 579, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, T.J. Orai channels-New insights, new ideas. J. Physiol. 2012, 590, 4155–4156. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.L.; Shuttleworth, T.J. A plasma membrane-targeted cytosolic domain of STIM1 selectively activates ARC channels, an arachidonate-regulated store-independent Orai channel. Channels 2012, 6, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cobos, J.C.; Zhang, X.; Zhang, W.; Ruhle, B.; Motiani, R.K.; Schindl, R.; Muik, M.; Spinelli, A.M.; Bisaillon, J.M.; Shinde, A.V.; et al. Store-Independent Orai1/3 Channels Activated by Intracrine LeukotrieneC4: Role in Neointimal Hyperplasia. Circ. Res. 2013, 112, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Jousset, H.; König, S.; Frieden, M. Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef] [PubMed]
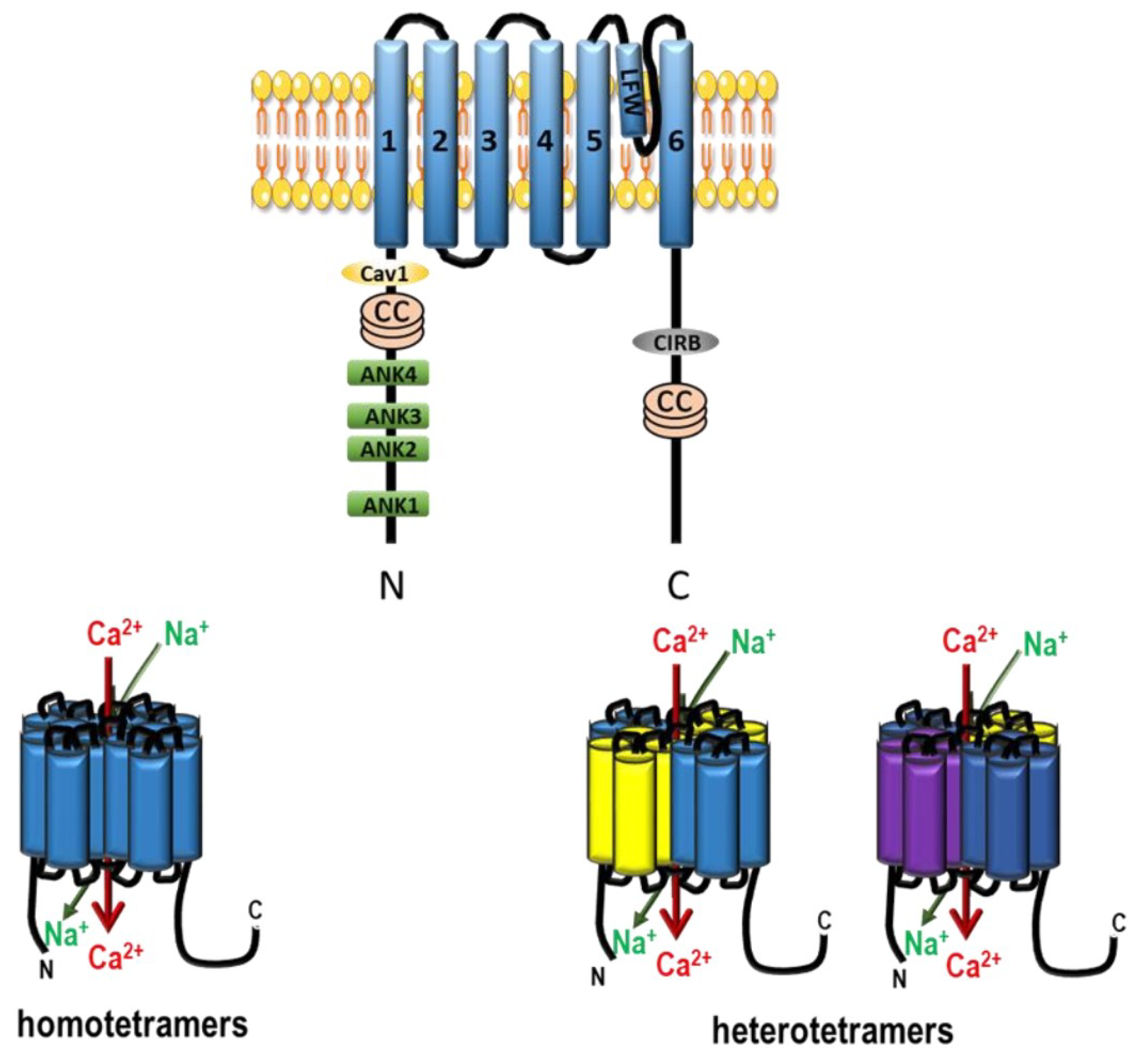
- Beech, D.J. Characteristics of transient receptor potential canonical calcium-permeable channels and their relevance to vascular physiology and disease. Circ. J. 2013, 77, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Golovina, V.A.; Platoshyn, O.; Bailey, C.L.; Wang, J.; Limsuwan, A.; Sweeney, M.; Rubin, L.J.; Yuan, J.X. Upregulated TRP and enhanced capacitative Ca(2+) entry in human pulmonary artery myocytes during proliferation. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H746–H755. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.C.; Gurney, A.M. Store-operated channels mediate Ca(2+) influx and contraction in rat pulmonary artery. Circ. Res. 2001, 89, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Remillard, C.V.; Yuan, J.X.-J. TRP channels, CCE, and the pulmonary vascular smooth muscle. Microcirculation. 2006, 13, 671–692. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-J.; Leung, G.P. H.; Zhang, W.-M.; Yang, X.-R.; Yip, K.-P.; Tse, C.-M.; Sham, J.S.K. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ. Res. 2004, 95, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, W.; Yang, K.; Xu, L.; Lai, N.; Tian, L.; Jiang, Q.; Duan, X.; Chen, M.; Wang, J. Bone morphogenetic protein 2 decreases TRPC expression, store-operated Ca2+ entry, and basal [Ca2+]i in rat distal pulmonary arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 2013, 304, C833–C843. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.C.; Airey, J.A.; Hume, J.R. The contribution of TRPC1 and STIM1 to capacitative Ca(2+) entry in pulmonary artery. Adv. Exp. Med. Biol. 2010, 661, 123–135. [Google Scholar] [PubMed]
- Ingueneau, C.; Huynh, U.D.; Marcheix, B.; Athias, A.; Gambert, P.; Nègre-Salvayre, A.; Salvayre, R.; Vindis, C. TRPC1 is regulated by caveolin-1 and is involved in oxidized LDL-induced apoptosis of vascular smooth muscle cells. J. Cell. Mol. Med. 2009, 13, 1620–1631. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, X.; Hewavitharana, T.; Soboloff, J.; Gill, D.L. Stim, ORAI and TRPC channels in the control of calcium entry signals in smooth muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.-Y.; Shen, B.; Ma, X.; Kwok, Y.-C.; Huang, Y.; Man, Y.-B.; Yu, S.; Yao, X. TRPC1 Associates With BKCa Channel to Form a Signal Complex in Vascular Smooth Muscle Cells. Circ. Res. 2009, 104, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McKeown, L.; Ojelabi, O.; Stacey, M.; Foster, R.; O’Regan, D.; Porter, K.E.; Beech, D.J. Nanomolar potency and selectivity of a Ca2+ release-activated Ca2+ channel inhibitor against store-operated Ca2+ entry and migration of vascular smooth muscle cells. Br. J. Pharmacol. 2011, 164, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Abdullaev, I.F.; Bisaillon, J.M.; Singer, H.A.; Trebak, M. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J. 2009, 23, 2425–2437. [Google Scholar] [CrossRef] [PubMed]
- Demura, Y.; Ishizaki, T.; Ameshima, S.; Okamura, S.; Hayashi, T.; Matsukawa, S.; Miyamori, I. The activation of nitric oxide synthase by copper ion is mediated by intracellular Ca2+ mobilization in human pulmonary arterial endothelial cells. Br. J. Pharmacol. 1998, 125, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Fagan, K.A.; Li, K.X.; Shaul, P.W.; Cooper, D.M.; Rodman, D.M. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. J. Biol. Chem. 2000, 275, 17979–17985. [Google Scholar] [CrossRef] [PubMed]
- Sundivakkam, P.C.; Freichel, M.; Singh, V.; Yuan, J.P.; Vogel, S.M.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. The Ca2+ Sensor Stromal Interaction Molecule 1 (STIM1) Is Necessary and Sufficient for the Store-Operated Ca2+ Entry Function of Transient Receptor Potential Canonical (TRPC) 1 and 4 Channels in Endothelial Cells. Mol. Pharmacol. 2012, 81, 510–526. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Girardin, N.; Frieden, M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A.; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Sundivakkam, P.C.; Kwiatek, A.M.; Sharma, T.T.; Minshall, R.D.; Malik, A.B.; Tiruppathi, C. Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am. J. Physiol. Cell Physiol. 2009, 296, C403–C413. [Google Scholar] [CrossRef] [PubMed]
- Paffett, M.L.; Riddle, M.A.; Kanagy, N.L.; Resta, T.C.; Walker, B.R. Altered Protein Kinase C Regulation of Pulmonary Endothelial Store- and Receptor-Operated Ca2+ Entry after Chronic Hypoxia. J. Pharmacol. Exp. Ther. 2010, 334, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Benham, C.D.; Davis, J.B.; Randall, A.D. Vanilloid and TRP channels: a family of lipid-gated cation channels. Neuropharmacology 2002, 42, 873–888. [Google Scholar] [CrossRef]
- Fleig, A.; Penner, R. The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol. Sci. 2004, 25, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE Signal Transduct. Knowl. Environ. 2005, 2005. [Google Scholar] [CrossRef] [PubMed]
