Why Differentiation Therapy Sometimes Fails: Molecular Mechanisms of Resistance to Retinoids

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: Primary and Secondary Drug Resistance
2. Retinoids as Inducers of Cell Differentiation
3. Molecular Mechanisms of Resistance to Retinoids
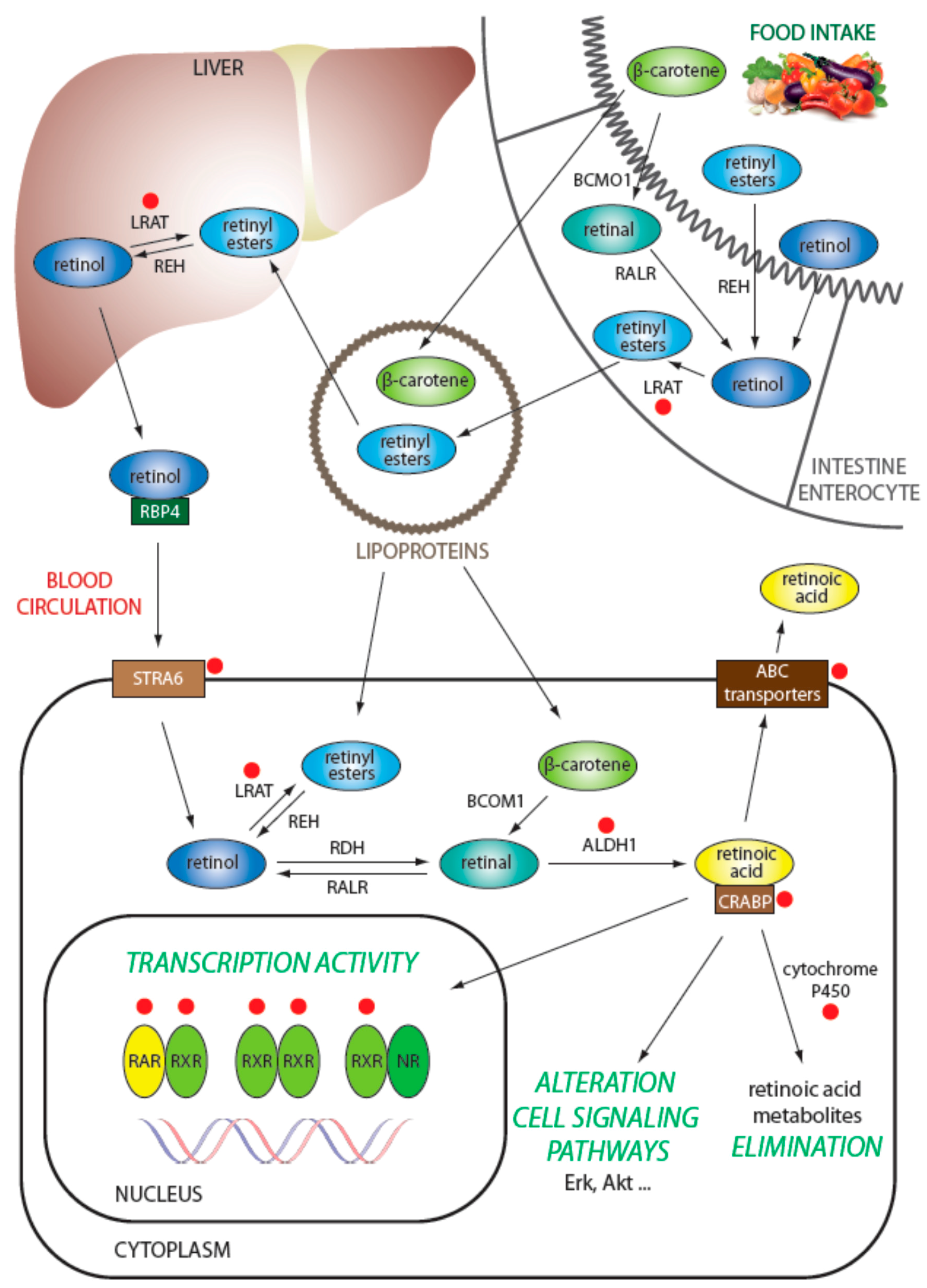
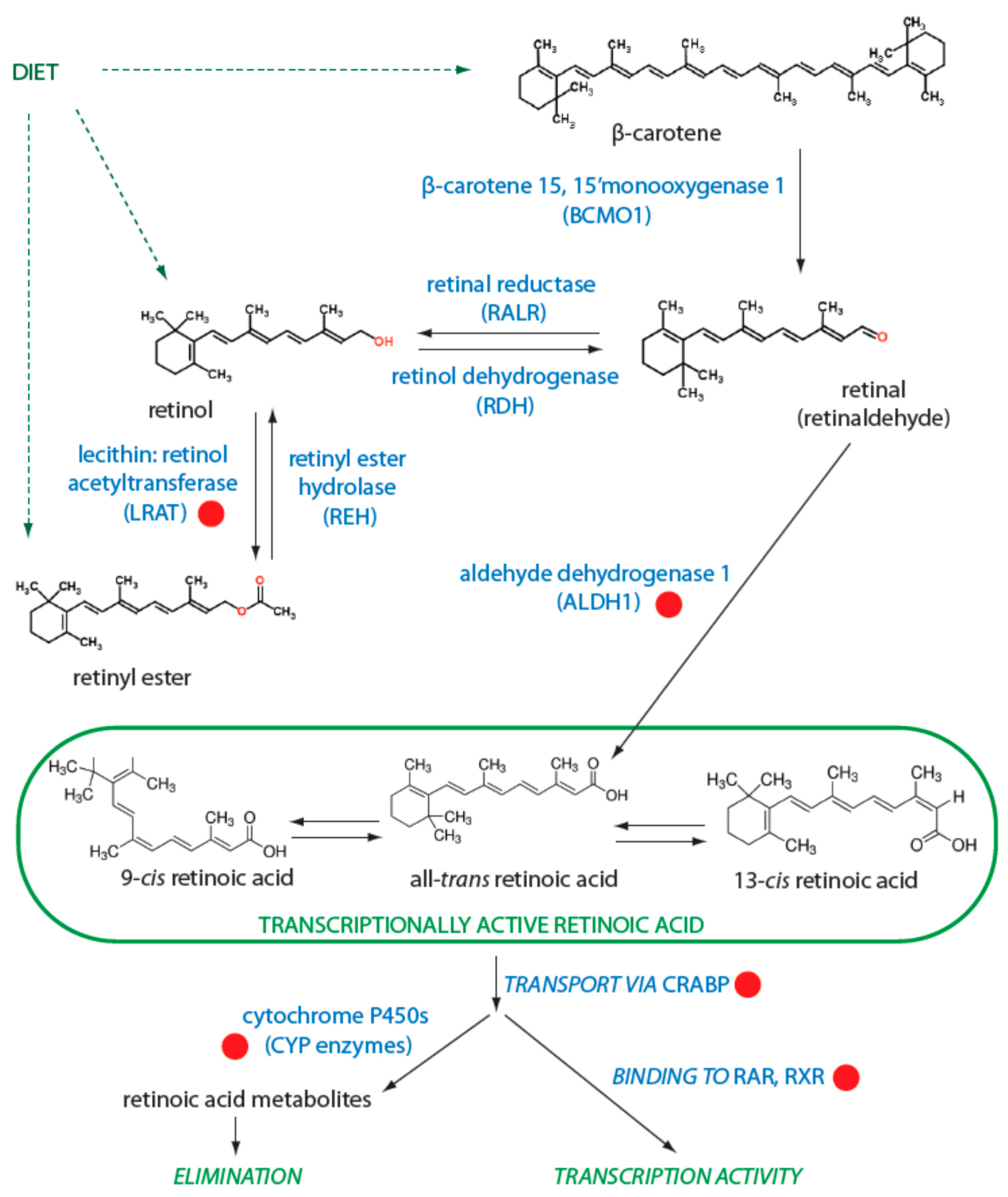
3.1. Regulation of Retinol Metabolism
3.2. Intracellular Transport of Retinoids
3.3. Degradation of Retinoids
3.4. Efflux of Retinoids from the Cell
3.5. Regulation of Transcription via Retinoid Receptors
3.6. Regulation of Transcription via Other Molecules in the Transcriptional Complex
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ALDH | aldehyde dehydrogenase |
| APL | acute promyelocytic leukemia |
| ATRA | all-trans retinoic acid |
| BCRP | breast cancer resistance protein |
| CRABP | cellular retinoic acid binding protein |
| CYP | cytochrome P450 |
| FXR | farnesoid X receptor |
| HAT | histone acetyltransferase |
| HDAC | histone deacetylase |
| LRAT | lecithin-retinol acyltransferase |
| LXR | liver X receptor |
| MDR | multidrug resistance |
| NBL | neuroblastoma |
| Pgp | P-glycoprotein |
| PPAR | peroxisome proliferator-activated receptor |
| RAR | retinoic acid receptor |
| RARE | retinoic acid response element |
| RBP4 | retinol-binding protein |
| REH | retinyl ester hydrolase |
| RXR | retinoid X receptor |
| RXRE | retinoid X response element |
| STRA | receptors stimulated by retinoic acid |
| THR | thyroid hormone receptor |
| XAB2 | xeroderma pigmentosum group A-binding protein 2 |
| 9cisRA | 9-cis retinoic acid |
| 13cisRA | 13-cis retinoic acid |
References
- Nowak, D.; Stewart, D.; Koeffler, H.P. Differentiation therapy of leukemia: 3 decades of development. Blood 2009, 113, 3655–3665. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Biagi, C.; Zama, D.; Vendemini, F.; Martoni, A.; Morello, W.; Gasperini, P.; Pession, A. Retinoids in pediatric onco-hematology: The model of acute promyelocytic leukemia and neuroblastoma. Adv. Ther. 2012, 29, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Haar, C.P.; Hebbar, P.; Wallace, G.C.; Das, A.; Vandergrift, W.A.; Smith, J.A.; Giglio, P.; Patel, S.J.; Ray, S.K.; Banik, N.L. Drug Resistance in Glioblastoma: A Mini Review. Neurochem. Res. 2012, 37, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Douer, D.; Estey, E.; Santillana, S.; Bennett, J.M.; Lopez-Bernstein, G.; Boehm, K.; Williams, T. Treatment of newly diagnosed and relapsed acute promyelocytic leukemia with intravenous liposomal all-trans retinoic acid. Blood 2001, 97, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Freemantle, S.J.; Spinella, M.J.; Dmitrovsky, E. Retinoids in cancer therapy and chemoprevention: Promise meets resistance. Oncogene 2003, 22, 7305–7315. [Google Scholar] [CrossRef] [PubMed]
