Pulmonary Arterial Hypertension and Hereditary Haemorrhagic Telangiectasia

Abstract
:1. Pulmonary Hypertension
2. Pulmonary Arterial Hypertension
3. Hereditary Haemorrhagic Telangiectasia
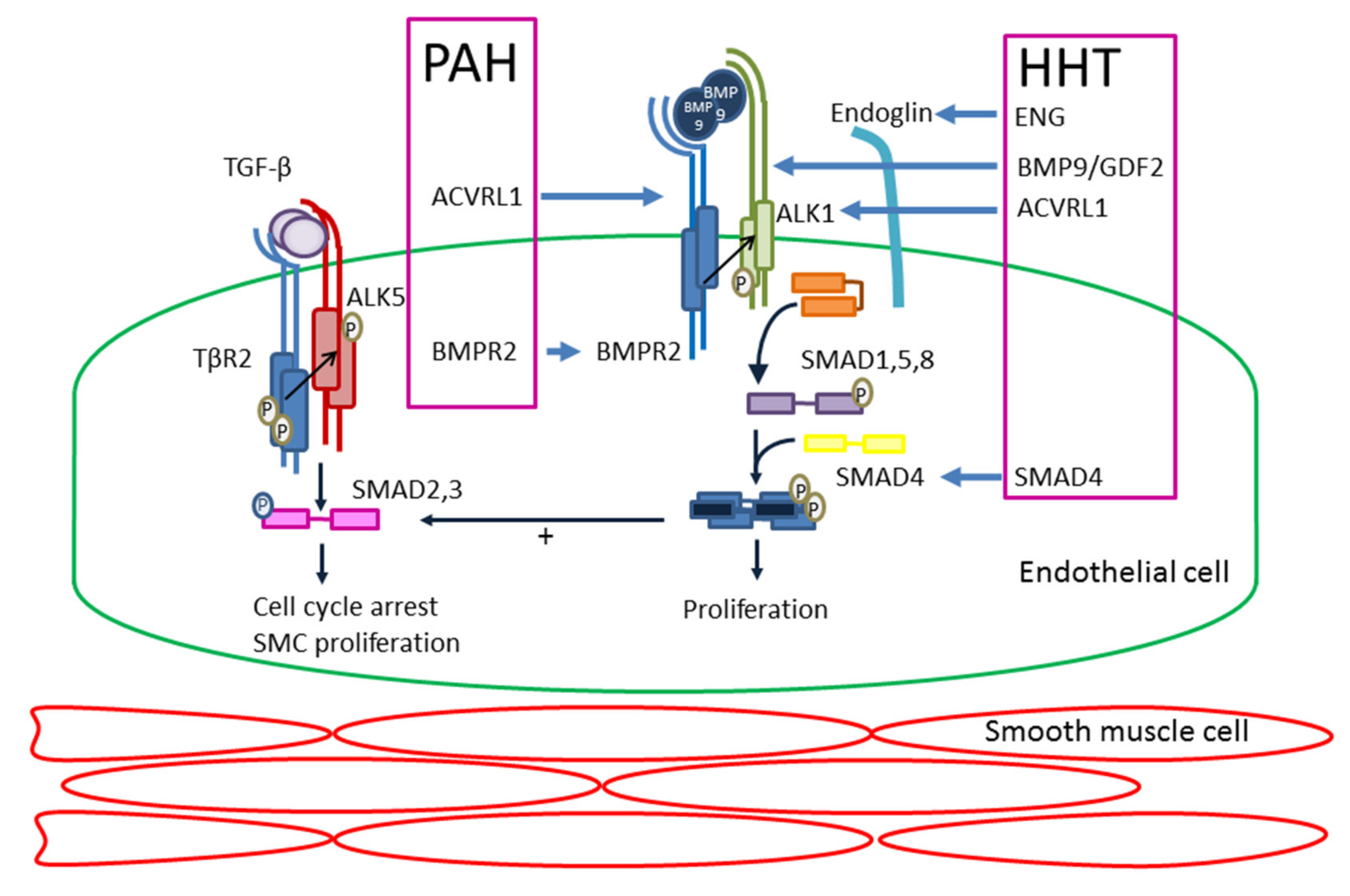
4. Molecular Mechanism
5. Heritable Pulmonary Arterial Hypertension (PAH) and Hereditary Haemorrhagic Telangiectasia (HHT)
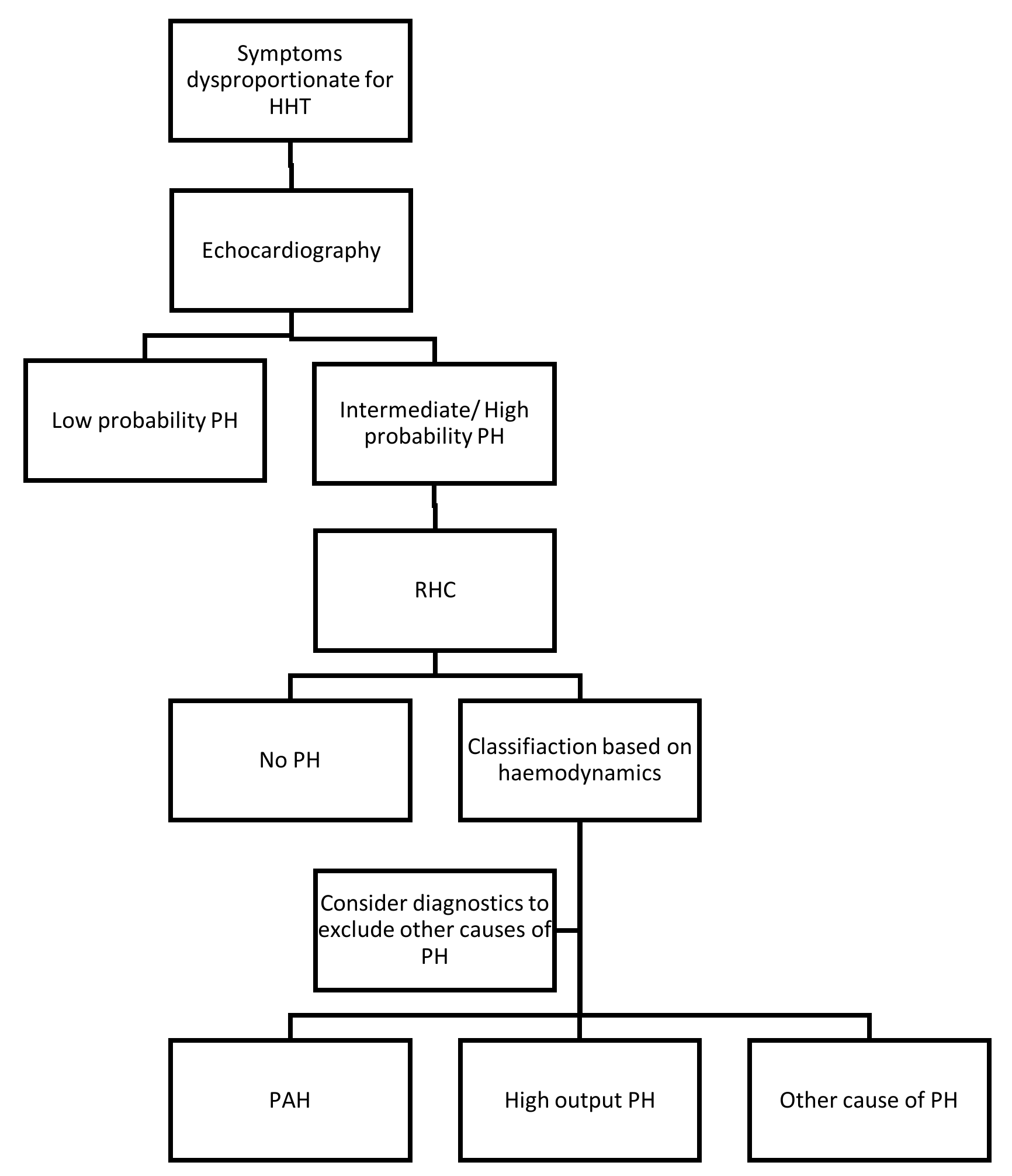
6. Diagnosis of Pulmonary (Arterial) Hypertension in Hereditary Haemorrhagic Telangiectasia (HHT)
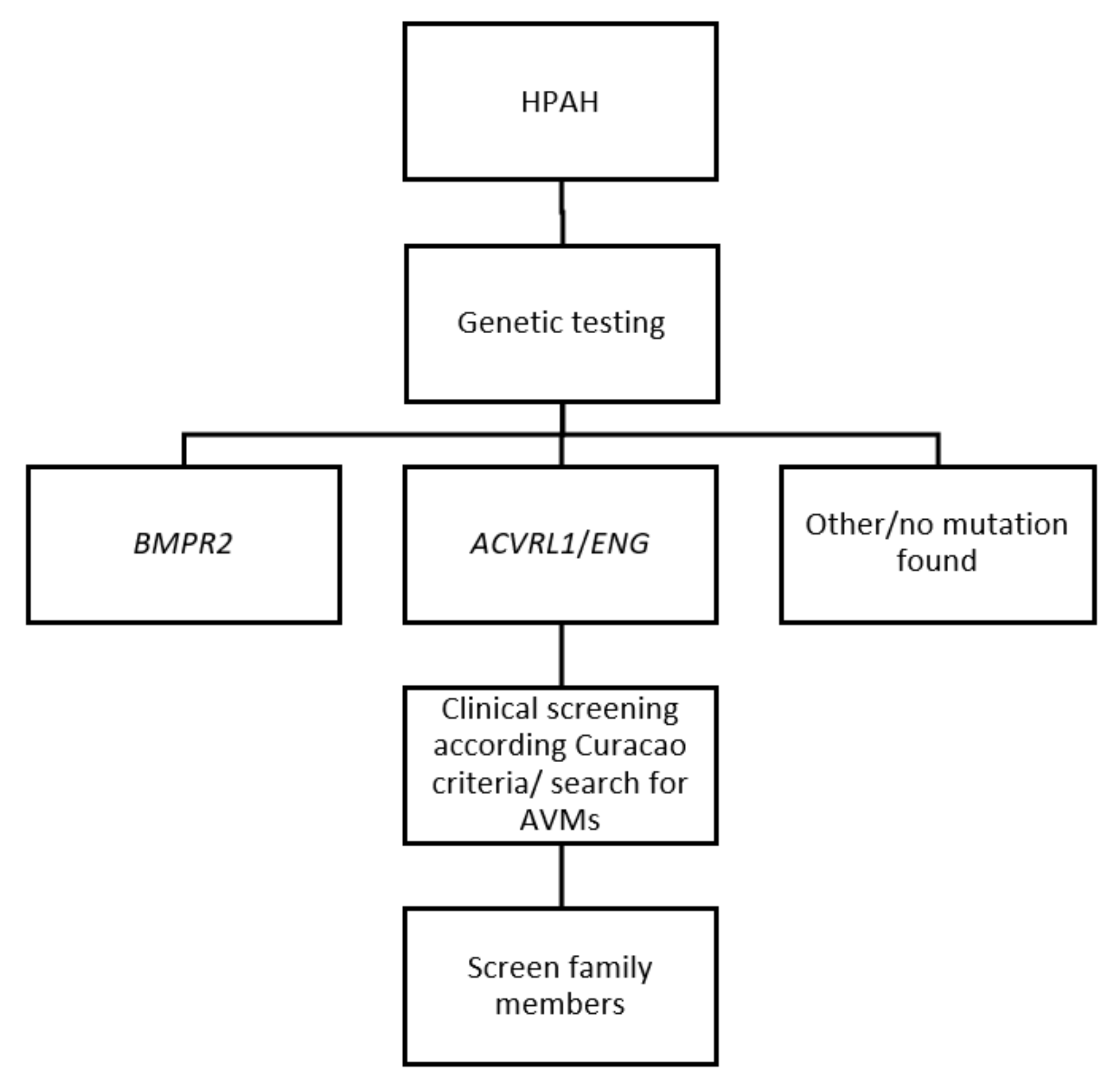
7. Diagnosis of Hereditary Haemorrhagic Telangiectasia (HHT) in Heritable Pulmonary Arterial Hypertension (HPAH)
8. Management of Heritable Pulmonary Arterial Hypertension (PAH) in Hereditary Haemorrhagic Telangiectasia (HHT)
9. Future Therapies
10. Pulmonary Hypertension as Complications of Hereditary Haemorrhagic Telangiectasia (HHT)
11. Conclusions
Funding
Conflicts of Interest
References
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Soubrier, F.; Chung, W.K.; Machado, R.; Grunig, E.; Aldred, M.; Geraci, M.; Loyd, J.E.; Elliott, C.G.; Trembath, R.C.; Newman, J.H.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62, D13–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A., III; Hamid, R.; Loyd, J.E. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Graf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Tillet, E.; Bailly, S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 5, 456. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fan, R.; Ji, R.; Zou, W.; Penny, D.J.; Varghese, N.P.; Fan, Y. Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulm. Med. 2016, 16, 17. [Google Scholar] [CrossRef] [PubMed]
- Velthuis, S.; Buscarini, E.; Gossage, J.R.; Snijder, R.J.; Mager, J.J.; Post, M.C. Clinical implications of pulmonary shunting on saline contrast echocardiography. J. Am. Soc. Echocardiogr. 2015, 28, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Trerotola, S.O.; Pyeritz, R.E. PAVM embolization: An update. AJR Am. J. Roentgenol. 2010, 195, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Buscarini, E.; Plauchu, H.; Garcia Tsao, G.; White, R.I., Jr.; Sabba, C.; Miller, F.; Saurin, J.C.; Pelage, J.P.; Lesca, G.; Marion, M.J.; et al. Liver involvement in hereditary hemorrhagic telangiectasia: Consensus recommendations. Liver Int. 2006, 26, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Vorselaars, V.M.; Velthuis, S.; Snijder, R.J.; Vos, J.A.; Mager, J.J.; Post, M.C. Pulmonary hypertension in hereditary haemorrhagic telangiectasia. World J. Cardiol. 2015, 7, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Willemse, R.B.; Mager, J.J.; Westermann, C.J.; Overtoom, T.T.; Mauser, H.; Wolbers, J.G. Bleeding risk of cerebrovascular malformations in hereditary hemorrhagic telangiectasia. J. Neurosurg. 