Manipulation of the Growth Hormone-Insulin-Like Growth Factor (GH-IGF) Axis: A Treatment Strategy to Reverse the Effects of Early Life Developmental Programming



{kind=link}
Abstract
:1. Introduction
2. Animal Models of Manipulation of the GH-IGF Axis
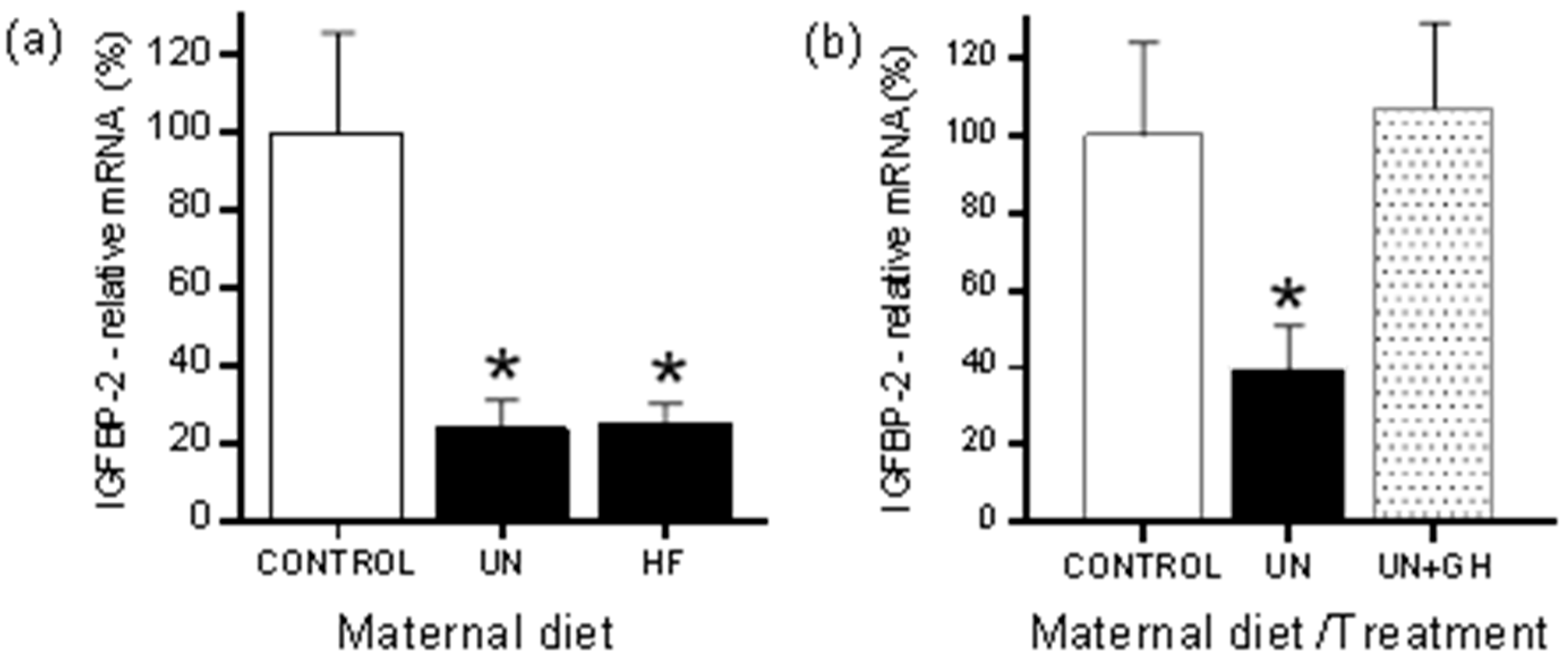
2.1. Rodents
2.2. Sheep
2.3. Other Models
3. Potential Role of Epigenetics
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Osmond, C.; Barker, D.J.; Slattery, J.M. Risk of death from cardiovascular disease and chronic bronchitis determined by place of birth in England and Wales. J. Epidemiol. Commun. Health 1990, 44, 139–141. [Google Scholar] [CrossRef]
- Barker, D.J.; Gluckman, P.D.; Godfrey, K.M.; Harding, J.E.; Owens, J.A.; Robinson, J.S. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993, 341, 938–941. [Google Scholar] [CrossRef]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Cardoso, R.C.; Puttabyatappa, M. Developmental Programming, a Pathway to Disease. Endocrinology 2016, 157, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Woodall, S.M.; Johnston, B.M.; Breier, B.H.; Gluckman, P.D. Chronic maternal undernutrition in the rat leads to delayed postnatal growth and elevated blood pressure of offspring. Pediatr. Res. 1996, 40, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Long, N.M.; Rule, D.C.; Zhu, M.J.; Nathanielsz, P.W.; Ford, S.P. Maternal obesity upregulates fatty acid and glucose transporters and increases expression of enzymes mediating fatty acid biosynthesis in fetal adipose tissue depots. J. Anim. Sci. 2012, 90, 2201–2210. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.E.; Cianfarani, S.; Czernichow, P.; Johannsson, G.; Rapaport, R.; Rogol, A. Management of the child born small for gestational age through to adulthood. a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J. Clin. Endocrinol. Metab. 2007, 92, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leger, J.; Noel, M.; Limal, J.M.; Czernichow, P. Growth factors and intrauterine growth retardation. II. Serum growth hormone, insulin-like growth factor (IGF) I, and IGF-binding protein 3 levels in children with intrauterine growth retardation compared with normal control subjects: Prospective study from birth to two years of age. Study Group of IUGR. Pediatr. Res. 1996, 40, 101–107. [Google Scholar] [PubMed]
- Vickers, M.H.; Sloboda, D.M. Strategies for reversing the effects of metabolic disorders induced as a consequence of developmental programming. Front. Physiol. 2012, 3, 242. [Google Scholar] [CrossRef] [PubMed]
- Kappeler, L.; de Magalhaes Filho, C.; Leneuve, P.; Xu, J.; Brunel, N.; Chatziantoniou, C.; Le Bouc, Y.; Holzenberger, M. Early postnatal nutrition determines somatotropic function in mice. Endocrinology 2009, 150, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.B.; Backeljauw, P.; Bidlingmaier, M.; Biller, B.M.; Boguszewski, M.; Burman, P.; Butler, G.; Chihara, K.; Christiansen, J.; Cianfarani, S.; et al. GH safety workshop position paper: A critical appraisal of recombinant human GH therapy in children and adults. Eur. J. Endocrinol. 2016, 174, P1–P9. [Google Scholar] [CrossRef] [PubMed]
- Carel, J.C.; Butler, G. Safety of recombinant human growth hormone. Endocr. Dev. 2010, 18, 40–54. [Google Scholar] [PubMed]