- Guibert, C.; Ducret, T.; Savineau, J.-P. Expression and physiological roles of TRP channels in smooth muscle cells. Adv. Exp. Med. Biol. 2011, 704, 687–706. [Google Scholar] [PubMed]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 2008, 102, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Waldron, B.J.; Brayden, J.E. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ. Res. 2004, 95, 922–929. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yao, G.; Savoia, C.; Touyz, R.M. Transient receptor potential melastatin 7 ion channels regulate magnesium homeostasis in vascular smooth muscle cells: role of angiotensin II. Circ. Res. 2005, 96, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; He, Y.; Montezano, A.C. I.; Yao, G.; Chubanov, V.; Gudermann, T.; Callera, G.E. Differential regulation of transient receptor potential melastatin 6 and 7 cation channels by ANG II in vascular smooth muscle cells from spontaneously hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R73–R78. [Google Scholar] [CrossRef] [PubMed]
- Muraki, K.; Iwata, Y.; Katanosaka, Y.; Ito, T.; Ohya, S.; Shigekawa, M.; Imaizumi, Y. TRPV2 is a component of osmotically sensitive cation channels in murine aortic myocytes. Circ. Res. 2003, 93, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wang, X.; Varty, L.; Rizzo, C.A.; Yang, R.; Correll, C.C.; Phelps, P.T.; Egan, R.W.; Hey, J.A. Functional TRPV4 channels are expressed in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L272–L278. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Muraki, K.; Ohya, S.; Yamamura, H.; Hatano, N.; Itoh, Y.; Imaizumi, Y. TRPV4-like non-selective cation currents in cultured aortic myocytes. J. Pharmacol. Sci. 2008, 108, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Dumas, S.J.; Bru-Mercier, G.; Courboulin, A.; Quatredeniers, M.; Rücker-Martin, C.; Antigny, F.; Nakhleh, M.K.; Ranchoux, B.; Gouadon, E.; Vinhas, M.-C.; et al. NMDA-Type Glutamate Receptor Activation Promotes Vascular Remodeling and Pulmonary Arterial Hypertension. Circulation 2018, 137, 2371–2389. [Google Scholar] [CrossRef] [PubMed]
- Vyklicky, V.; Korinek, M.; Smejkalova, T.; Balik, A.; Krausova, B.; Kaniakova, M.; Lichnerova, K.; Cerny, J.; Krusek, J.; Dittert, I.; et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol. Res. 2014, 63, S191–S203. [Google Scholar] [PubMed]
- Zito, K.; Scheuss, V. NMDA Receptor Function and Physiological Modulation. Encycl. Neurosci. 2009, 1157–1164. [Google Scholar] [CrossRef]
- Coste, B.; Murthy, S.E.; Mathur, J.; Schmidt, M.; Mechioukhi, Y.; Delmas, P.; Patapoutian, A. Piezo1 ion channel pore properties are dictated by C-terminal region. Nat. Commun. 2015, 6, 7223. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically-activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Imashuku, S.; Muramatsu, H.; Sugihara, T.; Okuno, Y.; Wang, X.; Yoshida, K.; Kato, A.; Kato, K.; Tatsumi, Y.; Hattori, A.; et al. PIEZO1 gene mutation in a Japanese family with hereditary high phosphatidylcholine hemolytic anemia and hemochromatosis-induced diabetes mellitus. Int. J. Hematol. 2016, 104, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Albuisson, J.; Murthy, S.E.; Bandell, M.; Coste, B.; Louis-dit-Picard, H.; Mathur, J.; Fénéant-Thibault, M.; Tertian, G.; de Jaureguiberry, J.-P.; Syfuss, P.-Y.; et al. Dehydrated Hereditary Stomatocytosislinked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat. Commun. 2013, 4, 1884. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; Alper, S.L.; De Franceschi, L.; Auriemma, C.; Russo, R.; De Falco, L.; Vallefuoco, F.; Esposito, M.R.; Vandorpe, D.H.; Shmukler, B.E.; et al. Iolascon, A. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood 2013, 121, 3925–3935. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chennupati, R.; Kaur, H.; Iring, A.; Wettschureck, N.; Offermanns, S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 2016, 126, 4527–4536. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-H.; Ranade, S.; Weyer, A.D.; Dubin, A.E.; Baba, Y.; Qiu, Z.; Petrus, M.; Miyamoto, T.; Reddy, K.; Lumpkin, E.A.; et al. Piezo2 is required for Merkel cell mechanotransduction. Nature 2014, 509, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Mechanosensation by PIEZO1 in blood pressure control: Hypertension. Nat. Rev. Nephrol. 2017, 13, 3–3. [Google Scholar] [CrossRef] [PubMed]
- Retailleau, K.; Duprat, F. Polycystins and partners: Proposed role in mechanosensitivity. J. Physiol. 2014, 592, 2453–2471. [Google Scholar] [CrossRef] [PubMed]
- Canessa, C.M.; Schild, L.; Buell, G.; Thorens, B.; Gautschi, I.; Horisberger, J.D.; Rossier, B.C. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994, 367, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Zhao, D.; Wang, Q.; Gu, Y.; Ma, H.; Zhang, Z. Role of the epithelial sodium channel in salt-sensitive hypertension. Acta Pharmacol. Sin. 2011, 32, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Drummond, H.A. Vascular ENaC proteins are required for renal myogenic constriction. Am. J. Physiol.-Ren. Physiol. 2005, 289, F891–F901. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, F.; Mohan, S.; Champaneri, B.; Gu, Y. Functional ENaC Channels Expressed in Endothelial Cells: A New Candidate for Mediating Shear Force. Microcirculation 2009, 16, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Drummond, H.A.; Gebremedhin, D.; Harder, D.R. Degenerin/epithelial Na+ channel proteins: components of a vascular mechanosensor. Hypertension. 2004, 44, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Remillard, C.V.; Fantozzi, I.; Sison, T.; Yuan, J.X.-J. Identification of functional voltage-gated Na+ channels in cultured human pulmonary artery smooth muscle cells. Pflugers Arch. 2005, 451, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, P.; Fraser, S.P.; Patterson, L.; Ahmad, Z.; Burcu, H.; Ottaviani, D.; Diss, J.K.J.; Box, C.; Eccles, S.A.; Djamgoz, M.B.A. Angiogenic Functions of Voltage-gated Na+ Channels in Human Endothelial Cells. J. Biol. Chem. 2011, 286, 16846–16860. [Google Scholar] [CrossRef] [PubMed]