2000, 92, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S.; et al. HHT foundation international-guidelines working group: International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Plauchu, H.; de Chadarevian, J.P.; Bideau, A.; Robert, J.M. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am. J. Med. Genet. 1989, 32, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Awan, I.; Cahilog, Z.; Abdulla, F.N.; Guttmacher, A.E. Reported cardiac phenotypes in hereditary hemorrhagic telangiectasia emphasize burdens from arrhythmias, anemia and its treatments, but suggest reduced rates of myocardial infarction. Int. J. Cardiol. 2016, 215, 179–185. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.; Wooderchak-Donahue, W.; VanSant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front. Genet. 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, E.; Westermann, C.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef]
- Bayrak-Toydemir, P.; McDonald, J. Available online: http://arup.utah.edu/database/HHT/ (accessed on 14 October 2018).
- De Gussem, E.M.; Edwards, C.P.; Hosman, A.E.; Westermann, C.J.; Snijder, R.J.; Faughnan, M.E.; Mager, J.J. Life expectancy of parents with hereditary haemorrhagic telangiectasia. Orphanet J. Rare Dis. 2016, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Andrejecsk, J.W.; Hosman, A.E.; Botella, L.M.; Shovlin, C.L.; Arthur, H.M.; Dupuis-Girod, S.; Buscarini, E.; Hughes, C.C.W.; Lebrin, F.; Mummery, C.L.; et al. Executive summary of the 12th HHT international scientific conference. Angiogenesis 2018, 21, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Letteboer, T.G.; Mager, J.J.; Snijder, R.J.; Koeleman, B.P.; Lindhout, D.; Ploos van Amstel, J.K.; Westermann, C.J. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J. Med. Genet. 2006, 43, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Benzinou, M.; Clermont, F.F.; Letteboer, T.G.; Kim, J.H.; Espejel, S.; Harradine, K.A.; Arbelaez, J.; Luu, M.T.; Roy, R.; Quigley, D.; et al. Mouse and human strategies identify PTPN14 as a modifier of angiogenesis and hereditary haemorrhagic telangiectasia. Nat. Commun. 2012, 3, 616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, K.; Freimuth, J.; Meyer, D.S.; Lee, M.M.; Tochimoto-Okamoto, A.; Benzinou, M.; Clermont, F.F.; Wu, G.; Roy, R.; Letteboer, T.G.; et al. Genetic variants of Adam17 differentially regulate TGFβ signaling to modify vascular pathology in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7723–7728. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Goumans, M.J.; Lebrin, F.; Valdimarsdottir, G. Controlling the angiogenic switch: A balance between two distinct TGF-β receptor signaling pathways. Trends Cardiovasc. Med. 2003, 13, 301–307. [Google Scholar] [CrossRef]