- Donzeau, A.; Bouhours-Nouet, N.; Fauchard, M.; Decrequy, A.; Mathieu, E.; Boux de Casson, F.; Gascoin, G.; Coutant, R. Birth Weight Is Associated With the IGF-1 Response to GH in Children: Programming of the Anabolic Action of GH? J. Clin. Endocrinol. Metab. 2015, 100, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Bateson, P.; Gluckman, P.; Hanson, M. The biology of developmental plasticity and the Predictive Adaptive Response hypothesis. J. Physiol. 2014, 592, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Elias, S.G.; Keinan-Boker, L.; Peeters, P.H.; van Gils, C.H.; Kaaks, R.; Grobbee, D.E.; van Noord, P.A. Long term consequences of the 1944–1945 Dutch famine on the insulin-like growth factor axis. Int. J. Cancer 2004, 108, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Van Abeelen, A.F.; Veenendaal, M.V.; Painter, R.C.; de Rooij, S.R.; Dijkgraaf, M.G.; Bossuyt, P.M.; Elias, S.G.; Grobbee, D.E.; Uiterwaal, C.S.; Roseboom, T.J. Survival effects of prenatal famine exposure. Am. J. Clin. Nutr. 2012, 95, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nagasaka, M.; Iwatani, S.; Koda, T.; Kurokawa, D.; Yamana, K.; Nishida, K.; Taniguchi-Ikeda, M.; Uchino, E.; Shirai, C.; et al. Prevalence of small for gestational age (SGA) and short stature in children born SGA who qualify for growth hormone treatment at 3 years of age: Population-based study. Pediatr. Int. 2016, 58, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Ciresi, A.; Amato, M.C.; Giordano, C. Reduction in insulin sensitivity and inadequate beta-cell capacity to counteract the increase in insulin resistance in children with idiopathic growth hormone deficiency during 12 months of growth hormone treatment. J. Endocrinol. Investig. 2015, 38, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutfield, W.S.; Wilton, P.; Bennmarker, H.; Albertsson-Wikland, K.; Chatelain, P.; Ranke, M.B.; Price, D.A. Incidence of diabetes mellitus and impaired glucose tolerance in children and adolescents receiving growth-hormone treatment. Lancet 2000, 355, 610–613. [Google Scholar] [CrossRef]
- Baronio, F.; Mazzanti, L.; Girtler, Y.; Tamburrino, F.; Fazzi, A.; Lupi, F.; Longhi, S.; Radetti, G. The Influence of Growth Hormone Treatment on Glucose Homeostasis in GrowthHormone-Deficient Children: A Six-Year Follow-Up Study. Horm. Res. Paediatr. 2016, 86, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Sas, T.; Mulder, P.; Hokken-Koelega, A. Body composition, blood pressure, and lipid metabolism before and during long-term growth hormone (GH) treatment in children with short stature born small for gestational age either with or without GH deficiency. J. Clin. Endocrinol. Metab. 2000, 85, 3786–3792. [Google Scholar] [PubMed]
- Cutfield, W.S.; Lindberg, A.; Rapaport, R.; Wajnrajch, M.P.; Saenger, P. Safety of growth hormone treatment in children born small for gestational age: The US trial and KIGS analysis. Horm. Res. 2006, 65, S153–S159. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, R.; Tanaka, T.; Nishinaga, H.; Ogawa, Y.; Yokoya, S. Evaluation of growth hormone treatment efficacy in short Japanese children born small for gestational age: Five-year treatment outcome and impact on puberty. Clin. Pediatr. Endocrinol. 2017, 26, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Lafeber, H.N. Nutritional management and growth hormone treatment of preterm infants born small for gestational age. Acta Paediatr. Suppl. 1997, 423, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Sejrsen, K.; Purup, S.; Vestergaard, M.; Weber, M.S.; Knight, C.H. Growth hormone and mammary development. Domest. Anim. Endocrinol. 1999, 17, 117–129. [Google Scholar] [CrossRef]