- Traub, O.; Ishida, T.; Ishida, M.; Tupper, J.C.; Berk, B.C. Shear Stress-mediated Extracellular Signal-regulated Kinase Activation Is Regulated by Sodium in Endothelial Cells: POTENTIAL ROLE FOR A VOLTAGE-DEPENDENT SODIUM CHANNEL. J. Biol. Chem. 1999, 274, 20144–20150. [Google Scholar] [CrossRef] [PubMed]
- Bulley, S.; Neeb, Z.P.; Burris, S.K.; Bannister, J.P.; Thomas-Gatewood, C.M.; Jangsangthong, W.; Jaggar, J.H. TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ. Res. 2012, 111, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Huang, L.; Zhao, D.; He, J.Z.; Sharma, P.; Liu, J.; Gramolini, A.O.; Ward, M.E.; Cho, H.C.; Backx, P.H. Swelling-activated Cl− currents and intracellular CLC-3 are involved in proliferation of human pulmonary artery smooth muscle cells. J. Hypertens. 2014, 32, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Matchkov, V.V.; Larsen, P.; Bouzinova, E.V.; Rojek, A.; Boedtkjer, D.M. B.; Golubinskaya, V.; Pedersen, F.S.; Aalkjaer, C.; Nilsson, H. Bestrophin-3 (vitelliform macular dystrophy 2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ. Res. 2008, 103, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J.V. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.-S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.-M.; Raouf, R.; Shin, Y.K.; Oh, U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Billig, G.M.; Pál, B.; Fidzinski, P.; Jentsch, T.J. Ca2+-activated Cl− currents are dispensable for olfaction. Nat. Neurosci. 2011, 14, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Futtner, C.R.; Harfe, B.D. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev. Biol. 2008, 321, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Heinze, C.; Seniuk, A.; Sokolov, M.V.; Huebner, A.K.; Klementowicz, A.E.; Szijártó, I.A.; Schleifenbaum, J.; Vitzthum, H.; Gollasch, M.; Ehmke, H.; et al. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J. Clin. Investig. 2014, 124, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.-M.; Gao, M.; Guo, K.-M.; Wang, M.; Li, X.-Y.; Zeng, X.-L.; Xiao-Fei, L.; Du, Y.-H.; Wang, G.-L.; Zhou, J.-G.; et al. TMEM16A Contributes to Endothelial Dysfunction by Facilitating Nox2 NADPH Oxidase–Derived Reactive Oxygen Species Generation in Hypertension. Hypertension 2017, 69, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Manoury, B.; Tamuleviciute, A.; Tammaro, P. TMEM16A/Anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J. Physiol. 2010, 588, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, N.; Forrest, A.S.; Ayon, R.J.; Wiwchar, M.; Angermann, J.E.; Pritchard, H.A.T.; Singer, C.A.; Valencik, M.L.; Britton, F.; Greenwood, I.A. Molecular and functional significance of Ca2+-activated Cl− channels in pulmonary arterial smooth muscle. Pulm. Circ. 2015, 5, 244–268. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Dannhoffer, L.; Antigny, F.; Vachel, L.; Jayle, C.; Vandebrouck, C.; Becq, F.; Norez, C. A functional tandem between transient receptor potential canonical channels 6 and calcium-dependent chloride channels in human epithelial cells. Eur. J. Pharmacol. 2015, 765, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Birnbaumer, L.; Singh, B.B. TRPC1 regulates calcium-activated chloride channels in salivary gland cells. J. Cell. Physiol. 2015, 230, 2848–2856. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, A.R.; Vaeth, M.; Wagner, L.E.; Eckstein, M.; Hecht, L.; Yang, J.; Crottes, D.; Seidl, M.; Shin, H.P.; Weidinger, C.; et al. Store-operated Ca2+ entry regulates Ca2+-activated chloride channels and eccrine sweat gland function. J. Clin. Investig. 2016, 126, 4303–4318. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J. Role of calcium-activated chloride current in regulating pulmonary vasomotor tone. Am. J. Physiol. 1997, 272, L959–L968. [Google Scholar] [CrossRef] [PubMed]
- Robert, R.; Savineau, J.-P.; Norez, C.; Becq, F.; Guibert, C. Expression and function of cystic fibrosis transmembrane conductance regulator in rat intrapulmonary arteries. Eur. Respir. J. 2007, 30, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Totani, L.; Plebani, R.; Piccoli, A.; Di Silvestre, S.; Lanuti, P.; Recchiuti, A.; Cianci, E.; Dell’Elba, G.; Sacchetti, S.; Patruno, S.; et al. Mechanisms of endothelial cell dysfunction in cystic fibrosis. BBA Mol. Basis Dis. 2017, 1863, 3243–3253. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial Primary Pulmonary Hypertension (Gene PPH1) Is Caused by Mutations in the Bone Morphogenetic Protein Receptor–II Gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Young, K.A.; Ivester, C.; West, J.; Carr, M.; Rodman, D.M. BMP signaling controls PASMC K V channel expression in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L841–L848. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Powell, F.; Haddad, G.H.; Yuan, J.X.-J. Hypoxia Selectively Inhibits KCNA5 Channels in Pulmonary Artery Smooth Muscle Cells. Ann. N. Y. Acad. Sci. 2009, 1177, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Tang, L.-L.; Wei, J.-K.; Xu, X.-F.; Gu, W.; Fu, L.-C.; Zhang, L.-Y.; Du, L.-Z. Decreased Kv1.5 expression in intrauterine growth retardation rats with exaggerated pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L856–L865. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, V.; Joshi, S.; Ghaly, Y.; Mizera, R.; Zaloudikova, M.; Brennan, S.; Novotna, J.; Herget, J.; Gurney, A.M. Role of Kv7 channels in responses of the pulmonary circulation to hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L48–L57. [Google Scholar] [CrossRef] [PubMed]
- Morecroft, I.; Murray, A.; Nilsen, M.; Gurney, A.M.; MacLean, M.R. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br. J. Pharmacol. 2009, 157, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Weigand, L.; Wang, W.; Sylvester, J.T.; Shimoda, L.A. Chronic hypoxia inhibits K v channel gene expression in rat distal pulmonary artery. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L1049–L1058. [Google Scholar] [CrossRef] [PubMed]
