Inhibitory Antibodies against Activin A and TGF-β Reduce Self-Supported, but Not Soluble Factors-Induced Growth of Human Pulmonary Arterial Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension

 ,
,
Abstract
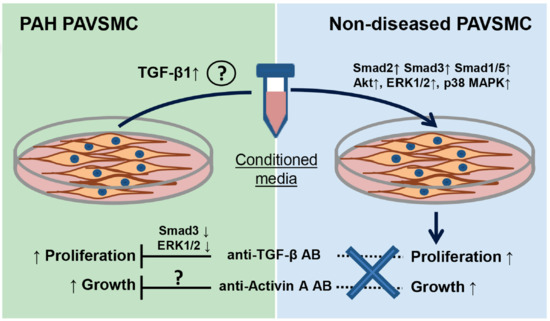
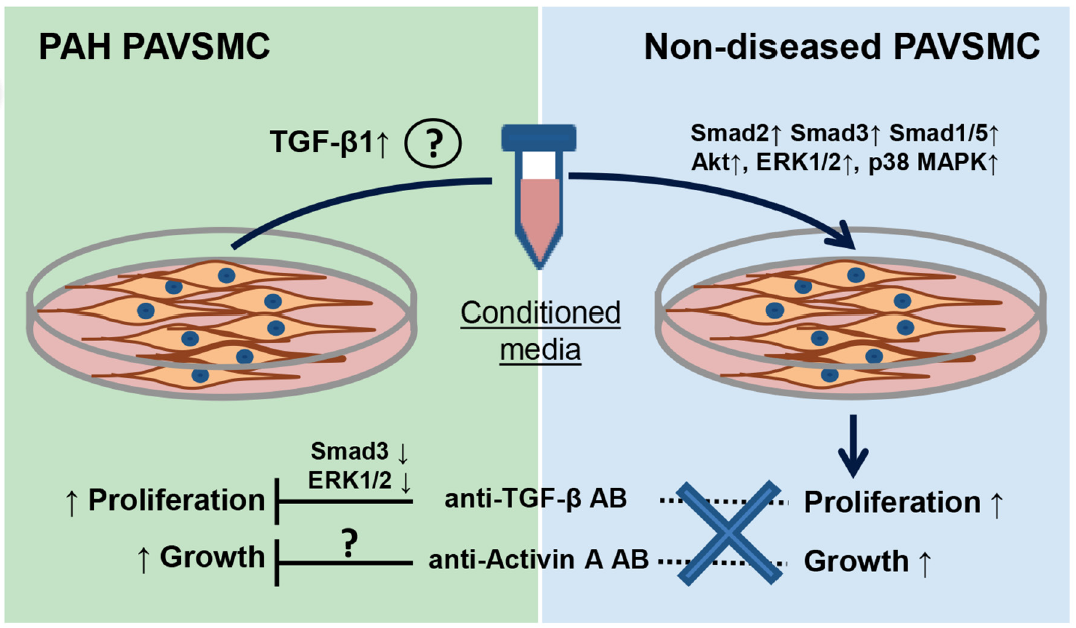
:
1. Introduction
2. Results
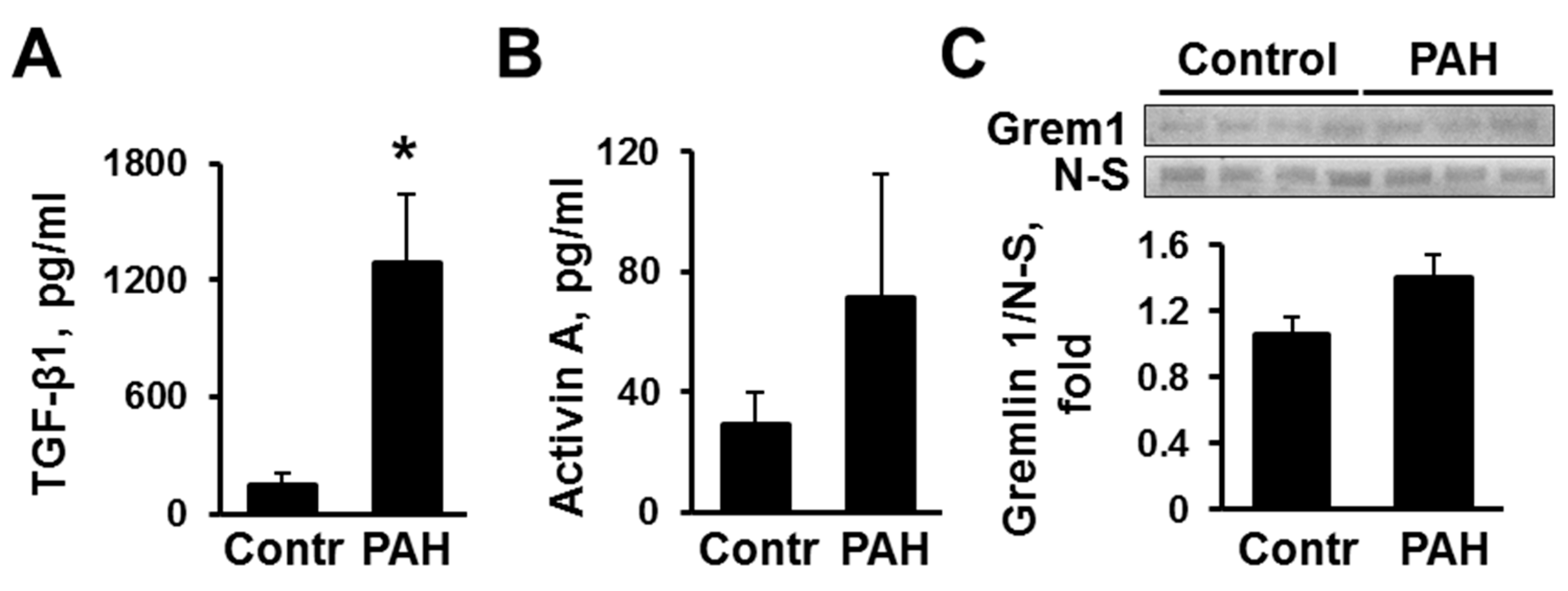
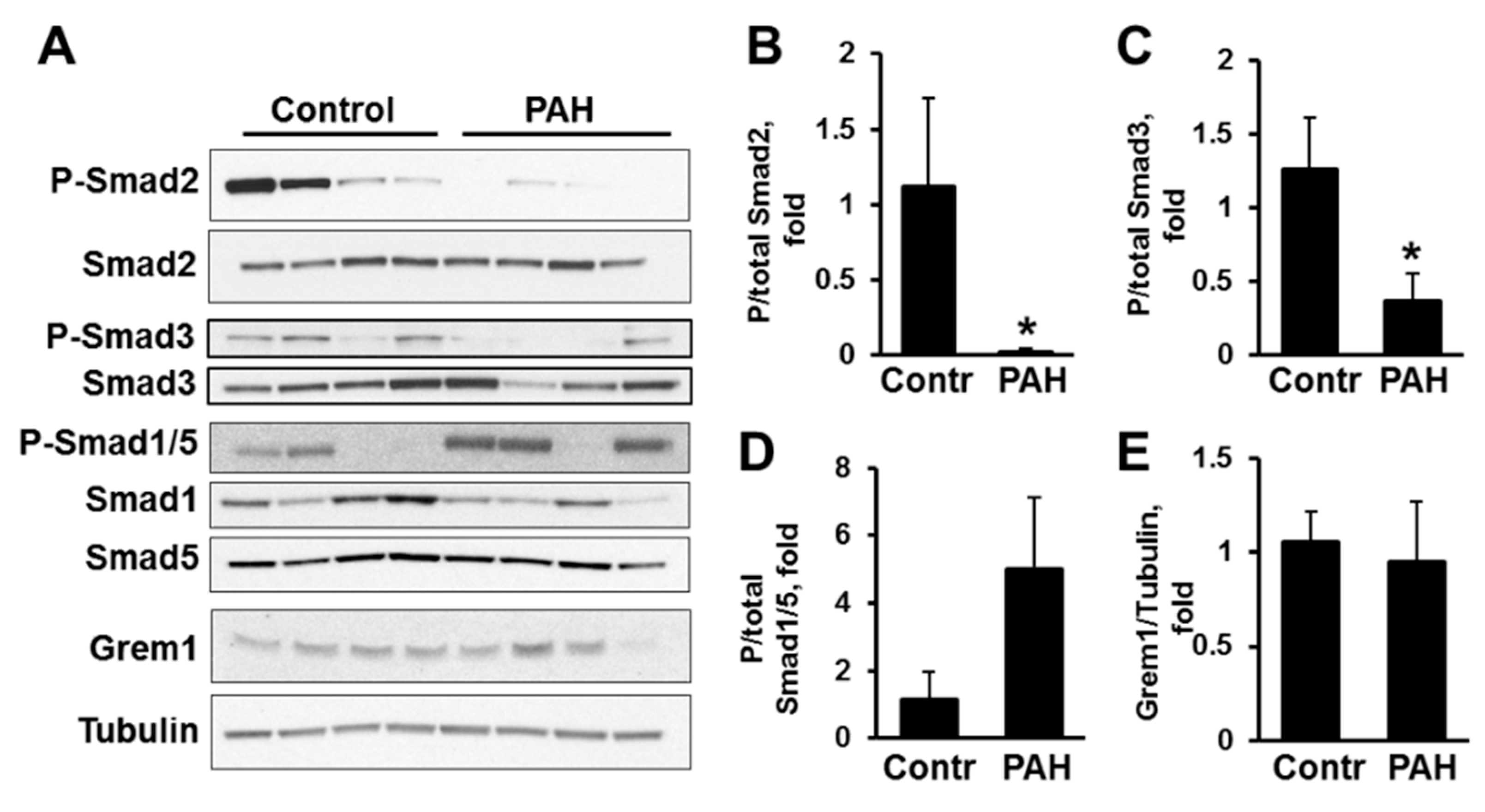
2.1. Human PAH PAVSMC (Pulmonary Arterial Hypertension Pulmonary Arterial Vascular Smooth Muscle Cells) Have Increased Secretion of TGF-β1 and Reduced Smad2 and Smad3 Phosphorylation Compared to Controls
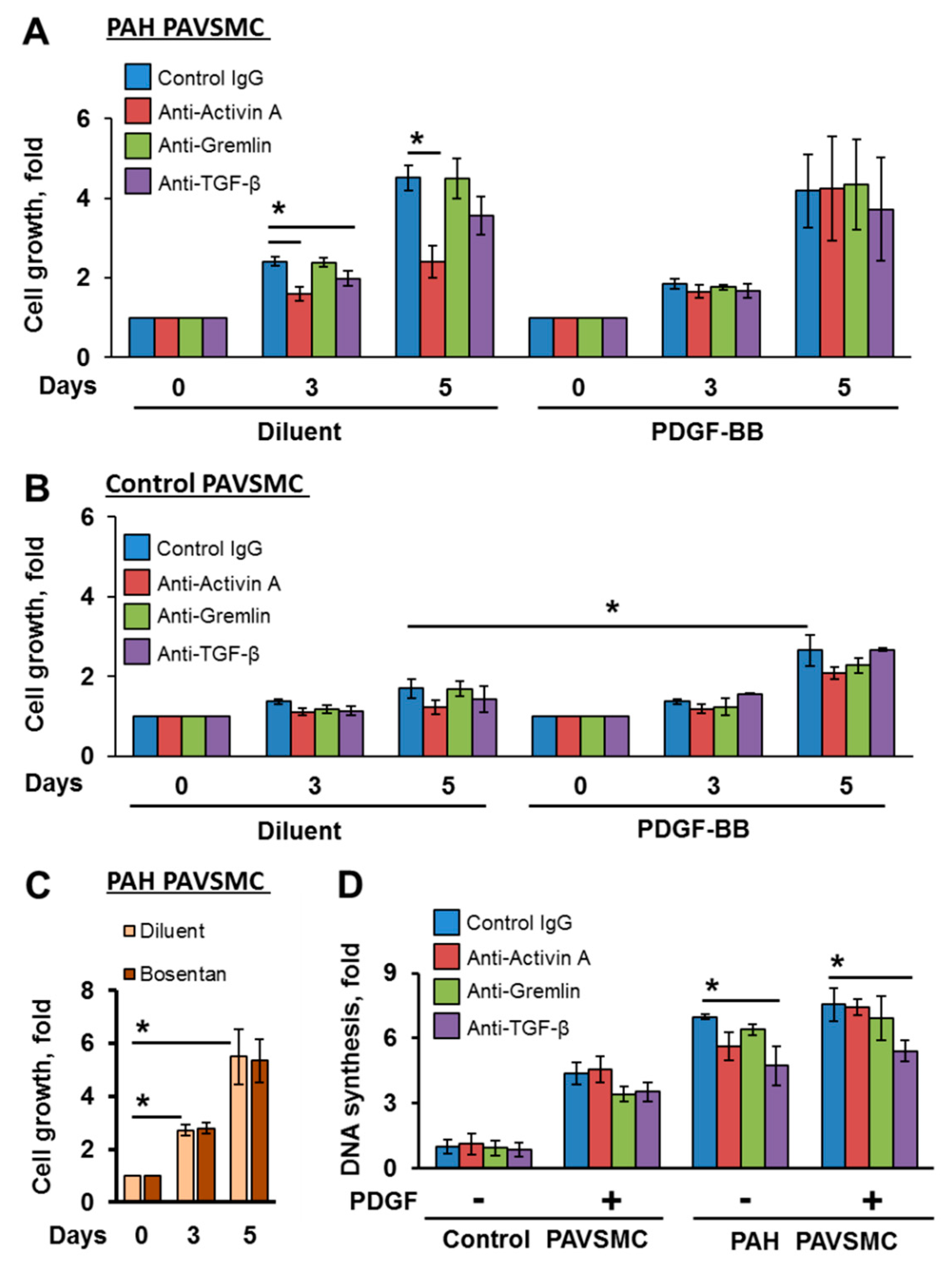
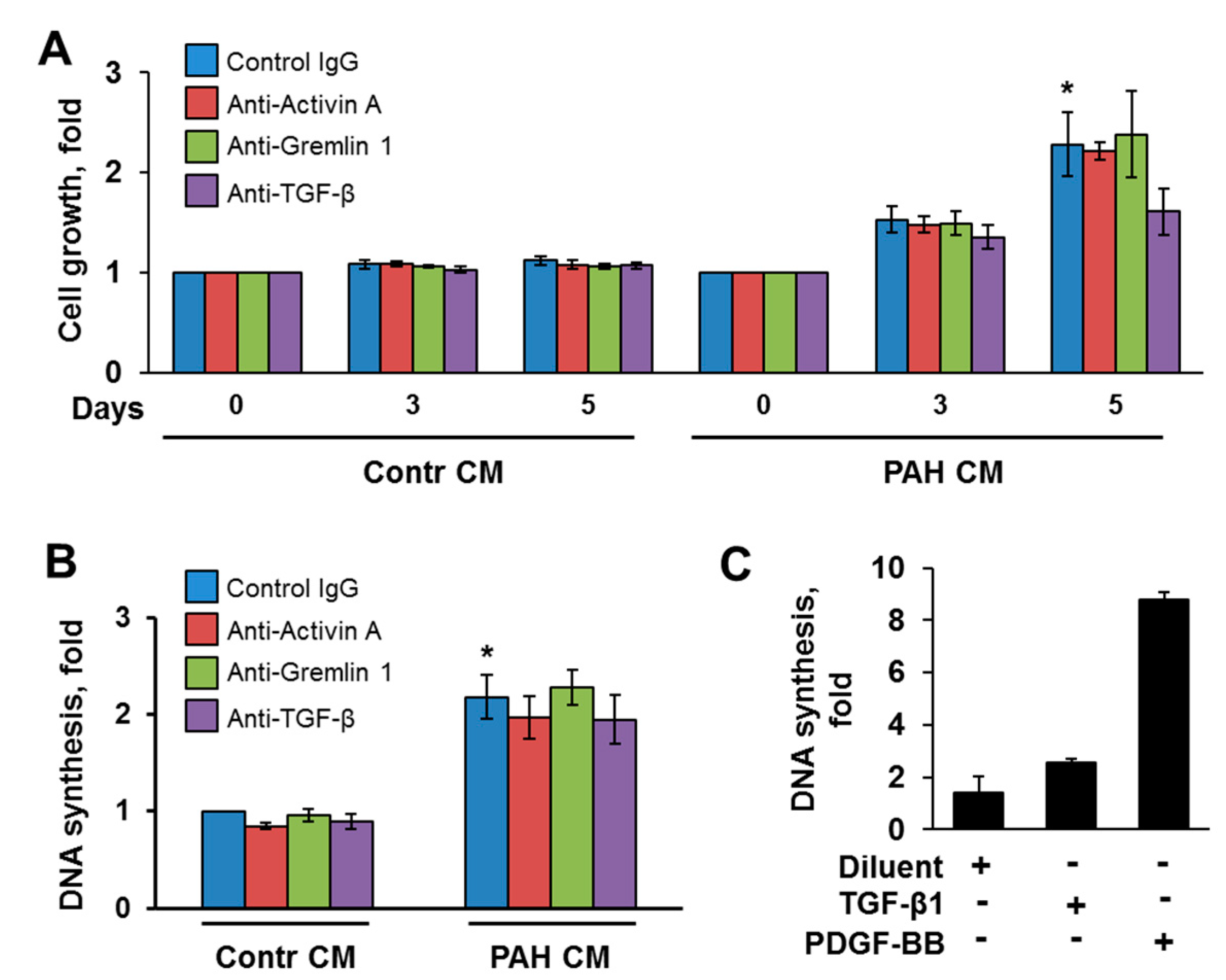
2.2. Inhibitory Antibodies against Activin A and TGF-β, but Not Gremlin 1, Reduce Unstimulated Growth of Human PAH PAVSMC
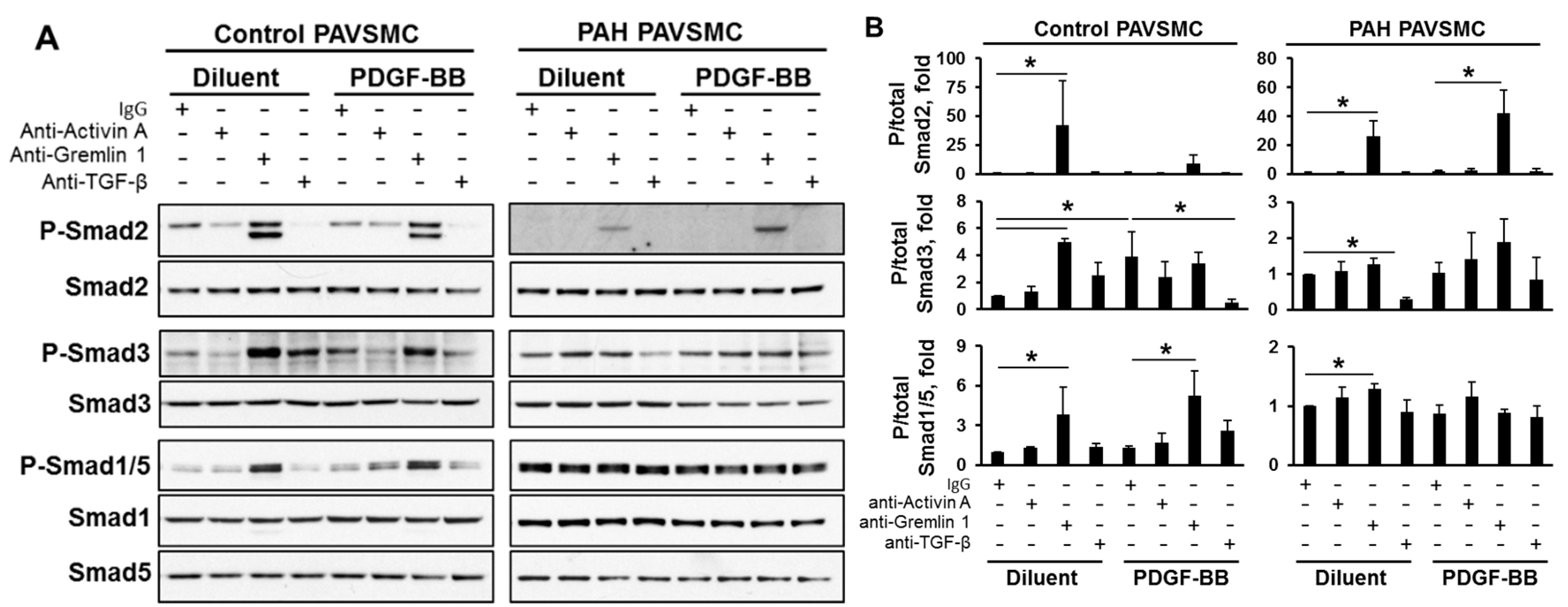
2.3. Effects of Inhibitory Anti-Activin A, Anti-Gremlin 1 and Anti-TGF-β Antibodies on Canonical and Non-Canonical Downstream Targets of TGF-β Network
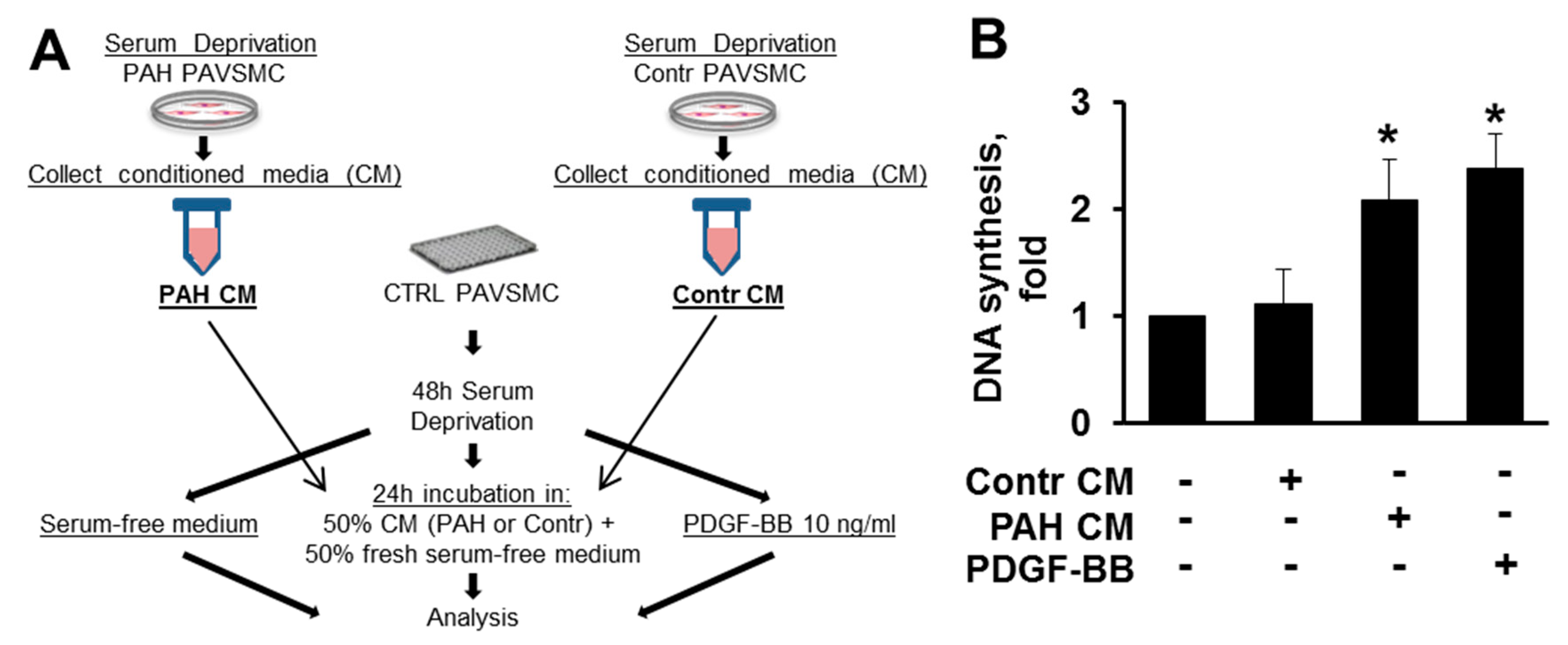
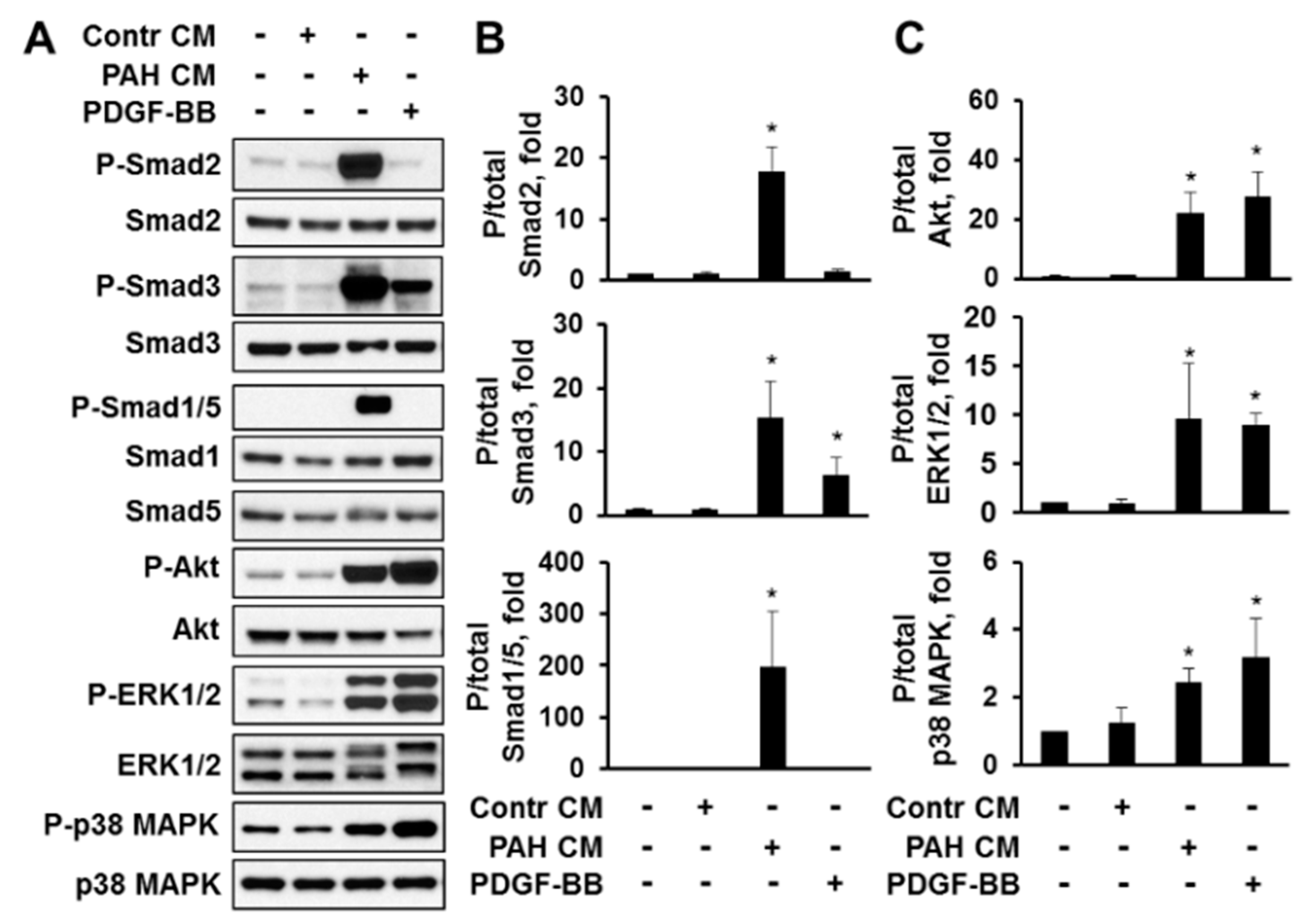
2.4. Factors, Secreted by Human PAH PAVSMC, Promote Proliferation and Up-Regulate Multiple Signaling Pathways in Non-Diseased Human PAVSMC
2.5. Inhibitory Antibodies against Activin A, Gremlin 1 and TGF-β Have No Effect on Proliferation of Non-Diseased PAVSMC Induced by PAH PAVSMC-Secreted Factors
3. Discussion
4. Materials and Methods
4.1. Human Cell Cultures
4.2. Analysis of TGF-β, Activin A and Gremlin 1 Secretion
4.3. Inhibitory Antibodies
4.4. Cell Growth and Proliferation Assays
4.5. Immunoblot Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMP | Bone morphogenetic protein |
| BrdU | Bromodeoxyuridine |
| BSA | Bovine serum albumin |
| CM | Conditioned medium |
| ELISA | Enzyme-linked immunosorbent assay |
| ERK | Extracellular signal-regulated kinases |
| ET-1 | Endothelin-1 |
| LAP | Latency-associated protein |
| MAPK | Mitogen-activated protein kinase |
| PA | Pulmonary artery |
| PAEC | Pulmonary arterial endothelial cells |
| PAH | Pulmonary arterial hypertension |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PAVSMC | Pulmonary artery vascular smooth muscle cell |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet-derived growth factor receptor |
| PI3K | Phosphoinositide 3-kinase |
| R-Smad | Regulated Smad |
| RTK | Receptor tyrosine kinases |
| RV | Right ventricle |