- Gunn, A.J.; Gunn, T.R.; Rabone, D.L.; Breier, B.H.; Blum, W.F.; Gluckman, P.D. Growth hormone increases breast milk volumes in mothers of preterm infants. Pediatrics 1996, 98, 279–282. [Google Scholar] [PubMed]
- Maningat, P.D.; Sen, P.; Rijnkels, M.; Hadsell, D.L.; Bray, M.S.; Haymond, M.W. Short-term administration of rhGH increases markers of cellular proliferation but not milk protein gene expression in normal lactating women. Physiol. Genom. 2011, 43, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.M.; Hilson, J.A.; Kjolhede, C.L. Obesity may impair lactogenesis II. J. Nutr. 2001, 131, 3009s–3011s. [Google Scholar] [PubMed]
- Bao, Z.; Lin, J.; Ye, L.; Zhang, Q.; Chen, J.; Yang, Q.; Yu, Q. Modulation of Mammary Gland Development and Milk Production by Growth Hormone Expression in GH Transgenic Goats. Front. Physiol. 2016, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Ikenasio, B.A.; Breier, B.H. Adult growth hormone treatment reduces hypertension and obesity induced by an adverse prenatal environment. J. Endocrinol. 2002, 175, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Reynolds, C.M.; Gray, C.; Vickers, M.H.; Preweaning, G.H. Treatment Normalizes Body Growth Trajectory and Reverses Metabolic Dysregulation in Adult Offspring After Maternal Undernutrition. Endocrinology 2015, 156, 3228–3238. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.M.; Li, M.; Gray, C.; Vickers, M.H. Pre-Weaning Growth Hormone Treatment Ameliorates Bone Marrow Macrophage Inflammation in Adult Male Rat Offspring following Maternal Undernutrition. PLoS ONE 2013, 8, e68262. [Google Scholar] [CrossRef] [PubMed]
- Podlutsky, A.; Valcarcel-Ares, M.N.; Yancey, K.; Podlutskaya, V.; Nagykaldi, E.; Gautam, T.; Miller, R.A.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z. The GH/IGF-1 axis in a critical period early in life determines cellular DNA repair capacity by altering transcriptional regulation of DNA repair-related genes: Implications for the developmental origins of cancer. Geroscience 2017, 39, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Ikenasio, B.A.; Breier, B.H. IGF-I treatment reduces hyperphagia, obesity, and hypertension in metabolic disorders induced by fetal programming. Endocrinology 2001, 142, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Gluckman, P.D.; Coveny, A.H.; Hofman, P.L.; Cutfield, W.S.; Gertler, A.; Breier, B.H.; Harris, M. Neonatal leptin treatment reverses developmental programming. Endocrinology 2005, 146, 4211–4216. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Gluckman, P.D.; Coveny, A.H.; Hofman, P.L.; Cutfield, W.S.; Gertler, A.; Breier, B.H.; Harris, M. The effect of neonatal leptin treatment on postnatal weight gain in male rats is dependent on maternal nutritional status during pregnancy. Endocrinology 2008, 149, 1906–1913. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.; Baxter, L.; Cave, C.B.; Milne, R. Recombinant growth hormone for idiopathic short stature in children and adolescents. Cochrane Database Syst. Rev. 2007, 3, Cd004440. [Google Scholar]