- Gore, B.; Izikki, M.; Mercier, O.; Dewachter, L.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; Lebrin, F.; et al. Key role of the endothelial TGF-β/ALK1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS ONE 2014, 9, e100310. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I. receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Mallet, C.; Feige, J.J.; Bailly, S. Activin receptor-like kinase 1 is implicated in the maturation phase of angiogenesis. Blood 2002, 100, 4495–4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, T.; Fujii, M.; Sugizaki, T.; Ishii, M.; Miyazawa, K.; Aburatani, H.; Miyazono, K. Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-beta in human umbilical vein endothelial cells. J. Cell. Physiol. 2002, 193, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hampson, I.N.; Hampson, L.; Kumar, P.; Bernabeu, C.; Kumar, S. CD105 antagonizes the inhibitory signaling of transforming growth factor beta1 on human vascular endothelial cells. FASEB J. 2000, 14, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tual-Chalot, S.; Mahmoud, M.; Allinson, K.R.; Redgrave, R.E.; Zhai, Z.; Oh, S.P.; Fruttiger, M.; Arthur, H.M. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS ONE 2014, 9, e98646. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Jones, P.L.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef] [PubMed]
- Van der Bruggen, C.E.; Happe, C.M.; Dorfmuller, P.; Trip, P.; Spruijt, O.A.; Rol, N.; Hoevenaars, F.P.; Houweling, A.C.; Girerd, B.; Marcus, J.T.; et al. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial hypertension: A view on the right ventricle. Circulation 2016, 133, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, D.; Ihida-Stansbury, K.; Jones, P.L.; Martin, J.F. Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum. Mol. Genet. 2009, 18, 2791–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fulop, G.T.; Langa, C.; et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrell, N.W.; Bloch, D.B.; ten Dijke, P.; Goumans, M.J.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signalling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, R.E.; Flanagan, J.A.; Sankelo, M.; Abdalla, S.A.; Rowell, J.; Machado, R.D.; Elliott, C.G.; Robbins, I.M.; Olschewski, H.; McLaughlin, V.; et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 2003, 40, 865–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalla, S.A.; Gallione, C.J.; Barst, R.J.; Horn, E.M.; Knowles, J.A.; Marchuk, D.A.; Letarte, M.; Morse, J.H. Primary pulmonary hypertension in families with hereditary haemorrhagic telangiectasia. Eur. Respir. J. 2004, 23, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, R.E.; Berger, R.; Haworth, S.G.; Tulloh, R.; Mache, C.J.; Morrell, N.W.; Aldred, M.A.; Trembath, R.C. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005, 111, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Smoot, L.B.; Obler, D.; McElhinney, D.B.; Boardman, K.; Wu, B.L.; Lip, V.; Mullen, M.P. Clinical features of pulmonary arterial hypertension in young people with an ALK1 mutation and hereditary haemorrhagic telangiectasia. Arch. Dis. Child. 2009, 94, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Girerd, B.; Montani, D.; Coulet, F.; Sztrymf, B.; Yaici, A.; Jais, X.; Tregouet, D.; Reis, A.; Drouin-Garraud, V.; Fraisse, A.; et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am. J. Respir. Crit. Care Med. 2010, 181, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Lyle, M.A.; Fenstad, E.R.; McGoon, M.D.; Frantz, R.P.; Krowka, M.J.; Kane, G.C.; Swanson, K.L. Pulmonary Hypertension in the setting of Hereditary Hemorrhagic Telangiectasia. Chest 2015, 149, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Price, L.C.; Girerd, B.; Chinet, T.; Lacombe, P.; Simonneau, G.; Humbert, M. Fatal rupture of pulmonary arteriovenous malformation in hereditary haemorrhagic telangiectasis and severe PAH. Eur. Respir. Rev. 2009, 18, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chida, A.; Shintani, M.; Yagi, H.; Fujiwara, M.; Kojima, Y.; Sato, H.; Imamura, S.; Yokozawa, M.; Onodera, N.; Horigome, H.; et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am. J. Cardiol. 2012, 110, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Yagi, H.; Matsuoka, R.; Akimoto, K.; Furutani, M.; Imamura, S.; Uehara, R.; Nakayama, T.; Takao, A.; Nakazawa, M.; et al. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ. J. 2008, 72, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaouat, A.; Coulet, F.; Favre, C.; Simonneau, G.; Weitzenblum, E.; Soubrier, F.; Humbert, M. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 2004, 59, 446–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mache, C.J.; Gamillscheg, A.; Popper, H.H.; Haworth, S.G. Early-life pulmonary arterial hypertension with subsequent development of diffuse pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia type 1. Thorax 2008, 63, 85–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.J.; Yang, Q.H.; Liu, D.; Liu, Q.Q.; Eyries, M.; Wen, L.; Wu, W.H.; Jiang, X.; Yuan, P.; Zhang, R.; et al. Clinical and genetic characteristics of Chinese patients with hereditary haemorrhagic telangiectasia-associated pulmonary hypertension. Eur. J. Clin. Investig. 