Intrapancreatic Parenchymal Injection of Cells as a Useful Tool for Allowing a Small Number of Proliferative Cells to Grow In Vivo

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
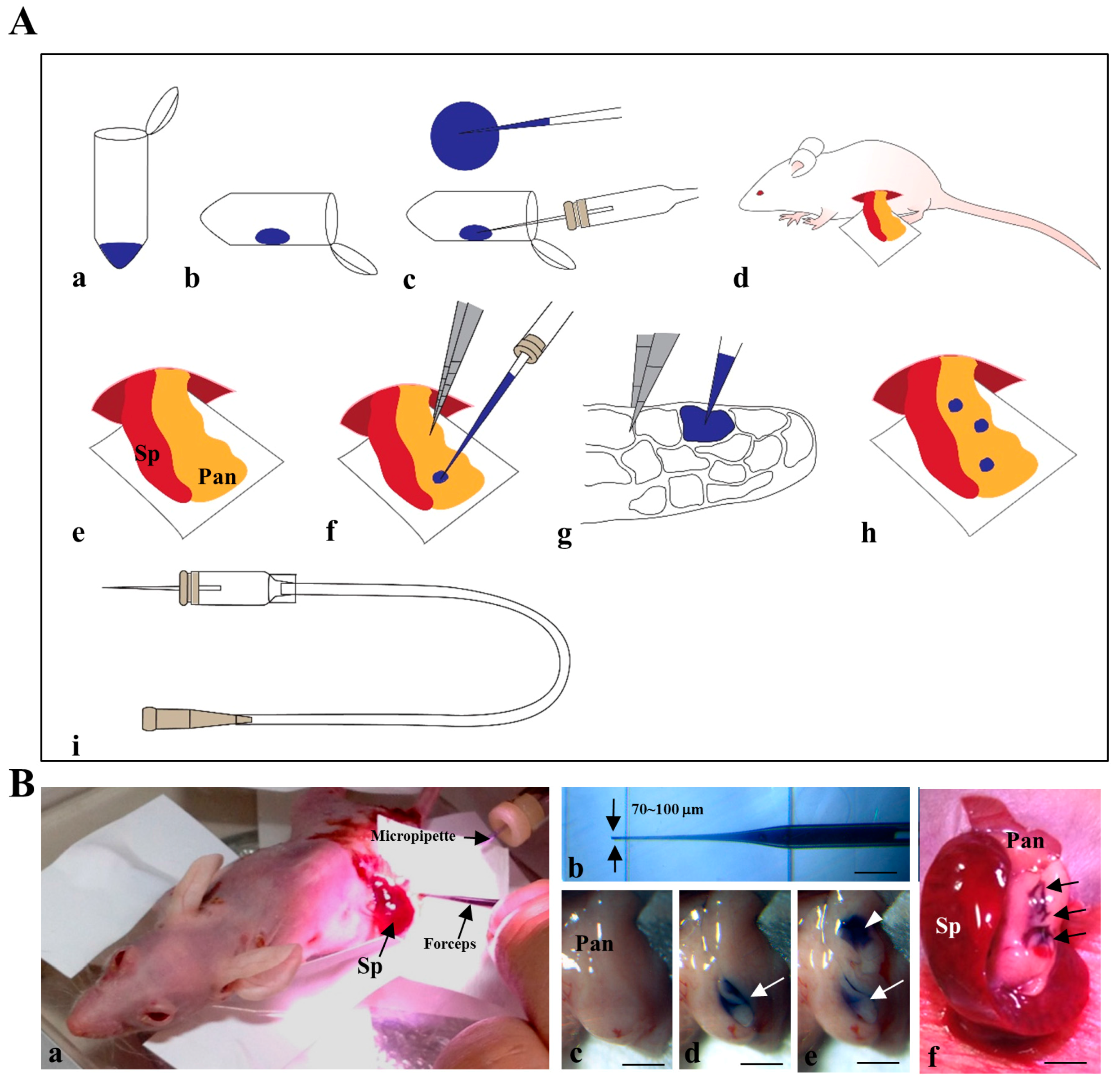