- Hogg, D.S.; Davies, A.R.L.; McMurray, G.; Kozlowski, R.Z. K(V)2.1 channels mediate hypoxic inhibition of I(KV) in native pulmonary arterial smooth muscle cells of the rat. Cardiovasc. Res. 2002, 55, 349–360. [Google Scholar] [CrossRef]
- Marino, M.; Bény, J.-L.; Peyter, A.-C.; Diaceri, G.; Tolsa, J.-F. Perinatal hypoxia enhances cyclic adenosine monophosphate-mediated BKCa channel activation in adult murine pulmonary artery. J. Cardiovasc. Pharmacol. 2011, 57, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, C.; Tang, B.; Nagy, B.M.; Papp, R.; Jain, P.P.; Marsh, L.M.; Meredith, A.L.; Ghanim, B.; Klepetko, W.; Kwapiszewska, G.; et al. Docosahexaenoic acid causes rapid pulmonary arterial relaxation via KCa channel-mediated hyperpolarisation in pulmonary hypertension. Eur. Respir. J. 2016, 48, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Olschewski, A.; Veale, E.L.; Nagy, B.M.; Nagaraj, C.; Kwapiszewska, G.; Antigny, F.; Lambert, M.; Humbert, M.; Czirják, G.; Enyedi, P.; et al. TASK-1 (KCNK3) channels in the lung: from cell biology to clinical implications. Eur. Respir. J. 2017, 50, 1700754. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, C.; Tang, B.; Balint, Z.; Wygrecka, M.; Hrzenjak, A.; Kwapiszewska, G.; Stacher, E.; Lindenmann, J.; Weir, E.K.; Olschewski, H.; et al. Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur. Respir. J. 2013, 41, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Mermer, P.; Goldenberg, A.; Pfeil, U.; Paddenberg, R.; Weissmann, N.; Lochnit, G.; Kummer, W. TASK-1 potassium channel is not critically involved in mediating hypoxic pulmonary vasoconstriction of murine intra-pulmonary arteries. PLoS ONE 2017, 12, e0174071. [Google Scholar] [CrossRef] [PubMed]
- Pandit, L.M.; Lloyd, E.E.; Reynolds, J.O.; Lawrence, W.S.; Reynolds, C.; Wehrens, X.H.T.; Bryan, R.M. TWIK-2 channel deficiency leads to pulmonary hypertension through a rho-kinase-mediated process. Hypertension 2014, 64, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, I.F.; Davidson, A.B.; Reeves, J.T.; Grover, R.F. Inhibition of Hypoxic Pulmonary Vasoconstriction by Calcium Antagonists in Isolated Rat Lungs. Circulation 1976, 38, 99–104. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Sham, J.S.K.; Shimoda, T.H.; Sylvester, J.T. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L884–L894. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, S.J.; Chen, Y.F.; Meng, Q.C.; Durand, J.; Oparil, S.; Elton, T.S. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J. Appl. Physiol. 1994, 77, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Luke, T.; Maylor, J.; Undem, C.; Sylvester, J.T.; Shimoda, L.A. Kinase-dependent activation of voltage-gated Ca 2+ channels by ET-1 in pulmonary arterial myocytes during chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1128–L1139. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Humbert, M.; Jaïs, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Qiu, Z.; Wei, L.; Yu, X.; Gao, X.; Jiang, S.; Tian, H.; Jiang, C.; Zhu, D. The MicroRNA-328 Regulates Hypoxic Pulmonary Hypertension by Targeting at Insulin Growth Factor 1 Receptor and L-Type Calcium Channel-α1C. Hypertension 2012, 59, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Yamamura, A.; Zimnicka, A.M.; Voiriot, G.; Smith, K.A.; Tang, H.; Ayon, R.J.; Choudhury, M.S.R.; Ko, E.A.; Wang, J.; et al. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca 2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L154–L164. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Li, Y.; Kaku, T.; Inagaki, S.; Hashimoto, Y.; Kimura, K.; Miyamoto, S.; Hadama, T.; Ono, K. Remodeling excitation–contraction coupling of hypertrophied ventricular myocytes is dependent on T-type calcium channels expression. Biochem. Biophys. Res. Commun. 2006, 345, 766–773. [Google Scholar] [CrossRef] [PubMed]
- de Man, F.S.; Tu, L.; Handoko, M.L.; Rain, S.; Ruiter, G.; François, C.; Schalij, I.; Dorfmüller, P.; Simonneau, G.; Fadel, E.; et al. Dysregulated Renin–Angiotensin–Aldosterone System Contributes to Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Ferron, L.; Capuano, V.; Deroubaix, E.; Coulombe, A.; Renaud, J.-F. Functional and molecular characterization of a T-type Ca2+ channel during fetal and postnatal rat heart development. J. Mol. Cell. Cardiol. 2002, 34, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Ferron, L.; Capuano, V.; Ruchon, Y.; Deroubaix, E.; Coulombe, A.; Renaud, J.-F. Angiotensin II signaling pathways mediate expression of cardiac T-type calcium channels. Circ. Res. 2003, 93, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Ruchon, Y.; Ferron, L.; Sankhe, S.; Renaud, J.-F.; Capuano, V. T-type Ca2+ signalling downregulates MEK1/2 phosphorylation and cross-talk with the RAAS transcriptional response in cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 53, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Song, M.Y.; Makino, A.; Yuan, J.X.-J. STIM2 Contributes to Enhanced Store-operated Ca Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension. Pulm. Circ. 2011, 1, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, R.A.; Wan, J.; Song, S.; Smith, K.A.; Gu, Y.; Tauseef, M.; Tang, H.; Makino, A.; Mehta, D.; Yuan, J.X.-J. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am. J. Physiol. Cell Physiol. 2015, 308, C581–C593. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sysol, J.R.; Singla, S.; Zhao, S.; Yamamura, A.; Valdez-Jasso, D.; Abbasi, T.; Shioura, K.M.; Sahni, S.; Reddy, V.; et al. Nicotinamide Phosphoribosyltransferase Promotes Pulmonary Vascular Remodeling and Is a Therapeutic Target in Pulmonary Arterial Hypertension. Circulation 2017, 135, 1532–1546. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wang, J.; Shimoda, L.A.; Sylvester, J.T. Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+ responses to hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L104–L113. [Google Scholar] [CrossRef] [PubMed]