- Al-Dimassi, S.; Abou-Antoun, T.; El-Sibai, M. Cancer cell resistance mechanisms: A mini review. Clin. Transl. Oncol. 2014, 16, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Felsted, R.L.; Gupta, S.K.; Glover, C.J.; Fischkoff, S.A.; Gallagher, R.E. Cell surface membrane protein changes during the differentiation of cultured human promyelocytic leukemia HL-60 cells. Cancer Res. 1983, 43, 2754–2761. [Google Scholar] [PubMed]
- Sidell, N.; Altman, A.; Haussler, M.R.; Seeger, R.C. Effects of retinoic acid (RA) on the growth and phenotypic expression of several human neuroblastoma cell lines. Exp. Cell Res. 1983, 148, 21–30. [Google Scholar] [CrossRef]
- Chou, J.Y. Effects of retinoic acid on differentiation of choriocarcinoma cells in vitro. J. Clin. Endocrinol. Metab. 1982, 54, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, E.; Lehto, V.P.; Badley, R.A.; Virtanen, I. Formation of vinculin plaques precedes other cytoskeletal changes during retinoic acid-induced teratocarcinoma cell differentiation. Exp. Cell Res. 1983, 144, 191–197. [Google Scholar] [CrossRef]
- Veselska, R.; Zitterbart, K.; Auer, J.; Neradil, J. Differentiation of HL-60 myeloid leukemia cells induced by all-trans retinoic acid is enhanced in combination with caffeic acid. Int. J. Mol. Med. 2004, 14, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Redova, M.; Chlapek, P.; Loja, T.; Zitterbart, K.; Hermanova, M.; Sterba, J.; Veselska, R. Influence of LOX/COX inhibitors on cell differentiation induced by all-trans retinoic acid in neuroblastoma cell lines. Int. J. Mol. Med. 2010, 25, 271–280. [Google Scholar] [PubMed]
- Chlapek, P.; Redova, M.; Zitterbart, K.; Hermanova, M.; Sterba, J.; Veselska, R. Enhancement of ATRA-induced differentiation of neuroblastoma cells with LOX/COX inhibitors: An expression profiling study. J. Exp. Clin. Cancer Res. 2010, 29, 45. [Google Scholar] [CrossRef] [PubMed]
- Chlapek, P.; Neradil, J.; Redova, M.; Zitterbart, K.; Sterba, J.; Veselska, R. The ATRA-induced differentiation of medulloblastoma cells is enhanced with LOX/COX inhibitors: An analysis of gene expression. Cancer Cell Int. 2014, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, S.M.; Blaner, W.S. Retinol and retinyl esters: Biochemistry and physiology. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; Li, Y.; Brun, P.-J.; Yuen, J.J.; Lee, S.-A.; Clugston, R.D. Vitamin A Absorption, Storage and Mobilization. Subcell. Biochem. 2016, 81, 95–125. [Google Scholar] [CrossRef] [PubMed]