| TGF-β | Transforming growth factor β |
References
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010, 122, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the reveal registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Manes, A.; Negro, L.; Palazzini, M.; Bacchi-Reggiani, M.L.; Branzi, A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur. Heart J. 2009, 30, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Frost, A.E.; Badesch, D.B.; Barst, R.J.; Benza, R.L.; Elliott, C.G.; Farber, H.W.; Krichman, A.; Liou, T.G.; Raskob, G.E.; Wason, P. The changing picture of patients with pulmonary arterial hypertension in the united states: How reveal differs from historic and non-us contemporary registries. Chest 2011, 139, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S13–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullamsetti, S.S.; Savai, R.; Seeger, W.; Goncharova, E.A. Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. Targeting cell growth and proliferation signaling hubs. Am. J. Respir. Crit. Care Med. 2017, 195, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Aschner, Y.; Downey, G.P. Transforming growth factor-β: Master regulator of the respiratory system in health and disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.-M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A selective transforming growth factor-β ligand trap attenuates pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Attisano, L.; Wrana, J.L. Signal transduction by the TGF-β superfamily. Science 2002, 296, 1646–1647. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGF-β signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Upton, P.D.; Morrell, N.W. The transforming growth factor-β–bone morphogenetic protein type signalling pathway in pulmonary vascular homeostasis and disease. Exp. Physiol. 2013, 98, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W. Mutations of the TGF-β type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Fagan, K.; Steudel, W.; Fouty, B.; Lane, K.; Harral, J.; Hoedt-Miller, M.; Tada, Y.; Ozimek, J.; Tuder, R. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ. Res. 2004, 94, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Ciuclan, L.; Sheppard, K.; Dong, L.; Sutton, D.; Duggan, N.; Hussey, M.; Simmons, J.; Morrell, N.W.; Jarai, G.; Edwards, M.; et al. Treatment with anti-gremlin 1 antibody ameliorates chronic hypoxia/SU induced pulmonary arterial hypertension in mice. Am. J. Pathol. 