- Rode, B.; Bailey, M.A.; Marthan, R.; Beech, D.J.; Guibert, C. ORAI Channels as Potential Therapeutic Targets in Pulmonary Hypertension. Physiology 2018, 33, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fantozzi, I.; Remillard, C.V.; Landsberg, J.W.; Kunichika, N.; Platoshyn, O.; Tigno, D.D.; Thistlethwaite, P.A.; Rubin, L.J.; Yuan, J.X.-J. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 13861–13866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Patel, H.H.; Murray, F.; Remillard, C.V.; Schach, C.; Thistlethwaite, P.A.; Insel, P.A.; Yuan, J.X.-J. Pulmonary artery smooth muscle cells from normal subjects and IPAH patients show divergent cAMP-mediated effects on TRPC expression and capacitative Ca2+ entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1202–L1210. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; To, W.K.; Meng, F.; Wang, Y.; Gu, Y. A Role for Receptor-Operated Ca2+ Entry in Human Pulmonary Artery Smooth Muscle Cells in Response to Hypoxia. Physiol. Res. 2010, 59, 909–918. [Google Scholar] [PubMed]
- Kumar, B.; Dreja, K.; Shah, S.S.; Cheong, A.; Xu, S.-Z.; Sukumar, P.; Naylor, J.; Forte, A.; Cipollaro, M.; McHugh, D.; et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ. Res. 2006, 98, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yang, X.-R.; Fu, Z.; Paudel, O.; Abramowitz, J.; Birnbaumer, L.; Sham, J.S.K. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertensions 2014, 63, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Malczyk, M.; Veith, C.; Fuchs, B.; Hofmann, K.; Storch, U.; Schermuly, R.T.; Witzenrath, M.; Ahlbrecht, K.; Fecher-Trost, C.; Flockerzi, V.; et al. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, A.; Almalouf, P.; Toba, M.; O’Neill, K.; Qian, X.; Francis, M.; Taylor, M.S.; Alexeyev, M.; McMurtry, I.F.; Oka, M.; et al. TRPC4 inactivation confers a survival benefit in severe pulmonary arterial hypertension. Am. J. Pathol. 2013, 183, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.; Xu, N.; Zhou, C.; Stevens, T. Transient Receptor Potential Channel 4 Encodes a Vascular Permeability Defect and High-Frequency Ca2+ Transients in Severe Pulmonary Arterial Hypertension. Am. J. Pathol. 2016, 186, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Linde, C.I.; Karashima, E.; Raina, H.; Zulian, A.; Wier, W.G.; Hamlyn, J.M.; Ferrari, P.; Blaustein, M.P.; Golovina, V.A. Increased arterial smooth muscle Ca2+ signaling, vasoconstriction, and myogenic reactivity in Milan hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H611–H620. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fu, Z.; Hu, J.; Huang, C.; Paudel, O.; Cai, S.; Liedtke, W.; Sham, J.S.K. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am. J. Physiol. Cell Physiol. 2013, 305, C704–C715. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Dahan, D.; Cardouat, G.; Gillibert-Duplantier, J.; Marthan, R.; Savineau, J.-P.; Ducret, T. Involvement of TRPV1 and TRPV4 channels in migration of rat pulmonary arterial smooth muscle cells. Pflugers Arch. 2012, 464, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Dahan, D.; Ducret, T.; Quignard, J.-F.; Marthan, R.; Savineau, J.-P.; Estève, E. Implication of the ryanodine receptor in TRPV4-induced calcium response in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L824–L833. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ayon, R.J.; Yamamura, A.; Yamamura, H.; Dash, S.; Babicheva, A.; Tang, H.; Sun, X.; Cordery, A.G.; Khalpey, Z.; et al. Capsaicin-induced Ca2+ signaling is enhanced via upregulated TRPV1 channels in pulmonary artery smooth muscle cells from patients with idiopathic PAH. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L309–L325. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Wang, J.; Guo, M.; Zhou, Q.; Lu, W. TRPM8 genetic variations associated with COPD risk in the Chinese Han population. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 2563–2571. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L. Smooth muscle acid-sensing ion channel 1: pathophysiological implication in hypoxic pulmonary hypertension. Exp. Physiol. 2015, 100, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Plomaritas, D.R.; Herbert, L.M.; Yellowhair, T.R.; Resta, T.C.; Gonzalez Bosc, L.V.; Walker, B.R.; Jernigan, N.L. Chronic hypoxia limits H2O2-induced inhibition of ASIC1-dependent store-operated calcium entry in pulmonary arterial smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L419–L430. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Herbert, L.M.; Walker, B.R.; Resta, T.C. Chronic hypoxia upregulates pulmonary arterial ASIC1: a novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am. J. Physiol. Cell Physiol. 2012, 302, C931–C940. [Google Scholar] [CrossRef] [PubMed]
- Nitta, C.H.; Osmond, D.A.; Herbert, L.M.; Beasley, B.F.; Resta, T.C.; Walker, B.R.; Jernigan, N.L. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H41–H52. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Bosc, L.V.; Plomaritas, D.R.; Herbert, L.M.; Giermakowska, W.; Browning, C.; Jernigan, N.L. ASIC1-mediated calcium entry stimulates NFATc3 nuclear translocation via PICK1 coupling in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L48–L58. [Google Scholar] [CrossRef] [PubMed]
- Hendy, G.N.; Guarnieri, V.; Canaff, L. Calcium-sensing receptor and associated diseases. Prog. Mol. Biol. Transl. Sci. 2009, 89, 31–95. [Google Scholar] [PubMed]
- Smith, K.A.; Ayon, R.J.; Tang, H.; Makino, A.; Yuan, J.X.-J. Calcium-Sensing Receptor Regulates Cytosolic [Ca2+] and Plays a Major Role in the Development of Pulmonary Hypertension. Front. Physiol. 2016, 7, 517. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, A.; Yamamura, H.; Yuan, J.X.-J. Enhanced Ca2+-sensing receptor function in pulmonary hypertension. Yakugaku Zasshi 2013, 133, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Yamamura, A.; Yamamura, H.; Song, S.; Fraidenburg, D.R.; Chen, J.; Gu, Y.; Pohl, N.M.; Zhou, T.; Jiménez-Pérez, L.; et al. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L846–L859. [Google Scholar] [CrossRef] [PubMed]