- Moise, A.R.; Noy, N.; Palczewski, K.; Blaner, W.S. Delivery of retinoid-based therapies to target tissues. Biochemistry 2007, 46, 4449–4458. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Wojtowicz, K.; Zabel, M. The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance. Biomed. Pharmacother. 2013, 67, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Lanvers, C.; Hempel, G.; Blaschke, G.; Boos, J. Chemically induced isomerization and differential uptake modulate retinoic acid disposition in HL-60 cells. FASEB J. 1998, 12, 1627–1633. [Google Scholar] [PubMed]
- Armstrong, J.L.; Redfern, C.P.F.; Veal, G.J. 13-cis retinoic acid and isomerisation in paediatric oncology—Is changing shape the key to success? Biochem. Pharmacol. 2005, 69, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol. Ther. 2017, 173, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Uray, I.P.; Dmitrovsky, E.; Brown, P.H. Retinoids and Rexinoids in Cancer Prevention: From Laboratory to Clinic. Semin. Oncol. 2016, 43, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Iskakova, M.; Karbyshev, M.; Piskunov, A.; Rochette-Egly, C. Nuclear and extranuclear effects of vitamin A. Can. J. Physiol. Pharmacol. 2015, 93, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Niles, R.M. Recent advances in the use of vitamin A (retinoids) in the prevention and treatment of cancer. Nutrition 2000, 16, 1084–1089. [Google Scholar] [CrossRef]
- Huang, S.; Laoukili, J.; Epping, M.T.; Koster, J.; Hölzel, M.; Westerman, B.A.; Nijkamp, W.; Hata, A.; Asgharzadeh, S.; Seeger, R.C.; et al. ZNF423 is critically required for retinoic acid-induced differentiation and is a marker of neuroblastoma outcome. Cancer Cell 2009, 15, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Napoli, J.L. All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. FASEB J. 2008, 22, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Onisko, B.; Napoli, J.L. The Nuclear Transcription Factor RARα Associates with Neuronal RNA Granules and Suppresses Translation. J. Biol. Chem. 2008, 283, 20841–20847. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Kao, Y.-L.; Joshi, S.; Jeetendran, S.; Dipette, D.; Singh, U.S. Activation of Rac1 by phosphatidylinositol 3-kinase in vivo: Role in activation of mitogen-activated protein kinase (MAPK) pathways and retinoic acid-induced neuronal differentiation of SH-SY5Y cells. J. Neurochem. 2005, 93, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Masia, S.; Alvarez, S.; de Lera, A.R.; Barettino, D. Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol. Endocrinol. 2007, 21, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-H.; Gudas, L.J. Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol. 2011, 6, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Amann, P.M.; Czaja, K.; Bazhin, A.V.; Rühl, R.; Skazik, C.; Heise, R.; Marquardt, Y.; Eichmüller, S.B.; Merk, H.F.; Baron, J.M. Knockdown of lecithin retinol acyltransferase increases all-trans retinoic acid levels and restores retinoid sensitivity in malignant melanoma cells. Exp. Dermatol. 2014, 23, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Amann, P.M.; Czaja, K.; Bazhin, A.V.; Rühl, R.; Eichmüller, S.B.; Merk, H.F.; Baron, J.M. LRAT overexpression diminishes intracellular levels of biologically active retinoids and reduces retinoid antitumor efficacy in the murine melanoma B16F10 cell line. Skin Pharmacol. Physiol. 2015, 28, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Kurlandsky, S.B.; Duell, E.A.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Auto-regulation of retinoic acid biosynthesis through regulation of retinol esterification in human keratinocytes. J. Biol. Chem. 1996, 271, 15346–15352. [Google Scholar] [CrossRef] [PubMed]
- Hartomo, T.B.; Van Huyen Pham, T.; Yamamoto, N.; Hirase, S.; Hasegawa, D.; Kosaka, Y.; Matsuo, M.; Hayakawa, A.; Takeshima, Y.; Iijima, K.; et al. Involvement of aldehyde dehydrogenase 1A2 in the regulation of cancer stem cell properties in neuroblastoma. Int. J. Oncol. 2015, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Moreb, J.S.; Mohuczy, D.; Muhoczy, D.; Ostmark, B.; Zucali, J.R. RNAi-mediated knockdown of aldehyde dehydrogenase class-1A1 and class-3A1 is specific and reveals that each contributes equally to the resistance against 4-hydroperoxycyclophosphamide. Cancer Chemother. Pharmacol. 2007, 59, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Moreb, J.S.; Ucar-Bilyeu, D.A.; Khan, A. Use of retinoic acid/aldehyde dehydrogenase pathway as potential targeted therapy against cancer stem cells. Cancer Chemother. Pharmacol. 2017, 79, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-S.; Wang, Q.; Ma, J.-X.; Yang, X.-H.; Wu, M.-L.; Zhang, K.-L.; Kong, Q.-Y.; Chen, X.-Y.; Sun, Y.; Chen, N.-N.; et al. CRABP-II methylation: A critical determinant of retinoic acid resistance of medulloblastoma cells. Mol. Oncol. 2012, 6, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Cornic, M.; Delva, L.; Castaigne, S.; Lefebvre, P.; Balitrand, N.; Degos, L.; Chomienne, C. In Vitro all-trans retinoic acid (ATRA) sensitivity and cellular retinoic acid binding protein (CRABP) levels in relapse leukemic cells after remission induction by ATRA in acute promyelocytic leukemia. Leukemia 1994, 8 (Suppl. 2), S16–S19. [Google Scholar] [PubMed]
- Delva, L.; Cornic, M.; Balitrand, N.; Guidez, F.; Micléa, J.M.; Delmer, A.; Teillet, F.; Fenaux, P.; Castaigne, S.; Degos, L. Resistance to all-trans retinoic acid (ATRA) therapy in relapsing acute promyelocytic leukemia: Study of in vitro ATRA sensitivity and cellular retinoic acid binding protein levels in leukemic cells. Blood 1993, 82, 2175–2181. [Google Scholar] [PubMed]