2013, 183, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Xing, D.; Li, P.; Aksut, B.; Ambalavanan, N.; Yang, Q.; Nozell, S.E.; Oparil, S.; Chen, Y.-F. Hypoxia induces downregulation of PPAR-γ in isolated pulmonary arterial smooth muscle cells and in rat lung via transforming growth factor-β signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L899–L907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-F.; Feng, J.-A.; Li, P.; Xing, D.; Zhang, Y.; Serra, R.; Ambalavanan, N.; Majid-Hassan, E.; Oparil, S. Dominant negative mutation of the TGF-β receptor blocks hypoxia-induced pulmonary vascular remodeling. J. Appl. Physiol. 2006, 100, 564–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukerji, S.S.; Katsman, E.A.; Wilber, C.; Haner, N.A.; Selman, W.R.; Hall, A.K. Activin is a neuronal survival factor that is rapidly increased after transient cerebral ischemia and hypoxia in mice. J. Cereb. Blood Flow Metab. 2007, 27, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.B.; Chabon, J.; Gebreab, L.; Poole, J.; Debella, E.; Davis, L.; Tanaka, T.; Sanders, L.; Dropcho, N.; Bandeira, A.; et al. TGF-β signaling promotes pulmonary hypertension caused by schistosoma mansoni. Circulation 2013, 128. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R. Fk506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, G.; de Jesus Perez, V.A.; Alastalo, T.-P.; Alvira, C.M.; Guignabert, C.; Bekker, J.M.; Schellong, S.; Urashima, T.; Wang, L.; Morrell, N.W. An antiproliferative BMP-2/PPARγ/apoE axis in human and murine smcs and its role in pulmonary hypertension. J. Clin. Investig. 2008, 118, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Sysol, J.R.; Natarajan, V.; Machado, R.F. PDGF induces SphK1 expression via Egr-1 to promote pulmonary artery smooth muscle cell proliferation. Am. J. Physiol. Cell Physiol. 2016, 310, C983–992. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, Y.; Su, X.; Zhu, Y.; Liu, L.; Pan, Y.; Zhu, B.; Yang, L.; Gao, L.; Li, M. Activation of ampk inhibits PDGF-induced pulmonary arterial smooth muscle cells proliferation and its potential mechanisms. Pharmacol. Res. 2016, 107, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Kim, K.-H.; Lee, W.-R.; An, H.-J.; Lee, S.-J.; Han, S.-M.; Lee, K.-G.; Park, Y.-Y.; Kim, K.-S.; Lee, Y.