2013, 43, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Girerd, B.; Montani, D.; Jais, X.; Eyries, M.; Yaici, A.; Sztrymf, B.; Savale, L.; Parent, F.; Coulet, F.; Godinas, L.; et al. Genetic counselling in a national referral centre for pulmonary hypertension. Eur. Respir. J. 2016, 47, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Vorselaars, V.; Velthuis, S.; van Gent, M.; Westermann, C.; Snijder, R.; Mager, J.; Post, M. Pulmonary Hypertension in a Large Cohort with Hereditary Hemorrhagic Telangiectasia. Respiration 2017, 94, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Miyake, R.; Fujino, T.; Abe, K.; Hosokawa, K.; Ohtani, K.; Morisaki, H.; Yamada, O.; Higo, T.; Ide, T. Pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia successfully treated with sildenafil. Int. J. Cardiol. 2016, 214, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Revuz, S.; Decullier, E.; Ginon, I.; Lamblin, N.; Hatron, P.Y.; Kaminsky, P.; Carette, M.F.; Lacombe, P.; Simon, A.C.; Riviere, S.; et al. Pulmonary hypertension subtypes associated with hereditary haemorrhagic telangiectasia: Haemodynamic profiles and survival probability. PLoS ONE 2017, 12, e0184227. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xiong, C.M.; Gu, Q.; Wang, X.T.; Cheng, X.L.; Huang, L.; Yang, T.; Luo, Q.; Zhao, Z.H.; Ni, X.H.; et al. The clinical characteristics and long-term prognosis of pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ogo, T.; Tahara, N.; Fukui, S.; Tsuji, A.; Ueda, J.; Fukumoto, Y.; Nakanishi, N.; Ogawa, H.; Yasuda, S. Thalidomide for Hereditary Hemorrhagic Telangiectasia with Pulmonary Arterial Hypertension. Circ. J. 2018, 82, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Gomberg-Maitland, M.; Miller, D.P.; Frost, A.; Frantz, R.P.; Foreman, A.J.; Badesch, D.B.; McGoon, M.D. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012, 141, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Pugh, M.E.; Hemnes, A.R. Pulmonary hypertension in women. Expert Rev. Cardiovasc. Ther. 2010, 8, 1549–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaniv, E.; Preis, M.; Shevro, J.; Nageris, B.; Hadar, T. Anti-estrogen therapy for hereditary hemorrhagic telangiectasia—A long-term clinical trial. Rhinology 2011, 49, 214–216. [Google Scholar] [PubMed]
- Albinana, V.; Bernabeu-Herrero, M.E.; Zarrabeitia, R.; Bernabeu, C.; Botella, L.M. Estrogen therapy for hereditary haemorrhagic telangiectasia (HHT): Effects of raloxifene, on Endoglin and ALK1 expression in endothelial cells. Thromb. Haemost. 