- Vickers, M.H.; Hofman, P.L.; Gluckman, P.D.; Lobie, P.E.; Cutfield, W.S. Combination therapy with acipimox enhances the effect of growth hormone treatment on linear body growth in the normal and small-for-gestational-age rat. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1212–E1219. [Google Scholar] [CrossRef] [PubMed]
- Casanueva, F.F.; Villanueva, L.; Dieguez, C.; Diaz, Y.; Cabranes, J.A.; Szoke, B.; Scanlon, M.F.; Schally, A.V.; Fernandez-Cruz, A. Free fatty acids block growth hormone (GH) releasing hormone-stimulated GH secretion in man directly at the pituitary. J. Clin. Endocrinol. Metab. 1987, 65, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Moller, N.; Christiansen, J.S.; Jorgensen, J.O. Pharmacological antilipolysis restores insulin sensitivity during growth hormone exposure. Diabetes 2001, 50, 2301–2308. [Google Scholar] [CrossRef] [PubMed]
- Segerlantz, M.; Bramnert, M.; Manhem, P.; Laurila, E.; Groop, L.C. Inhibition of lipolysis during acute GH exposure increases insulin sensitivity in previously untreated GH-deficient adults. Eur. J. Endocrinol. 2003, 149, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Mauras, N.; Ross, J.L.; Gagliardi, P.; Yu, Y.M.; Hossain, J.; Permuy, J.; Damaso, L.; Merinbaum, D.; Singh, R.J.; Gaete, X.; et al. Randomized Trial of Aromatase Inhibitors, Growth Hormone, or Combination in Pubertal Boys with Idiopathic, Short Stature. J. Clin. Endocrinol. Metab. 2016, 101, 4984–4993. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Balen, H.V.; Kamp, G.A.; Oostdijk, W. Benefit of postponing normal puberty for improving final height. Eur. J. Endocrinol. 2004, 151, S41–S45. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.; Li, M.; Reynolds, C.M.; Vickers, M.H. Pre-Weaning Growth Hormone Treatment Reverses Hypertension and Endothelial Dysfunction in Adult Male Offspring of Mothers Undernourished during Pregnancy. PLoS ONE 2013, 8, e53505. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.; Li, M.; Reynolds, C.M.; Vickers, M.H. Let-7 miRNA profiles are associated with the reversal of left ventricular hypertrophy and hypertension in adult male offspring from mothers undernourished during pregnancy following pre-weaning growth hormone treatment. Endocrinology 2014, 155, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Woodall, S.M.; Breier, B.H.; Johnston, B.M.; Bassett, N.S.; Barnard, R.; Gluckman, P.D. Administration of growth hormone or IGF-I to pregnant rats on a reduced diet throughout pregnancy does not prevent fetal intrauterine growth retardation and elevated blood pressure in adult offspring. J. Endocrinol. 1999, 163, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Kopchick, J.; Chen, X.Z.; Li, Y.; Steger, R.W.; Yun, J.S.; Wagner, T.E.; Bartke, A. Differential in vivo activities of bovine growth hormone analogues. Transgenic Res. 1998, 7, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Hull, K.L.; Harvey, S. Growth hormone and reproduction: A review of endocrine and autocrine/paracrine interactions. Int. J. Endocrinol. 2014, 2014, 234014. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G.; Simerly, R.B. Minireview: Leptin and development of hypothalamic feeding circuits. Endocrinology 2004, 145, 2621–2626. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G.; Draper, S.J.; Simerly, R.B. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 2004, 304, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Watanobe, H.; Habu, S. Leptin regulates growth hormone-releasing factor, somatostatin, and alpha-melanocyte-stimulating hormone but not neuropeptide Y release in rat hypothalamus in vivo: Relation with growth hormone secretion. J. Neurosci. 2002, 22, 6265–6271. [Google Scholar] [PubMed]