-S.; et al. Apamin inhibits PDGF-BB-induced vascular smooth muscle cell proliferation and migration through suppressions of activated Akt and Erk signaling pathway. Vascul. Pharmacol. 2015, 70, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Morrell, N.W.; Hoeper, M.M.; Olschewski, H.; Peacock, A.J.; Barst, R.J.; Shapiro, S.; Golpon, H.; Toshner, M.; Grimminger, F. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am. J. Respir. Crit. Care Med. 2010, 182, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, E.A.; Ammit, A.J.; Irani, C.; Carroll, R.G.; Eszterhas, A.J.; Panettieri, R.A.; Krymskaya, V.P. PI3K is required for proliferation and migration of human pulmonary vascular smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L354–L363. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Montani, D.; Dorfmüller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; Mazmanian, M.; Fadel, E.; Mussot, S.; Mercier, O. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Alvira, C.M.; Alastalo, T.P.; Sawada, H.; Hansmann, G.; Zhao, M.; Wang, L.; El-Bizri, N.; Rabinovitch, M. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes PDGF receptor-β-dependent pulmonary arterial muscularization. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L1082–L1090. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J. PDGF signaling in pulmonary arterial hypertension. J. Clin. Investig. 2005, 115, 2691–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W.; et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Investig. 2005, 115, 2811–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.I.; Wright, J.H.; Johnson, M.M.; Bauer, R.L.; Sorg, K.; Yuen, S.; Hayes, B.J.; Nguyen, L.; Riehle, K.J.; Campbell, J.S. Role of Smad3 in platelet-derived growth factor-C-induced liver fibrosis. Am. J. Physiol. Cell Physiol. 2016, 310, C436–C445. [Google Scholar] [CrossRef] [PubMed]
- Zabini, D.; Granton, E.; Hu, Y.; Miranda, M.Z.; Weichelt, U.; Breuils Bonnet, S.; Bonnet, S.; Morrell, N.W.; Connelly, K.A.; Provencher, S. Loss of Smad3 promotes vascular remodeling in pulmonary arterial hypertension via mrtf disinhibition. Am. J. Respir. Crit. Care Med. 2018, 197, 244–260. [Google Scholar] [CrossRef] [PubMed]