- Hadri, L.; Kratlian, R.G.; Benard, L.; Maron, B.A.; Dorfmüller, P.; Ladage, D.; Guignabert, C.; Ishikawa, K.; Aguero, J.; Ibanez, B.; et al. Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation 2013, 128, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Girerd, B.; Günther, S.; Riant, F.; Tournier-Lasserve, E.; Magy, L.; Maazi, N.; Guignabert, C.; Savale, L.; Sitbon, O.; et al. Pulmonary arterial hypertension in familial hemiplegic migraine with ATP1A2 channelopathy. Eur. Respir. J. 2014, 43, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Tabeling, C.; Yu, H.; Wang, L.; Ranke, H.; Goldenberg, N.M.; Zabini, D.; Noe, E.; Krauszman, A.; Gutbier, B.; Yin, J.; et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 2015, 112, E1614–E1623. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Norez, C.; Dannhoffer, L.; Bertrand, J.; Raveau, D.; Corbi, P.; Jayle, C.; Becq, F.; Vandebrouck, C. Transient Receptor Potential Canonical Channel 6 Links Ca 2+ Mishandling to Cystic Fibrosis Transmembrane Conductance Regulator Channel Dysfunction in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 44, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Telles, C.J.; Decker, S.E.; Motley, W.W.; Peters, A.W.; Mehr, A.P.; Frizzell, R.A.; Forrest, J.N. Functional and molecular identification of a TASK-1 potassium channel regulating chloride secretion through CFTR channels in the shark rectal gland: implications for cystic fibrosis. Am. J. Physiol. Cell Physiol. 2016, 311, C884–C894. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.S.; Joyce, T.C.; Huebner, M.L.; Ayon, R.J.; Wiwchar, M.; Joyce, J.; Freitas, N.; Davis, A.J.; Ye, L.; Duan, D.D.; et al. Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am. J. Physiol. Cell Physiol. 2012, 303, C1229–C1243. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xia, Y.; Paudel, O.; Yang, X.-R.; Sham, J.S.K. Chronic hypoxia-induced upregulation of Ca2+-activated Cl− channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J. Physiol. 2012, 590, 3507–3521. [Google Scholar] [CrossRef] [PubMed]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thébaud, B.; Wu, X.-C.; Dyck, J.R.B.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Long, C.; Cui, W.; Wang, H. Iptakalim ameliorates monocrotaline-induced pulmonary arterial hypertension in rats. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, S.; Xie, W.; Li, Q.; Zhou, Y.; Wang, H. Iptakalim inhibited endothelin-1-induced proliferation of human pulmonary arterial smooth muscle cells through the activation of KATP channel. Vascul. Pharmacol. 2008, 48, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, Y.; Nagaraj, C.; Morty, R.E.; Gabor, S.; Stacher, E.; Voswinckel, R.; Weissmann, N.; Leithner, K.; Olschewski, H.; et al. Endothelin-1 Inhibits Background Two-Pore Domain Channel TASK-1 in Primary Human Pulmonary Artery Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2009, 41, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Weatherald, J.; Chaumais, M.-C.; Savale, L.; Jaïs, X.; Seferian, A.; Canuet, M.; Bouvaist, H.; Magro, P.; Bergeron, A.; Guignabert, C.; et al. Long-term outcomes of dasatinib-induced pulmonary arterial hypertension: a population-based study. Eur. Respir. J. 2017, 50, 1700217. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Phan, C.; Seferian, A.; Huertas, A.; Tu, L.; Thuillet, R.; Sattler, C.; Le Hiress, M.; Tamura, Y.; Jutant, E.-M.; et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J. Clin. Investig. 2016, 126, 3207–3218. [Google Scholar] [CrossRef] [PubMed]
- Revermann, M.; Schloss, M.; Mieth, A.; Babelova, A.; Schröder, K.; Neofitidou, S.; Buerkl, J.; Kirschning, T.; Schermuly, R.T.; Hofstetter, C.; et al. an attenuates pulmonary vascular remodeling. Intensive Care Med. 2011, 37, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, J.; Zhao, L.; Wang, Y.; Liu, J.; Shi, L.; Xu, M.; Wang, C. Sildenafil Inhibits Human Pulmonary Artery Smooth Muscle Cell Proliferation by Decreasing Capacitative Ca2+ Entry. J. Pharmacol. Sci. 2008, 108, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Singal, R.; Gupta, P.; Jain, N.; Gupta, S. Role of Flupirtine in the Treatment of Pain - Chemistry and its Effects. Maedica 2012, 7, 163–166. [Google Scholar] [PubMed]
- Chung, S.C.; McDonald, T.V.; Gardner, P. Inhibition by SK&F 96365 of Ca2+ current, IL-2 production and activation in T lymphocytes. Br. J. Pharmacol. 1994, 113, 861–868. [Google Scholar] [PubMed]
- Singh, A.; Hildebrand, M.E.; Garcia, E.; Snutch, T.P. The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharmacol. 2010, 160, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deshpande, M.; Payne, R. 2-Aminoethoxydiphenyl borate inhibits phototransduction and blocks voltage-gated potassium channels in Limulus ventral photoreceptors. Cell Calcium 2002, 32, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Missiaen, L.; Callewaert, G.; De Smedt, H.; Parys, J.B. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium 2001, 29, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 2001, 536, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Mori, Y.; Hara, Y.; Uchida, K.; Zhou, H.; Mikoshiba, K. 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Receptors Channels 2001, 7, 429–439. [Google Scholar] [PubMed]
- Morihara, H.; Obana, M.; Tanaka, S.; Kawakatsu, I.; Tsuchiyama, D.; Mori, S.; Suizu, H.; Ishida, A.; Kimura, R.; Tsuchimochi, I.; et al. 2-aminoethoxydiphenyl borate provides an anti-oxidative effect and mediates cardioprotection during ischemia reperfusion in mice. PLoS ONE 2017, 12, e0189948. [Google Scholar] [CrossRef] [PubMed]