- Allensworth-James, M.L.; Odle, A.; Haney, A.; Childs, G. Sex Differences in Somatotrope Dependency on Leptin Receptors in Young Mice: Ablation of LEPR Causes Severe Growth Hormone Deficiency and Abdominal Obesity in Males. Endocrinology 2015, 156, 3253–3264. [Google Scholar] [CrossRef] [PubMed]
- Oberbauer, A.M. Developmental programming: The role of growth hormone. J. Anim. Sci. Biotechnol. 2015, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Decourtye, L.; Mire, E.; Clemessy, M.; Heurtier, V.; Ledent, T.; Robinson, I.C.; Mollard, P.; Epelbaum, J.; Meaney, M.J.; Garel, S.; et al. IGF-1 Induces GHRH Neuronal Axon Elongation during Early Postnatal Life in Mice. PLoS ONE 2017, 12, e0170083. [Google Scholar]
- Kappeler, L.; De Magalhaes Filho, C.; Dupont, J.; Leneuve, P.; Cervera, P.; Perin, L.; Loudes, C.; Blaise, A.; Klein, R.; Epelbaum, J.; et al. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008, 6, e254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, M.; Raizada, M.K.; LeRoith, D. Insulin and insulin-like growth factor receptors in the nervous system. Mol. Neurobiol. 1989, 3, 71–100. [Google Scholar] [CrossRef] [PubMed]
- Plank, C.; Grillhosl, C.; Ostreicher, I.; Meissner, U.; Struwe, F.G.; Rauh, M.; Hartner, A.; Rascher, W.; Dotsch, J. Transient growth hormone therapy to rats with low protein-inflicted intrauterine growth restriction does not prevent elevated blood pressure in later life. Growth Factors 2008, 26, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.M.; Li, M.; Gray, C.; Vickers, M.H. Pre-weaning Growth Hormone Treatment Ameliorates Adipose Tissue Insulin Resistance and Inflammation in Adult Male Offspring Following Maternal Undernutrition. Endocrinology 2013, 154, 2676–2686. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.M.; Li, M.; Gray, C.; Vickers, M.H. Early-life growth hormone treatment to offspring of undernourished mothers alters metabolic parameters in primary adipocytes in adulthood. Growth Factors 2014, 32, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Breier, B.H.; Cutfield, W.S.; Hofman, P.L.; Gluckman, P.D. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E83–E87. [Google Scholar] [PubMed]
- Dunn, G.A.; Bale, T.L. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology 2009, 150, 4999–5009. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Sloboda, D.M.; Saffery, R.; Joo, E.; Vickers, M.H. Maternal nutritional history modulates the hepatic IGF-IGFBP axis in adult male rat offspring. Endocrine 2013, 46, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.E.; Bloomfield, F.H. Prenatal treatment of intrauterine growth restriction: Lessons from the sheep model. Pediatr. Endocrinol. Rev. 2004, 2, 182–192. [Google Scholar] [PubMed]