- BouchÉ, M.; Canipari, R.; Melchionna, R.; Willems, D.; Senni, M.I.; Molinaro, M. TGF-β autocrine loop regulates cell growth and myogenic differentiation in human rhabdomyosarcoma cells. FASEB J. 2000, 14, 1147–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Hyder, A.; Hinz, H.; Groth, S.; Lange, H.; El-Sayed, K.M.F.; Ehnert, S.; Nüssler, A.K.; Fändrich, F.; Gieseler, F. Pluripotency gene expression and growth control in cultures of peripheral blood monocytes during their conversion into programmable cells of monocytic origin (PCMO): Evidence for a regulatory role of autocrine activin and TGF-β. PLoS ONE 2015, 10, e0118097. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ding, Z.-Y.; Jin, G.-N.; Xiong, Y.-X.; Yu, B.; Sun, Y.-M.; Wang, W.; Liang, H.-F.; Zhang, B.; Chen, X.-P. Autocrine transforming growth factor-β/activin a-Smad signaling induces hepatic progenitor cells undergoing partial epithelial-mesenchymal transition states. Biochimie 2018, 148, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Cahill, E.; Costello, C.M.; Rowan, S.C.; Harkin, S.; Howell, K.; Leonard, M.O.; Southwood, M.; Cummins, E.P.; Fitzpatrick, S.F.; Taylor, C.T.; et al. Gremlin plays a key role in the pathogenesis of pulmonary hypertension. Circulation 2012, 125, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, T.V.; Goncharov, D.A.; Pena, A.; Kelly, N.; Vanderpool, R.; Baust, J.; Kobir, A.; Shufesky, W.; Mora, A.L.; Morelli, A.E.; et al. HIPPO–integrin-linked kinase cross-talk controls self-sustaining proliferation and survival in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, W.; Hayashi, Y.; Jester, J.V.; Birk, D.E.; Gao, M.; Liu, C.-Y.; Kao, W.W.Y.; Karin, M.; Xia, Y. A role for mek kinase 1 in TGF-β/activin-induced epithelium movement and embryonic eyelid closure. EMBO J. 2003, 22, 4443–4454. [Google Scholar] [CrossRef] [PubMed]
- Pena, A.; Kobir, A.; Goncharov, D.; Goda, A.; Kudryashova, T.V.; Ray, A.; Vanderpool, R.; Baust, J.; Chang, B.; Mora, A.L. Pharmacological inhibition of mtor kinase reverses right ventricle remodeling and improves right ventricle structure and function in rats. Am. J. Respir. Cell Mol. Biol. 2017, 57, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Gore, B.; Izikki, M.; Mercier, O.; Dewachter, L.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; Lebrin, F.; et al. Key role of the endothelial TGF-β/Alk1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS ONE 2014, 9, e100310. [Google Scholar] [CrossRef] [PubMed]
- Battegay, E.J.; Raines, E.W.; Seifert, R.A.; Bowen-Pope, D.F.; Ross, R. TGF-β induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 1990, 63, 515–524. [Google Scholar] [CrossRef]