2010, 103, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Pfarr, N.; Fischer, C.; Ehlken, N.; Becker-Grunig, T.; Lopez-Gonzalez, V.; Gorenflo, M.; Hager, A.; Hinderhofer, K.; Miera, O.; Nagel, C.; et al. Hemodynamic and genetic analysis in children with idiopathic, heritable, and congenital heart disease associated pulmonary arterial hypertension. Respir. Res. 2013, 14, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pousada, G.; Baloira, A.; Fontan, D.; Nunez, M.; Valverde, D. Mutational and clinical analysis of the ENG gene in patients with pulmonary arterial hypertension. BMC Genet. 2016, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Faughnan, M.E.; Granton, J.T.; Young, L.H. The pulmonary vascular complications of hereditary haemorrhagic telangiectasia. Eur. Respir. J. 2009, 33, 1186–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [PubMed]
- Devlin, H.L.; Hosman, A.E.; Shovlin, C.L. Antiplatelet and anticoagulant agents in hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2013, 368, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Bonderman, D.; Nowotny, R.; Skoro-Sajer, N.; Adlbrecht, C.; Lang, I.M. Bosentan therapy for pulmonary arterial hypertension associated with hereditary haemorrhagic telangiectasia. Eur. J. Clin. Investig. 2006, 36, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.A.; Jang, S.Y.; Ki, C.S.; Kang, I.S.; Kim, D.K. Successful bosentan therapy for pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia. Heart Vessels 2011, 26, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Tighe, H.C.; Davies, R.J.; Gibbs, J.S.; Jackson, J.E. Embolisation of pulmonary arteriovenous malformations: No consistent effect on pulmonary artery pressure. Eur. Respir. J. 2008, 32, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Vorselaars, V.M.; Velthuis, S.; Mager, J.J.; Snijder, R.J.; Bos, W.J.; Vos, J.A.; van Strijen, M.J.; Post, M.C. Direct haemodynamic effects of pulmonary arteriovenous malformation embolisation. Neth. Heart J. 2014, 22, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Plauchu, H.; Bayle, J.Y.; Barthelet, M.; Revel, D.; Cordier, J.F. Pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. Am. J. Respir. Crit. Care Med. 2004, 169, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Remy-Jardin, M.; Dumont, P.; Brillet, P.Y.; Dupuis, P.; Duhamel, A.; Remy, J. Pulmonary arteriovenous malformations treated with embolotherapy: Helical CT evaluation of long-term effectiveness after 2-21-year follow-up. Radiology 2006, 239, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Albinana, V.; Sanz-Rodriguez, F.; Recio-Poveda, L.; Bernabeu, C.; Botella, L.M. Immunosuppressor FK506 increases endoglin and activin receptor-like kinase 1 expression and modulates transforming growth factor-beta1 signaling in endothelial cells. Mol. Pharmacol. 