- Koch, J.M.; Wilmoth, T.A.; Wilson, M.E. Periconceptional growth hormone treatment alters fetal growth and development in lambs. J. Anim. Sci. 2010, 88, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Costine, B.A.; Inskeep, E.K.; Wilson, M.E. Growth hormone at breeding modifies conceptus development and postnatal growth in sheep. J. Anim. Sci. 2005, 83, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.E.; Evans, P.C.; Gluckman, P.D. Maternal growth hormone treatment increases placental diffusion capacity but not fetal or placental growth in sheep. Endocrinology 1997, 138, 5352–5358. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.K.; Harding, J.E.; Breier, B.H.; Gluckman, P.D. Exogenous GH infusion to late-gestational fetal sheep does not alter fetal growth and metabolism. J. Endocrinol. 2000, 166, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.K.; Breier, B.B.; Bloomfield, F.H.; Jensen, E.C.; Gluckman, P.D.; Harding, J.E. Chronic pulsatile infusion of growth hormone to growth-restricted fetal sheep increases circulating fetal insulin-like growth factor-I levels but not fetal growth. J. Endocrinol. 2003, 177, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, F.H.; van Zijl, P.L.; Bauer, M.K.; Phua, H.H.; Harding, J.E. Effect of pulsatile growth hormone administration to the growth-restricted fetal sheep on somatotrophic axis gene expression in fetal and placental tissues. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E333–E339. [Google Scholar] [CrossRef] [PubMed]
- Curran, A.J.; Peacey, S.R.; Shalet, S.M. Is maternal growth hormone essential for a normal pregnancy? Eur. J. Endocrinol. 1998, 139, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Kadakia, R.; Josefson, J. The Relationship of Insulin-Like Growth Factor 2 to Fetal Growth and Adiposity. Horm. Res. Paediatr. 2016, 85, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.M.; Matsuzaki, M.; Milne, J.; Aitken, R. Late but not early gestational maternal growth hormone treatment increases fetal adiposity in overnourished adolescent sheep. Biol. Reprod. 2006, 75, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Lonberg, U.; Damm, P.; Andersson, A.M.; Main, K.M.; Chellakooty, M.; Lauenborg, J.; Skakkebaek, N.E.; Juul, A. Increase in maternal placental growth hormone during pregnancy and disappearance during parturition in normal and growth hormone-deficient pregnancies. Am. J. Obstet. Gynecol. 2003, 188, 247–251. [Google Scholar] [CrossRef] [PubMed]
- De Boo, H.A.; Eremia, S.C.; Bloomfield, F.H.; Oliver, M.H.; Harding, J.E. Treatment of intrauterine growth restriction with maternal growth hormone supplementation in sheep. Am. J. Obstet. Gynecol. 2008, 199, e551–e559. [Google Scholar] [CrossRef] [PubMed]
- Eremia, S.C.; de Boo, H.A.; Bloomfield, F.H.; Oliver, M.H.; Harding, J.E. Fetal and amniotic insulin-like growth factor-I supplements improve growth rate in intrauterine growth restriction fetal sheep. Endocrinology 2007, 148, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Wali, J.A.; de Boo, H.A.; Derraik, J.G.; Phua, H.H.; Oliver, M.H.; Bloomfield, F.H.; Harding, J.E. Weekly intra-amniotic IGF-1 treatment increases growth of growth-restricted ovine fetuses and up-regulates placental amino acid transporters. PLoS ONE 2012, 7, e37899. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.M.; Milne, J.S.; Aitken, R.P. Maternal growth hormone treatment from day 35 to 80 of gestation alters nutrient partitioning in favor of uteroplacental growth in the overnourished adolescent sheep. Biol. Reprod. 2004, 70, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Guimarey, L.M.; Oyhenart, E.E.; Quintero, F.A.; Fucini, M.C. Body weight recovery in intrauterine growth-retarded rats treated with growth hormone. Clin. Exp. Obstet. Gynecol. 2003, 30, 51–56. [Google Scholar] [PubMed]