- Xiao, L.; Du, Y.; Shen, Y.; He, Y.; Zhao, H.; Li, Z. TGF-β 1 induced fibroblast proliferation is mediated by the FGF-2/ERK pathway. Front. Biosci. 2012, 17, 2667–2674. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Li, N.; Teng, W.; Wang, M.; Zhang, Y.; Xiao, Z. TGF-β1 promotes scar fibroblasts proliferation and transdifferentiation via up-regulating microRNA-21. Sci. Rep. 2016, 6, 32231. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Zeisberg, M.; Renziehausen, A.; Raschke, B.; Becker, V.; Van Kooten, C.; Müller, G.A. TGF-β1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney Int. 2001, 59, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Ghouleh, I.A.; Sahoo, S.; Meijles, D.N.; Amaral, J.H.; de Jesus, D.S.; Sembrat, J.; Rojas, M.; Goncharov, D.A.; Goncharova, E.A.; Pagano, P.J. Endothelial NOX1 oxidase assembly in human pulmonary arterial hypertension; driver of gremlin1-mediated proliferation. Clin. Sci. 2017, 131, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Costello, C.M.; Cahill, E.; Martin, F.; Gaine, S.; McLoughlin, P. Role of gremlin in the lung. Am. J. Respir. Cell Mol. Biol. 2010, 42, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.-J.; ten Dijke, P. TGF-β signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewicz, A.; Kouri, F.M.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Sandu, R.; Dony, E.; Seeger, W.; Schermuly, R.T.; Eickelberg, O.; et al. The transforming growth factor-β/Smad2,3 signalling axis is impaired in experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazono, K. Positive and negative regulation of TGF-β signaling. J. Cell Sci. 2000, 113, 1101–1109. [Google Scholar] [PubMed]
- Yamaguchi, H.; Igarashi, M.; Hirata, A.; Susa, S.; Ohnuma, H.; Tominaga, M.; Daimon, M.; Kato, T. Platelet-derived growth factor BB-induced p38 mitogen- activated protein kinase activation causes cell growth, but not apoptosis, in vascular smooth muscle cells. Endocr. J. 2001, 48, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Megalou, A.J.; Glava, C.; Oikonomidis, D.L.; Vilaeti, A.; Agelaki, M.G.; Baltogiannis, G.G.; Papalois, A.; Vlahos, A.P.; Kolettis, T.M. Transforming growth factor-β inhibition attenuates pulmonary arterial hypertension in rats. Int. J. Clin. Exp. Med. 2010, 3, 332–340. [Google Scholar] [PubMed]