- Zhou, D.C.; Hallam, S.J.; Lee, S.J.; Klein, R.S.; Wiernik, P.H.; Tallman, M.S.; Gallagher, R.E. Constitutive expression of cellular retinoic acid binding protein II and lack of correlation with sensitivity to all-trans retinoic acid in acute promyelocytic leukemia cells. Cancer Res. 1998, 58, 5770–5776. [Google Scholar] [PubMed]
- Van der Leede, B.M.; van den Brink, C.E.; Pijnappel, W.W.; Sonneveld, E.; van der Saag, P.T.; van der Burg, B. Autoinduction of retinoic acid metabolism to polar derivatives with decreased biological activity in retinoic acid-sensitive, but not in retinoic acid-resistant human breast cancer cells. J. Biol. Chem. 1997, 272, 17921–17928. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.A.; Sidell, N.; Haussler, M.R. Saturation analysis of cellular retinoid binding proteins: Application to retinoic acid resistant human neuroblastoma cells and to human tumors. Biochem. Cell Biol. 1987, 65, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.H.; Buttrick, B.R.; Isoherranen, N. Therapeutic potential of the inhibition of the retinoic acid hydroxylases CYP26A1 and CYP26B1 by xenobiotics. Curr. Top. Med. Chem. 2013, 13, 1402–1428. [Google Scholar] [CrossRef] [PubMed]
- Muindi, J.; Frankel, S.R.; Miller, W.H.; Jakubowski, A.; Scheinberg, D.A.; Young, C.W.; Dmitrovsky, E.; Warrell, R.P. Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: Implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia. Blood 1992, 79, 299–303. [Google Scholar] [PubMed]
- Muindi, J.R.; Frankel, S.R.; Huselton, C.; DeGrazia, F.; Garland, W.A.; Young, C.W.; Warrell, R.P. Clinical pharmacology of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res. 1992, 52, 2138–2142. [Google Scholar] [PubMed]
- Diaz, P.; Huang, W.; Keyari, C.M.; Buttrick, B.; Price, L.; Guilloteau, N.; Tripathy, S.; Sperandio, V.G.; Fronczek, F.R.; Astruc-Diaz, F.; et al. Development and characterization of novel and selective inhibitors of cytochrome P450 CYP26A1, the human liver retinoic acid hydroxylase. J. Med. Chem. 2016, 59, 2579–2595. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-P.; Hsieh, C.-H.; Wu, Y.-S. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol. Pharm. 2011, 8, 1996–2011. [Google Scholar] [CrossRef] [PubMed]
- Peaston, A.E.; Gardaneh, M.; Franco, A.V.; Hocker, J.E.; Murphy, K.M.; Farnsworth, M.L.; Catchpoole, D.R.; Haber, M.; Norris, M.D.; Lock, R.B.; et al. MRP1 gene expression level regulates the death and differentiation response of neuroblastoma cells. Br. J. Cancer 2001, 85, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Cho, W.C.; Locatelli, F.; Fruci, D. Multidrug resistance and cancer stem cells in neuroblastoma and hepatoblastoma. Int. J. Mol. Sci. 2013, 14, 24706–24725. [Google Scholar] [CrossRef] [PubMed]