- Zitt, C.; Strauss, B.; Schwarz, E.C.; Spaeth, N.; Rast, G.; Hatzelmann, A.; Hoth, M. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J. Biol. Chem. 2004, 279, 12427–12437. [Google Scholar] [CrossRef] [PubMed]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.N.; Kappe, C.O.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.; Schernthaner, M.; Maechler, H.; Kappe, C.O.; Glasnov, T.N.; Hoefler, G.; Braune, M.; Wittchow, E.; Groschner, K. A TRPC3 blocker, ethyl-1-(4-(2,3,3-trichloroacrylamide)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate (Pyr3), prevents stent-induced arterial remodeling. J. Pharmacol. Exp. Ther. 2013, 344, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qi, Z.; Wang, Y. BTP2, a Store-Operated Calcium Channel Inhibitor, Attenuates Lung Ischemia-Reperfusion Injury in Rats. Inflammation 2017, 40, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Ohga, K.; Takezawa, R.; Arakida, Y.; Shimizu, Y.; Ishikawa, J. Characterization of YM-58483/BTP2, a novel store-operated Ca2+ entry blocker, on T cell-mediated immune responses in vivo. Int. Immunopharmacol. 2008, 8, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Sogkas, G.; Rau, E.; Atschekzei, F.; Syed, S.N.; Schmidt, R.E. The Pyrazole Derivative BTP2 Attenuates IgG Immune Complex-induced Inflammation. Inflammation 2018, 41, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 2013, 53, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Owsianik, G.; D’hoedt, D.; Voets, T.; Nilius, B. Structure-function relationship of the TRP channel superfamily. Rev. Physiol. Biochem. Pharmacol. 2006, 156, 61–90. [Google Scholar] [PubMed]
- van Kruchten, R.; Braun, A.; Feijge, M.A.H.; Kuijpers, M.J.E.; Rivera-Galdos, R.; Kraft, P.; Stoll, G.; Kleinschnitz, C.; Bevers, E.M.; Nieswandt, B.; et al. Antithrombotic potential of blockers of store-operated calcium channels in platelets. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; di Capite, J.; Singaravelu, K.; Parekh, A.B. Sustained activation of the tyrosine kinase Syk by antigen in mast cells requires local Ca2+ influx through Ca2+ release-activated Ca2+ channels. J. Biol. Chem. 2008, 283, 31348–31355. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Rovedatti, L.; Kaur, R.; Spencer, J.P.; Brown, J.T.; Morisset, V.D.; Biancheri, P.; Leakey, N.A. B.; Wilde, J.I.; Scott, L.; et al. Targeting gut T cell Ca2+ release-activated Ca2+ channels inhibits T cell cytokine production and T-box transcription factor T-bet in inflammatory bowel disease. J. Immunol. Baltim. 2009, 183, 3454–3462. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Shi, J.; Wu, M.; Engers, J.; Hopkins, C.R.; Lindsley, C.W.; Salovich, J.M.; Zhu, Y.; Tian, J.-B.; Zhu, M.X.; et al. Novel Chemical Inhibitor of TRPC4 Channels. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Miller, M.; Shi, J.; Zhu, Y.; Kustov, M.; Tian, J.; Stevens, A.; Wu, M.; Xu, J.; Long, S.; Yang, P.; et al. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J. Biol. Chem. 2011, 286, 33436–33446. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; McAlexander, M.A.; Bíró, T.; Szallasi, A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Manitpisitkul, P.; Brandt, M.; Flores, C.M.; Kenigs, V.; Moyer, J.A.; Romano, G.; Shalayda, K.; Mayorga, A.J. TRPV1 antagonist JNJ-39439335 (mavatrep) demonstrates proof of pharmacology in healthy men: a first-in-human, double-blind, placebo-controlled, randomized, sequential group study. Pain Rep. 2016, 1, e576. [Google Scholar] [CrossRef] [PubMed]
- McLeod, R.L.; Fernandez, X.; Correll, C.C.; Phelps, T.P.; Jia, Y.; Wang, X.; Hey, J.A. TRPV1 antagonists attenuate antigen-provoked cough in ovalbumin sensitized guinea pigs. Cough Lond. Engl. 2006, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Thorneloe, K.S.; Cheung, M.; Bao, W.; Alsaid, H.; Lenhard, S.; Jian, M.-Y.; Costell, M.; Maniscalco-Hauk, K.; Krawiec, J.A.; Olzinski, A.; et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci. Transl. Med. 2012, 4, 159ra148. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Everaerts, W.; Zhen, X.; Ghosh, D.; Vriens, J.; Gevaert, T.; Gilbert, J.P.; Hayward, N.J.; McNamara, C.R.; Xue, F.; Moran, M.M.; et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc. Natl. Acad. Sci. USA 2010, 107, 19084–19089. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, L.; Dinger, B.; Fidone, S.J. Chronic hypoxia-induced acid-sensitive ion channel expression in chemoafferent neurons contributes to chemoreceptor hypersensitivity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L985–L992. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Bernard, C.; Lambeau, G.; Lazdunski, M.; Darbon, H. Recombinant production and solution structure of PcTx1, the specific peptide inhibitor of ASIC1a proton-gated cation channels. Protein Sci. Publ. Protein Soc. 2003, 12, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Salinas, M.; Rash, L.D.; Baron, A.; Lambeau, G.; Escoubas, P.; Lazdunski, M. The receptor site of the spider toxin PcTx1 on the proton-gated cation channel ASIC1a. J. Physiol. 2006, 570, 339–354. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.A.; Rash, L.D.; Chassagnon, I.R.; King, G.F.; Widdop, R.E. PcTx1 affords neuroprotection in a conscious model of stroke in hypertensive rats via selective inhibition of ASIC1a. Neuropharmacology 2015, 99, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Bill, A.; Hall, M.L.; Borawski, J.; Hodgson, C.; Jenkins, J.; Piechon, P.; Popa, O.; Rothwell, C.; Tranter, P.; Tria, S.; et al. Small molecule-facilitated degradation of ANO1 protein: a new targeting approach for anticancer therapeutics. J. Biol. Chem. 2014, 289, 11029–11041. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Lee, H.K.; Park, J.; Jeon, D.-K.; Jo, S.; Jo, M.; Namkung, W. Ani9, A Novel Potent Small-Molecule ANO1 Inhibitor with Negligible Effect on ANO2. PLoS ONE 2016, 11, e0155771. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, H.; Wu, M.; Yang, H.; Kudo, M.; Peters, C.J.; Woodruff, P.G.; Solberg, O.D.; Donne, M.L.; Huang, X.; et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. USA 2012, 109, 16354–16359. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ramena, G.; Yin, Y.; Premkumar, L.; Elble, R.C. CLCA2 is a positive regulator of store-operated calcium entry and TMEM16A. PLoS ONE 2018, 13, e0196512. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Channel | Family | Aliases |