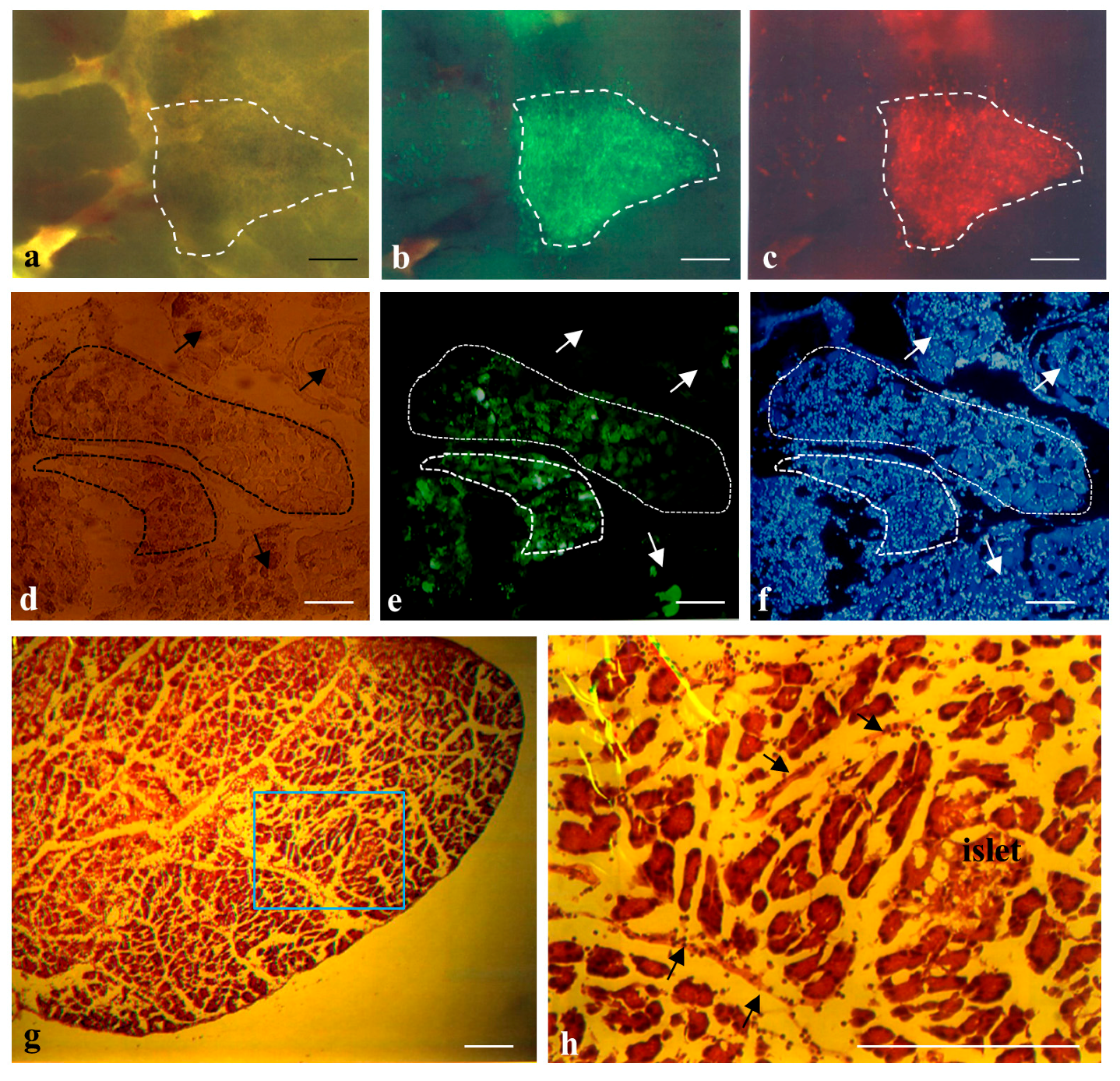
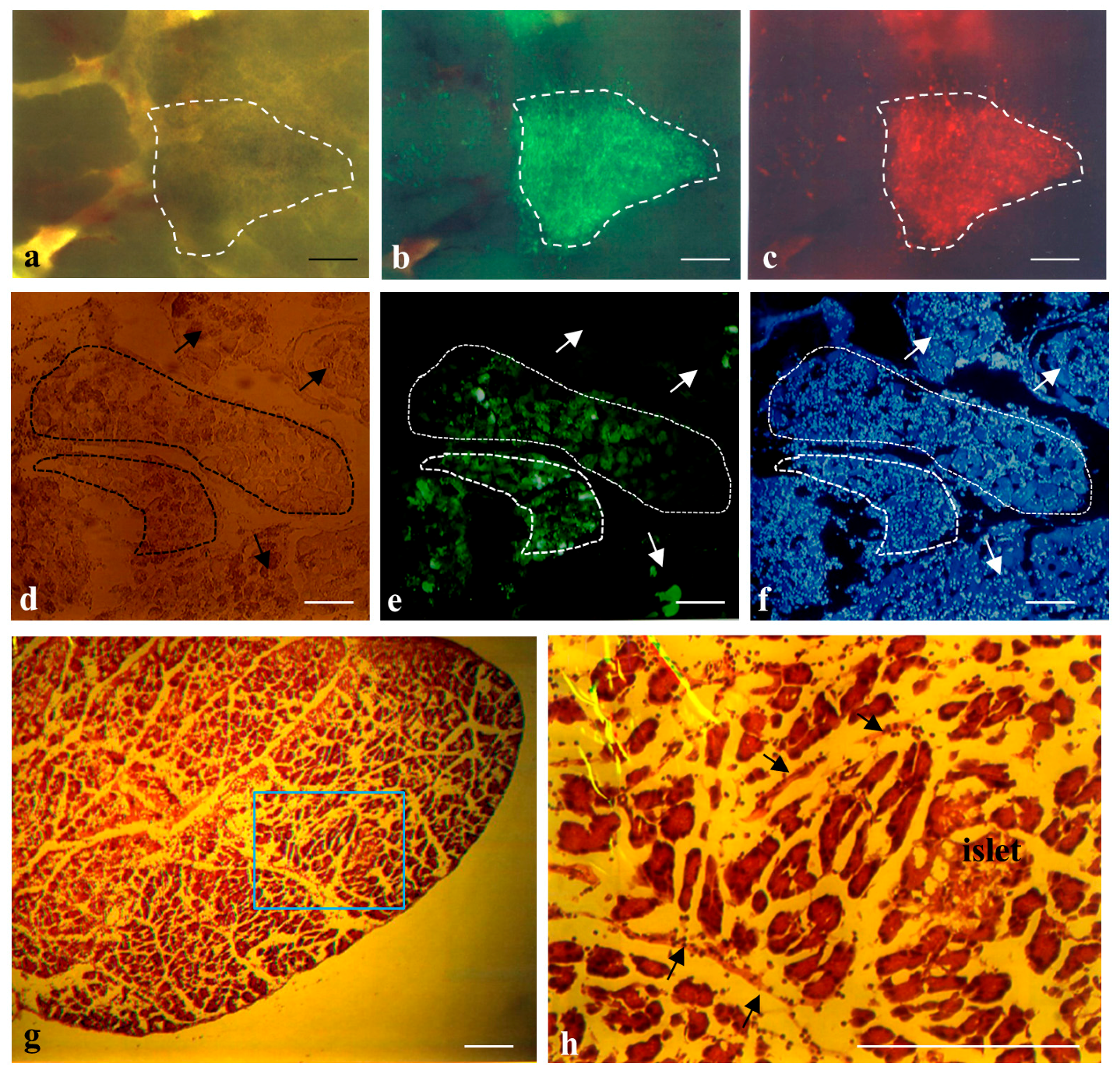
2.1. Cells Transplanted into