- Stromskaya, T.P.; Rybalkina, E.Y.; Zabotina, T.N.; Shishkin, A.A.; Stavrovskaya, A.A. Influence of RARalpha gene on MDR1 expression and P-glycoprotein function in human leukemic cells. Cancer Cell Int. 2005, 5, 15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tarapcsák, S.; Szalóki, G.; Telbisz, Á.; Gyöngy, Z.; Matúz, K.; Csősz, É.; Nagy, P.; Holb, I.J.; Rühl, R.; Nagy, L.; et al. Interactions of retinoids with the ABC transporters P-glycoprotein and Breast Cancer Resistance Protein. Sci. Rep. 2017, 7, 41376. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Nguyen, N.K.; Sukumar, S. Molecular Pathways: Current Role and Future Directions of the Retinoic Acid Pathway In Cancer Prevention and Treatment. Clin. Cancer Res. 2013, 19, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Duong, V.; Rochette-Egly, C. The molecular physiology of nuclear retinoic acid receptors. From health to disease. Biochim. Biophys. Acta 2011, 1812, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Moghal, N.; Neel, B.G. Evidence for impaired retinoic acid receptor-thyroid hormone receptor AF-2 cofactor activity in human lung cancer. Mol. Cell. Biol. 1995, 15, 3945–3959. [Google Scholar] [CrossRef] [PubMed]
- Altucci, L.; Gronemeyer, H. The promise of retinoids to fight against cancer. Nat. Rev. Cancer 2001, 1, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Pozzi, S.; Bistulfi, G.; Somenzi, G.; Rossetti, S.; Sacchi, N. Impaired Retinoic Acid (RA) Signal Leads to RARβ2 Epigenetic Silencing and RA Resistance. Mol. Cell. Biol. 2005, 25, 10591–10603. [Google Scholar] [CrossRef] [PubMed]
- Cras, A.; Darsin-Bettinger, D.; Balitrand, N.; Cassinat, B.; Soulié, A.; Toubert, M.-E.; Delva, L.; Chomienne, C. Epigenetic patterns of the retinoic acid receptor beta2 promoter in retinoic acid-resistant thyroid cancer cells. Oncogene 2007, 26, 4018–4024. [Google Scholar] [CrossRef] [PubMed]
- Cheung, B.; Hocker, J.E.; Smith, S.A.; Reichert, U.; Norris, M.D.; Haber, M.; Stewart, B.W.; Marshall, G.M. Retinoic acid receptors beta and gamma distinguish retinoid signals for growth inhibition and neuritogenesis in human neuroblastoma cells. Biochem. Biophys. Res. Commun. 1996, 229, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.-D.; Wu, H.; Zhang, H.-P.; Lu, N.; Ye, P.; Yu, F.-H.; Zhou, H.; Li, W.-G.; Cao, X.; Lin, Y.