2011, 79, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Chandakkar, P.; Zhao, H.; Papoin, J.; Chatterjee, P.K.; Christen, E.; Metz, C.N.; Blanc, L.; Campagne, F.; Marambaud, P. Tacrolimus rescues the signaling and gene expression signature of endothelial ALK1 loss-of-function and improves HHT vascular pathology. Hum. Mol. Genet. 2017, 26, 4786–4798. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Bill, M.; Aldred, M.A.; van de Veerdonk, M.C.; Vonk Noordegraaf, A.; Long-Boyle, J.; Dash, R.; Yang, P.C.; et al. Low-Dose FK506 (Tacrolimus) in End-Stage Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, V.N.; Apala, D.R.; Pannu, B.S.; Kotecha, A.; Brinjikji, W.; Leise, M.D.; Kamath, P.S.; Misra, S.; Begna, K.H.; Cartin-Ceba, R.; et al. Intravenous Bevacizumab for Refractory Hereditary Hemorrhagic Telangiectasia-Related Epistaxis and Gastrointestinal Bleeding. Mayo Clin. Proc. 2018, 93, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Arizmendez, N.P.; Rudmik, L.; Poetker, D.M. Intravenous bevacizumab for complications of hereditary hemorrhagic telangiectasia: A review of the literature. Int. Forum Allergy Rhinol. 2015, 5, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. American College of Gastroenterology. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–62. [Google Scholar] [CrossRef] [PubMed]
- Hosman, A.; Westermann, C.J.; Snijder, R.; Disch, F.; Mummery, C.L.; Mager, J.J. Follow-up of Thalidomide treatment in patients with Hereditary Haemorrhagic Telangiectasia. Rhinology 2015, 53, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, C.; Lanzarini, L.; Pagella, F.; Semino, L.; Corno, S.; Valacca, C.; Plauchu, H.; Lesca, G.; Barthelet, M.; Buscarini, E.; et al. Echocardiographic screening discloses increased values of pulmonary artery systolic pressure in 9 of 68 unselected patients affected with hereditary hemorrhagic telangiectasia. Genet. Med. 2006, 8, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sopena, B.; Perez-Rodriguez, M.T.; Portela, D.; Rivera, A.; Freire, M.; Martinez-Vazquez, C. High prevalence of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. Eur. J. Intern. Med. 2013, 24, e30–e34. [Google Scholar] [CrossRef] [PubMed]
- Naeije, R.; Vanderpool, R.; Dhakal, B.P.; Saggar, R.; Vachiery, J.L.; Lewis, G.D. Exercise-induced pulmonary hypertension: Physiological basis and methodological concerns. Am. J. Respir. Crit. Care Med. 2013, 187, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Sulaiman, N.L.; Govani, F.S.; Jackson, J.E.; Begbie, M.E. Elevated factor VIII in hereditary haemorrhagic telangiectasia (HHT): Association with venous thromboembolism. Thromb. Haemost. 2007, 98, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chizinga, M.; Rudkovskaia, A.A.; Henderson, K.; Pollak, J.; Garcia-Tsao, G.; Young, L.H.; Fares, W.H. Pulmonary Hypertension Prevalence and Prognosis in a Cohort of Patients with Hereditary Hemorrhagic Telangiectasia Undergoing Embolization of pAVMs. Am. J. Respir. Crit. Care Med. 2017, 196, 1353–1356. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study | Number of Patients | Genetics | Family Members |