- Ikeda, N.; Shoji, H.; Suganuma, H.; Ohkawa, N.; Kantake, M.; Murano, Y.; Sakuraya, K.; Shimizu, T. Effect of insulin-like growth factor-I during the early postnatal period in intrauterine growth-restricted rats. Pediatr. Int. 2016, 58, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.E.; Gordon, T.P.; Chikazawa, K.; Gust, D.; Tanner, J.M.; Rudman, C.G. Effects of growth hormone on neonatal growth in nursing rhesus monkeys. J. Clin. Endocrinol. Metab. 1991, 72, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Ouni, M.; Castell, A.L.; Linglart, A.; Bougneres, P. Genetic and Epigenetic Modulation of Growth Hormone Sensitivity Studied With the IGF-1 Generation Test. J. Clin. Endocrinol. Metab. 2015, 100, E919–E925. [Google Scholar] [CrossRef] [PubMed]
- Ouni, M.; Gunes, Y.; Belot, M.P.; Castell, A.L.; Fradin, D.; Bougneres, P. The IGF1 P2 promoter is an epigenetic QTL for circulating IGF1 and human growth. Clin. Epigenet. 2015, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, F.R.; Furukawa, L.N.; Oliveira, I.B.; Heimann, J.C. Glucose metabolism and hepatic Igf1 DNA methylation are altered in the offspring of dams fed a low-salt diet during pregnancy. Physiol. Behav. 2016, 154, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Massah, S.; Hollebakken, R.; Labrecque, M.P.; Kolybaba, A.M.; Beischlag, T.V.; Prefontaine, G.G. Epigenetic characterization of the growth hormone gene identifies SmcHD1 as a regulator of autosomal gene clusters. PLoS ONE 2014, 9, e97535. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.; Gourdji, D.; Laverriere, J.N. Site-specific methylation of the rat prolactin and growth hormone promoters correlates with gene expression. Mol. Cell Biol. 1996, 16, 3245–3254. [Google Scholar] [CrossRef] [PubMed]
- Duong, C.V.; Emes, R.D.; Wessely, F.; Yacqub-Usman, K.; Clayton, R.N.; Farrell, W.E. Quantitative, genome-wide analysis of the DNA methylome in sporadic pituitary adenomas. Endocr. Relat. Cancer 2012, 19, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Gaido, M.L.; Strobl, J.S. Inhibition of rat growth hormone promoter activity by site-specific DNA methylation. Biochim. Biophys. Acta 1989, 1008, 234–242. [Google Scholar] [CrossRef]
- Scully, K.M.; Jacobson, E.M.; Jepsen, K.; Lunyak, V.; Viadiu, H.; Carriere, C.; Rose, D.W.; Hooshmand, F.; Aggarwal, A.K.; Rosenfeld, M.G. Allosteric effects of Pit-1 DNA sites on long-term repression in cell type specification. Science 2000, 290, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.C.; Galati, J.C.; Burrows, S.; Beilin, L.J.; Li, X.; Pennell, C.E.; van Eekelen, J.; Mori, T.A.; Adams, L.A.; Craig, J.M. DNA methylation of the IGF2/H19 imprinting control region and adiposity distribution in young adults. Clin. Epigenet. 2012, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Walter, J.; Allen, N.D.; Reik, W. Developmental control of allelic methylation in the imprinted mouse Igf2 and H19 genes. Development 1994, 120, 2933–2943. [Google Scholar] [PubMed]
- Bergman, D.; Halje, M.; Nordin, M.; Engstrom, W. Insulin-like growth factor 2 in development and disease: A mini-review. Gerontology 2013, 59, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Pan, Y.X.; Chen, H. Gestational low protein diet in the rat mediates Igf2 gene expression in male offspring via altered hepatic DNA methylation. Epigenetics 2010, 5, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.C.; van Der Meulen, J.H.; Osmond, C.; Barker, D.J.; Bleker, O.P. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am. J. Clin. Nutr. 1999, 70, 811–816. [Google Scholar] [PubMed]