|---|---|---|---|
| KCNK1 | K2P1.1 | TWIK | TWIK-1 |
| KCNK2 | K2P2.1 | TREK | TREK-1 |
| KCNK3 | K2P3.1 | TASK | TASK-1 |
| KCNK4 | K2P4.1 | TREK | TRAAK |
| KCNK5 | K2P5.1 | TASK | TASK-2 |
| KCNK6 | K2P6.1 | TWIK | TWIK-2 |
| KCNK7 | K2P7.1 | TWIK | |
| KCNK9 | K2P9.1 | TASK | TASK-3 |
| KCNK10 | K2P10.1 | TREK | TREK-2 |
| KCNK12 | K2P12.1 | THIK | THIK-2 |
| KCNK13 | K2P13.1 | THIK | THIK-1 |
| KCNK15 | K2P15.1 | TASK | TASK-5 |
| KCNK16 | K2P16.1 | TALK | TALK-1 |
| KCNK17 | K2P17.1 | TALK | TALK-2, TASK-4 |
| KCNK18 | K2P18.1 | TRIK, TRESK |
| Channel | Permeability | mRNA | Protein | Function | Human PAH | Experimental PH Models | Clinical Features |
|---|---|---|---|---|---|---|---|
| Kv1.1 | K+ | ↓ | - | - | Lung from PAH patients | Link to plasma membrane depolarization, to voltage-gated-Ca2+ channel activation, to PA vasoconstriction and PASMC proliferation and apoptosis resistance In vivo Kv1.5 re-expression bygene transfert protect against PH in CH rats | |
| ↓ | ↓ | ↓ | Hypoxic rat PASMC | ||||
| Kv1.2 | K+ | ↓ | ↓ | ↓ | Hypoxic rat PASMC | ||
| Kv1.5 | K+ | ↓ | ↓ | ↓ | Lung from PAH patients | ||
| ↓ | ↓ | ↓ | Hypoxic rat PASMC | ||||
| ↓ | ↓ | ↓ | Lung from MCT rat | ||||
| Kv2.1 | K+ | ↓ | ↓ | ↓ | Hypoxic rat PASMC | ||
| ↓ | ↓ | ↓ | PASMC from CH rat | ||||
| Kv4.3 | K+ | ↓ | - | - | Lung from PAH patients | ||
| ↓ | ↓ | ↓ | Hypoxic rat PASMC | ||||
| Kv9.3 | K+ | ↓ | ↓ | ↓ | Hypoxic rat PASMC | ||
| KCNK3/TASK-1 | K+ | - | - | ↓ | Mutations in PAH patients | Link to plasma membrane depolarization, to voltage-gated-Ca2+ channel activation, to PA vasoconstriction and PASMC proliferation and apoptosis resistance. In rat, pharmacological activation with ONO-RS-082 compound reduces MCT phenotype | |
| ↓ | - | ↓ | Lung and PA from iPAH and hPAH patients | ||||
| ↓ | ↓ | ↓ | PASMC from PAH patients | PAEC and PASMC from MCT rat | |||
| ↓ | ↓ | ↓ | Lung from MCT rat | ||||
| BKCa | K+ | - | ↓ | ↓ | PASMC from CH rat | Alteration of PA relaxation | |
| Cav3.1 & Cav3.2 | Ca2+ | - | ↑ | - | PASMC from iPAH patients | Link to PA vasoconstriction and PASMC proliferation | |
| ↑ | ↑ | - | PASMC from CH rat and mice | ||||
| STIM2 | - | ↑ | - | PASMC from iPAH patients | Rat PASMC phenotypic transition | ||
| - | ↑ | - | Rat PASMC | ||||
| Orai2 | Ca2+ | - | ↑ | - | Rat PASMC | Rat PASMC phenotypic transition and PASMC proliferation | |
| SOCE | Ca2+ | - | - | ↑ | PASMC from iPAH patients | ||
| - | - | ↑ | Rat distal PASMC | ||||
| TRPC6 | Ca2+, Na+ | ↑ | ↑ | ↑ | PASMC from iPAH patients | Human and rodents PASMC proliferation In mice trpc6 gene deletion reduces PH due to CH exposure In human, a polymorphism in TRPC6 gene seems to predispose to PAH | |
| ↑ | ↑ | - | Lung and PASMC from iPAH patients | ||||
| - | ↑ | - | Milan Hypertensive rats | ||||
| ↑ | ↑ | - | Lung from CH rats | ||||
| TRPC1 | Ca2+, Na+ | ↑ | - | - | PASMC from CH mice | Rodents PASMC proliferation In mice trpc1 gene deletion reduces PH due to CH exposure | |
| ↑ | ↑ | ↑ | PASMC from CH / MCT rats | ||||
| TRPC3 | Ca2+, Na+ | ↑ | ↑ | ↑ | PASMC from iPAH patients | ||
| TRPV4 | Ca2+, Na+ | ↑ | ↑ | ↑ | PASMC from CH rat | PASMC migration and proliferation In mice, trpv4 gene deletion suppresses the development of PH due to CH | |
| TRPV1 | Ca2+, Na+ | ↑ | ↑ | - | PASMC from iPAH patients | PASMC proliferation and migration. | |
| ASIC | Na+, Ca2+ | - | ↑ | ↑ | PASMC from CH rat | Link to membrane depolarization and rat PA vasconstriction | |
| CaSR | Ca2+ | - | ↑ | ↑ | PASMC from iPAH patients | PASMC proliferation | |
| SERCA2a | Ca2+ | - | ↓ | - | Lung and PASMC from iPAH patients | Lung and PASMC from MCT rats | PASMC proliferation and migration. In vivo SERCA2a re-expression by gene therapy reduce MCT-PH phenotype |
| SCN1B | Na+ | ↑ | - | ↑ | Lung from iPAH patients | Contribute to abnormal vasoconstriction and pulmonary vascular remodeling | |
| NMDAR | Ca2+ | ↑ | ↑ | - | PA from iPAH patients | Link to PASMC proliferation, In rat NMDAR antagonist reduce PH, In mice, nmdar gene deletion reduces PH | |
| TMEM16A | Cl- | ↑ | ↑ | ↑ | Lung from MCT rat | Link to membrane depolarization and rat PA vasconstriction | |
| ↑ | ↑ | - | Lung from CH rat |
| All | Females | Males | ||
|---|---|---|---|---|
| Age of Diagnosis | Median | 29 | 35 | 22 |
| Max | 52 | 48 | 52 | |
| Min | 0.17 | 17 | 0.17 | |
| mPAP (mmHg) | Median | 76 | 67 | 94 |
| Max | 107 | 81 | 107 | |
| Min | 54 | 54 | 86 | |
| RAP (mmHg) | Median | 12 | 15.5 | 11 |
| Max | 29 | 29 | 16 | |
| Min | 3 | 7 | 3 | |
| CI (L/min) | Median | 2.7 | 2.7 | 2.36 |
| Max | 3.22 | 3.16 | 3.22 | |
| Min | 1.21 | 1.21 | 1.73 | |
| PVI (dyn/s/cm−5) | Median | 1826 | 1764 | 2874 |
| Max | 3977 | 3174 | 3977 | |
| Min | 1316 | 1316 | 1813 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambert, M.; Capuano, V.; Olschewski, A.; Sabourin, J.; Nagaraj, C.; Girerd, B.; Weatherald, J.; Humbert, M.; Antigny, F. Ion Channels in Pulmonary Hypertension: A Therapeutic Interest? Int. J. Mol. Sci. 2018, 19, 3162. https://doi.org/10.3390/ijms19103162
Lambert M, Capuano V, Olschewski A, Sabourin J, Nagaraj C, Girerd B, Weatherald J, Humbert M, Antigny F. Ion Channels in Pulmonary Hypertension: A Therapeutic Interest? International Journal of Molecular Sciences. 2018; 19(10):3162. https://doi.org/10.3390/ijms19103162
Chicago/Turabian StyleLambert, Mélanie, Véronique Capuano, Andrea Olschewski, Jessica Sabourin, Chandran Nagaraj, Barbara Girerd, Jason Weatherald, Marc Humbert, and Fabrice Antigny. 2018. "Ion Channels in Pulmonary Hypertension: A Therapeutic Interest?" International Journal of Molecular Sciences 19, no. 10: 3162. https://doi.org/10.3390/ijms19103162
APA StyleLambert, M., Capuano, V., Olschewski, A., Sabourin, J., Nagaraj, C., Girerd, B., Weatherald, J., Humbert, M., & Antigny, F. (2018). Ion Channels in Pulmonary Hypertension: A Therapeutic Interest? International Journal of Molecular Sciences, 19(10), 3162. https://doi.org/10.3390/ijms19103162