-Y.; et al. Oncogenic potential of retinoic acid receptor-gamma in hepatocellular carcinoma. Cancer Res. 2010, 70, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Z.P.; Zhang, D.; Soprano, D.R.; Soprano, K.J. Reduction of both RAR and RXR levels is required to maximally alter sensitivity of CA-OV3 ovarian tumor cells to growth suppression by all-trans-retinoic acid. Exp. Cell Res. 1997, 237, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Tari, A.M.; Lim, S.-J.; Hung, M.-C.; Esteva, F.J.; Lopez-Berestein, G. Her2/neu induces all-trans retinoic acid (ATRA) resistance in breast cancer cells. Oncogene 2002, 21, 5224–5232. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Li, Y.P.; Nobile, L.M.; Grills, G.; Carrera, I.; Paietta, E.; Tallman, M.S.; Wiernik, P.H.; Gallagher, R.E. Leukemic cellular retinoic acid resistance and missense mutations in the PML-RARalpha fusion gene after relapse of acute promyelocytic leukemia from treatment with all-trans retinoic acid and intensive chemotherapy. Blood 1998, 92, 1172–1183. [Google Scholar] [PubMed]
- Imaizumi, M.; Suzuki, H.; Yoshinari, M.; Sato, A.; Saito, T.; Sugawara, A.; Tsuchiya, S.; Hatae, Y.; Fujimoto, T.; Kakizuka, A.; et al. Mutations in the E-domain of RAR portion of the PML/RAR chimeric gene may confer clinical resistance to all-trans retinoic acid in acute promyelocytic leukemia. Blood 1998, 92, 374–382. [Google Scholar] [PubMed]
- Tomita, A.; Kiyoi, H.; Naoe, T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) in acute promyelocytic leukemia. Int. J. Hematol. 2013, 97, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Côté, S.; Zhou, D.; Bianchini, A.; Nervi, C.; Gallagher, R.E.; Miller, W.H. Altered ligand binding and transcriptional regulation by mutations in the PML/RARalpha ligand-binding domain arising in retinoic acid-resistant patients with acute promyelocytic leukemia. Blood 2000, 96, 3200–3208. [Google Scholar] [PubMed]
- Rochette-Egly, C. Dynamic combinatorial networks in nuclear receptor-mediated transcription. J. Biol. Chem. 2005, 280, 32565–32568. [Google Scholar] [CrossRef] [PubMed]
- Kruyt, F.A.; van der Veer, L.J.; Mader, S.; van den Brink, C.E.; Feijen, A.; Jonk, L.J.; Kruijer, W.; van der Saag, P.T. Retinoic acid resistance of the variant embryonal carcinoma cell line RAC65 is caused by expression of a truncated RAR alpha. Differ. Res. Biol. Divers. 1992, 49, 27–37. [Google Scholar] [CrossRef]
- Crowe, D.L.; Osaseri, U.E.; Shuler, C.F. Tumor suppressor function of a dominant negative retinoic acid receptor mutant. Mol. Carcinog. 1998, 22, 26–33. [Google Scholar] [CrossRef]