- Ravelli, G.P.; Stein, Z.A.; Susser, M.W. Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 1976, 295, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Hedbacker, K.; Birsoy, K.; Wysocki, R.W.; Asilmaz, E.; Ahima, R.S.; Farooqi, I.S.; Friedman, J.M. Antidiabetic effects of IGFBP2, a leptin-regulated gene. Cell Metab. 2010, 11, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, A.; Reisinger, R.; Lahm, H.; Kiess, W.; Blum, W.F.; Kolb, H.J.; Weber, M.M.; Wolf, E. Insulin-like growth factor-binding protein 2 in tumorigenesis: Protector or promoter? Cancer Res. 2001, 61, 8601–8610. [Google Scholar] [PubMed]
- Mirbahai, L.; Williams, T.D.; Zhan, H.; Gong, Z.; Chipman, J.K. Comprehensive profiling of zebrafish hepatic proximal promoter CpG island methylation and its modification during chemical carcinogenesis. BMC Genom. 2011, 12, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsmans, M.; Holvoet, P. MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc. Res. 2013, 100, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Stochholm, K.; Johannsson, G. Reviewing the safety of GH replacement therapy in adults. Growth Horm. IGF Res. 2015, 25, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Clemmons, D.R.; Rosenfeld, R.G. Does the GH-IGF axis play a role in cancer pathogenesis? Growth Horm. IGF Res. 2000, 10, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Schernhammer, E.S.; Holly, J.M.; Hunter, D.J.; Pollak, M.N.; Hankinson, S.E. Insulin-like growth factor-I, its binding proteins (IGFBP-1 and IGFBP-3), and growth hormone and breast cancer risk in The Nurses Health Study II. Endocr. Relat. Cancer 2006, 13, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Ogus, S.; Ke, Y.; Qiu, J.; Wang, B.; Chehab, F.F. Hyperleptinemia precipitates diet-induced obesity in transgenic mice overexpressing leptin. Endocrinology 2003, 144, 2865–2869. [Google Scholar] [CrossRef] [PubMed]
- Fouque, D.; Juillard, L.; Lasne, Y.; Tabakian, A.; Laville, M.; Joly, M.O. Acute leptin regulation in end-stage renal failure: The role of growth hormone and IGF-1. Kidney Int. 1998, 54, 932–937. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reynolds, C.M.; Perry, J.K.; Vickers, M.H. Manipulation of the Growth Hormone-Insulin-Like Growth Factor (GH-IGF) Axis: A Treatment Strategy to Reverse the Effects of Early Life Developmental Programming. Int. J. Mol. Sci. 2017, 18, 1729. https://doi.org/10.3390/ijms18081729
Reynolds CM, Perry JK, Vickers MH. Manipulation of the Growth Hormone-Insulin-Like Growth Factor (GH-IGF) Axis: A Treatment Strategy to Reverse the Effects of Early Life Developmental Programming. International Journal of Molecular Sciences. 2017; 18(8):1729. https://doi.org/10.3390/ijms18081729
Chicago/Turabian StyleReynolds, Clare M., Jo K. Perry, and Mark H. Vickers. 2017. "Manipulation of the Growth Hormone-Insulin-Like Growth Factor (GH-IGF) Axis: A Treatment Strategy to Reverse the Effects of Early Life Developmental Programming" International Journal of Molecular Sciences 18, no. 8: 1729. https://doi.org/10.3390/ijms18081729