- Zaiman, A.L.; Podowski, M.; Medicherla, S.; Gordy, K.; Xu, F.; Zhen, L.; Shimoda, L.A.; Neptune, E.; Higgins, L.; Murphy, A.; et al. Role of the TGF-β/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Li, J.; Zhuang, Q.; Zhang, X.; Yuan, A.; Shen, L.; Kang, K.; Qu, B.; Tang, Y.; Pu, J.; et al. MIR-125a-5p ameliorates monocrotaline-induced pulmonary arterial hypertension by targeting the TGF-β1 and IL-6/STAT3 signaling pathways. Exp. Mol. Med. 2018, 50, 45. [Google Scholar] [CrossRef] [PubMed]
- Yndestad, A.; Larsen, K.-O.; Øie, E.; Ueland, T.; Smith, C.; Halvorsen, B.; Sjaastad, I.; Skjønsberg, O.H.; Pedersen, T.M.; Anfinsen, O.-G.; et al. Elevated levels of activin a in clinical and experimental pulmonary hypertension. J. Appl. Physiol. 2009, 106, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Balsara, R.D.; Ploplis, V.A. Plasminogen activator inhibitor-1: The double edged sword in apoptosis. Thromb. Haemost. 2008, 100, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Clozel, M.; Breu, V.; Gray, G.A.; Kalina, B.; Löffler, B.; Burri, K.; Cassal, J.-M.; Hirth, G.; Müller, M.; Neidhart, W. Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J. Pharmacol. Exp. Ther. 1994, 270, 228–235. [Google Scholar] [PubMed]
- Sitbon, O.; Badesch, D.B.; Channick, R.N.; Frost, A.; Robbins, I.M.; Simonneau, G.; Tapson, V.F.; Rubin, L.J. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension: A 1-year follow-up study. Chest 2003, 124, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Dadrich, M.; Nicolay, N.H.; Flechsig, P.; Bickelhaupt, S.; Hoeltgen, L.; Roeder, F.; Hauser, K.; Tietz, A.; Jenne, J.; Lopez, R.; et al. Combined inhibition of TGF-β and PDGF signaling attenuates radiation-induced pulmonary fibrosis. Oncoimmunology 2016, 5, e1123366. [Google Scholar] [CrossRef] [PubMed]
- Frost, A.E.; Barst, R.J.; Hoeper, M.M.; Chang, H.-J.; Frantz, R.P.; Fukumoto, Y.; Galié, N.; Hassoun, P.M.; Klose, H.; Matsubara, H. Long-term safety and efficacy of imatinib in pulmonary arterial hypertension. J. Heart Lung Transplant. 2015, 34, 1366–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, H.; Kanzawa, T.; Kondo, Y.; Kondo, S. Inhibition of platelet-derived growth factor signalling induces autophagy in malignant glioma cells. Br. J. Cancer 2004, 90, 1069. [Google Scholar] [CrossRef] [PubMed]