- Somenzi, G.; Sala, G.; Rossetti, S.; Ren, M.; Ghidoni, R.; Sacchi, N. Disruption of retinoic acid receptor alpha reveals the growth promoter face of retinoic acid. PLoS ONE 2007, 2, e836. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma-Ishikawa, K.; Morio, T.; Yamada, T.; Sugawara, Y.; Ono, M.; Nagasawa, M.; Yasuda, A.; Morimoto, C.; Ohnuma, K.; Dang, N.H.; et al. Knockdown of XAB2 enhances all-trans retinoic acid-induced cellular differentiation in all-trans retinoic acid-sensitive and -resistant cancer cells. Cancer Res. 2007, 67, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Cheung, B.B.; Tan, O.; Koach, J.; Liu, B.; Shum, M.S.Y.; Carter, D.R.; Sutton, S.; Po’uha, S.T.; Chesler, L.; Haber, M.; et al. Thymosin-β4 is a determinant of drug sensitivity for Fenretinide and Vorinostat combination therapy in neuroblastoma. Mol. Oncol. 2015, 9, 1484–1500. [Google Scholar] [CrossRef] [PubMed]
- Schmoch, T.; Gal, Z.; Mock, A.; Mossemann, J.; Lahrmann, B.; Grabe, N.; Schmezer, P.; Lasitschka, F.; Beckhove, P.; Unterberg, A.; et al. Combined Treatment of ATRA with Epigenetic Drugs Increases Aggressiveness of Glioma Xenografts. Anticancer Res. 2016, 36, 1489–1496. [Google Scholar] [PubMed]
- Jensen, H.A.; Styskal, L.E.; Tasseff, R.; Bunaciu, R.P.; Congleton, J.; Varner, J.D.; Yen, A. The Src-family kinase inhibitor PP2 rescues inducible differentiation events in emergent retinoic acid-resistant myeloblastic leukemia cells. PLoS ONE 2013, 8, e58621. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, N.; Tsurumi, H.; Okuno, M.; Matsushima-Nishiwaki, R.; Shimizu, M.; Moriwaki, H. Retinoid X receptor alpha is highly phosphorylated in retinoic acid-resistant HL-60R cells and the combination of 9-cis retinoic acid plus MEK inhibitor induces apoptosis in the cells. Leuk. Res. 2008, 32, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Maris, J.M. MEKing Retinoids Work Better. Cancer Cell 2010, 18, 103–105. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chlapek, P.; Slavikova, V.; Mazanek, P.; Sterba, J.; Veselska, R. Why Differentiation Therapy Sometimes Fails: Molecular Mechanisms of Resistance to Retinoids. Int. J. Mol. Sci. 2018, 19, 132. https://doi.org/10.3390/ijms19010132
Chlapek P, Slavikova V, Mazanek P, Sterba J, Veselska R. Why Differentiation Therapy Sometimes Fails: Molecular Mechanisms of Resistance to Retinoids. International Journal of Molecular Sciences. 2018; 19(1):132. https://doi.org/10.3390/ijms19010132
Chicago/Turabian StyleChlapek, Petr, Viera Slavikova, Pavel Mazanek, Jaroslav Sterba, and Renata Veselska. 2018. "Why Differentiation Therapy Sometimes Fails: Molecular Mechanisms of Resistance to Retinoids" International Journal of Molecular Sciences 19, no. 1: 132. https://doi.org/10.3390/ijms19010132
APA StyleChlapek, P., Slavikova, V., Mazanek, P., Sterba, J., & Veselska, R. (2018). Why Differentiation Therapy Sometimes Fails: Molecular Mechanisms of Resistance to Retinoids. International Journal of Molecular Sciences, 19(1), 132. https://doi.org/10.3390/ijms19010132