|---|---|---|---|
| Trembath et al., 2001 [39] | 8 | ACVRL1 n = 8 | n = 5 (2 families) |
| Harrison et al., 2003 [40] | 14 | ACVRL1 n = 9, ENG n = 2, Unknown n = 3 * | n = 0 |
| Abdalla et al., 2004 [41] | 10 | ACVRL1 n = 10 | n = 0 |
| Chaouat et al., 2004 ¥ [50] | 1 | ENG n = 1 ** | NA |
| Harrison et al., 2005 [42] | 2 | ACVRL1 n = 1, ENG n = 1 | NA |
| Mache et al., 2008 ¥ [51] | 1 | ENG n = 1 | NA |
| Smoot et al., 2009 [43] | 3 | ACVRL1 n = 3 | n = 0 |
| Girerd et al., 2010 [44] | 9 | ACVRL1 n = 9 | n = 4 (1 family) |
| Lyle et al., 2015 [45] | 12 | ACVRL1 n = 4, Unknown n = 8 * | Unknown |
| Montani et al., 2009 ¥ [46] | 1 | ACVRL1 n = 1 | NA |
| Chida et al., 2012 [47] | 7 | ACVRL1 n = 7 | n = 0 |
| Fujiwara et al., 2008 [48] | 5 | ACVRL1 n = 5 | n = 0 |
| Chen et al., 2013 [52] | 12 | ACVRL1 n = 7, ENG n = 2, Unknown n = 3 * | n = 0 |
| Machado et al., 2015 [49] | 1 | ACVRL1 n = 1 | NA |
| Girerd et al., 2016 [53] | 10 | ACVRL1 n = 9, ENG n = 1 *** | n = 2 |
| Vorselaars et al., 2017 [54] | 2 | ACVRL1 n = 2 | n = 0 |
| Miyake et al., 2016 ¥ [55] | 1 | ACVRL1 n = 1 | NA |
| Revuz et al., 2017 [56] | 4 | ACVRL1 n = 4 | Unknown |
| Li et al., 2018 [57] | 9 | Unknown n = 9 * | Unknown |
| Nakamura et al., 2018 ¥ [58] | 1 | ACVRL1 n = 1 | NA |
| Haemodynamics | PAH | High Output PH |
|---|---|---|
| Mpap (mmHg) | ++ | + |
| PAWP (mmHg) | = (≤ 15) | =/+ |
| PVR (Wood units) | ++ (> 3) | = |
| CO (L/min) | − | ++ |
| DPG (mmHg) | − (< 7) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vorselaars, V.M.M.; Hosman, A.E.; Westermann, C.J.J.; Snijder, R.J.; Mager, J.J.; Goumans, M.-J.; Post, M.C. Pulmonary Arterial Hypertension and Hereditary Haemorrhagic Telangiectasia. Int. J. Mol. Sci. 2018, 19, 3203. https://doi.org/10.3390/ijms19103203
Vorselaars VMM, Hosman AE, Westermann CJJ, Snijder RJ, Mager JJ, Goumans M-J, Post MC. Pulmonary Arterial Hypertension and Hereditary Haemorrhagic Telangiectasia. International Journal of Molecular Sciences. 2018; 19(10):3203. https://doi.org/10.3390/ijms19103203
Chicago/Turabian StyleVorselaars, Veronique M. M., Anna E. Hosman, Cornelis J. J. Westermann, Repke J. Snijder, Johannes J. Mager, Marie-Jose Goumans, and Marco C. Post. 2018. "Pulmonary Arterial Hypertension and Hereditary Haemorrhagic Telangiectasia" International Journal of Molecular Sciences 19, no. 10: 3203. https://doi.org/10.3390/ijms19103203