- Bedi, A.; Chang, X.; Noonan, K.; Pham, V.; Bedi, R.; Fertig, E.J.; Considine, M.; Califano, J.A.; Borrello, I.; Chung, C.H.; et al. Inhibition of TGF-β enhances the in vivo antitumor efficacy of EGF receptor–targeted therapy. Mol. Cancer Ther. 2012, 11, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Bossé, Y.; Thompson, C.; Stankova, J.; Rola-Pleszczynski, M. Fibroblast growth factor 2 and transforming growth factor β1 synergism in human bronchial smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 2006, 34, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Provencher, S.; Guignabert, C.; Perros, F.; Boucherat, O.; Schermuly, R.T.; Hassoun, P.M.; Rabinovitch, M.; Nicolls, M.R.; Humbert, M. Translating research into improved patient care in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2017, 195, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.; Archer, S.L.; Ramirez, F.D.; Hibbert, B.; Paulin, R.; Boucherat, O.; Lacasse, Y.; Bonnet, S. Standards and methodological rigor in pulmonary arterial hypertension preclinical and translational research. Cric. Res. 2018, 122, 1021–1032. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Gender | Diagnosis |
|---|---|---|
| 40 | F | Non-diseased |
| 64 | F | Non-diseased |
| 38 | F | Non-diseased |
| 33 | F | Non-diseased |
| 37 | M | Non-diseased |
| 40 | F | IPAH |
| 53 | F | IPAH |
| 21 | M | IPAH |
| 45 | M | PAH |
| 50 | F | IPAH |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudryashova, T.V.; Shen, Y.; Pena, A.; Cronin, E.; Okorie, E.; Goncharov, D.A.; Goncharova, E.A. Inhibitory Antibodies against Activin A and TGF-β Reduce Self-Supported, but Not Soluble Factors-Induced Growth of Human Pulmonary Arterial Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2957. https://doi.org/10.3390/ijms19102957
Kudryashova TV, Shen Y, Pena A, Cronin E, Okorie E, Goncharov DA, Goncharova EA. Inhibitory Antibodies against Activin A and TGF-β Reduce Self-Supported, but Not Soluble Factors-Induced Growth of Human Pulmonary Arterial Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2018; 19(10):2957. https://doi.org/10.3390/ijms19102957
Chicago/Turabian StyleKudryashova, Tatiana V., Yuanjun Shen, Andressa Pena, Emily Cronin, Evelyn Okorie, Dmitry A. Goncharov, and Elena A. Goncharova. 2018. "Inhibitory Antibodies against Activin A and TGF-β Reduce Self-Supported, but Not Soluble Factors-Induced Growth of Human Pulmonary Arterial Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 19, no. 10: 2957. https://doi.org/10.3390/ijms19102957