the Pancreatic Parenchyma Are Trapped within Compartments of the Pancreas
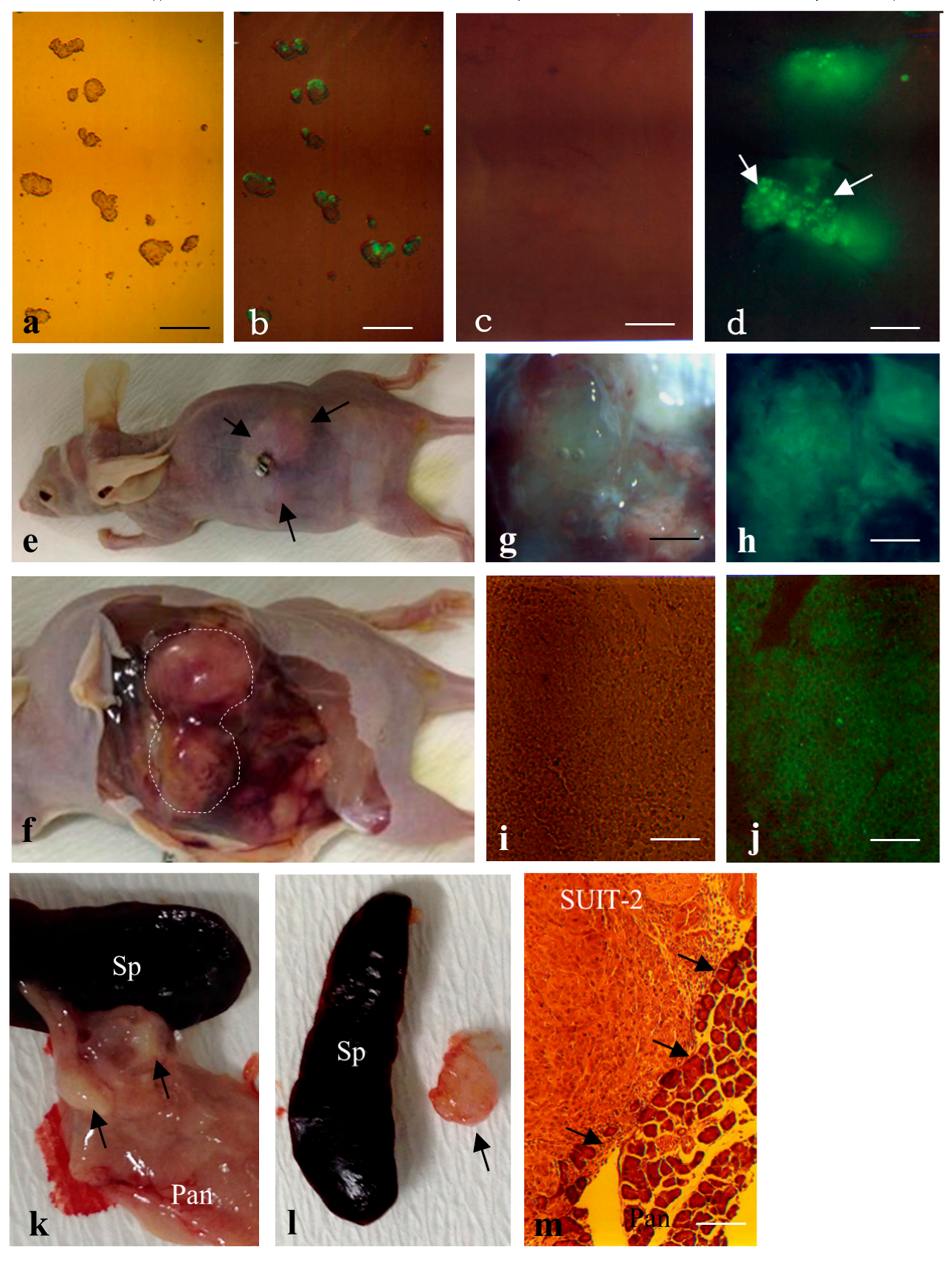
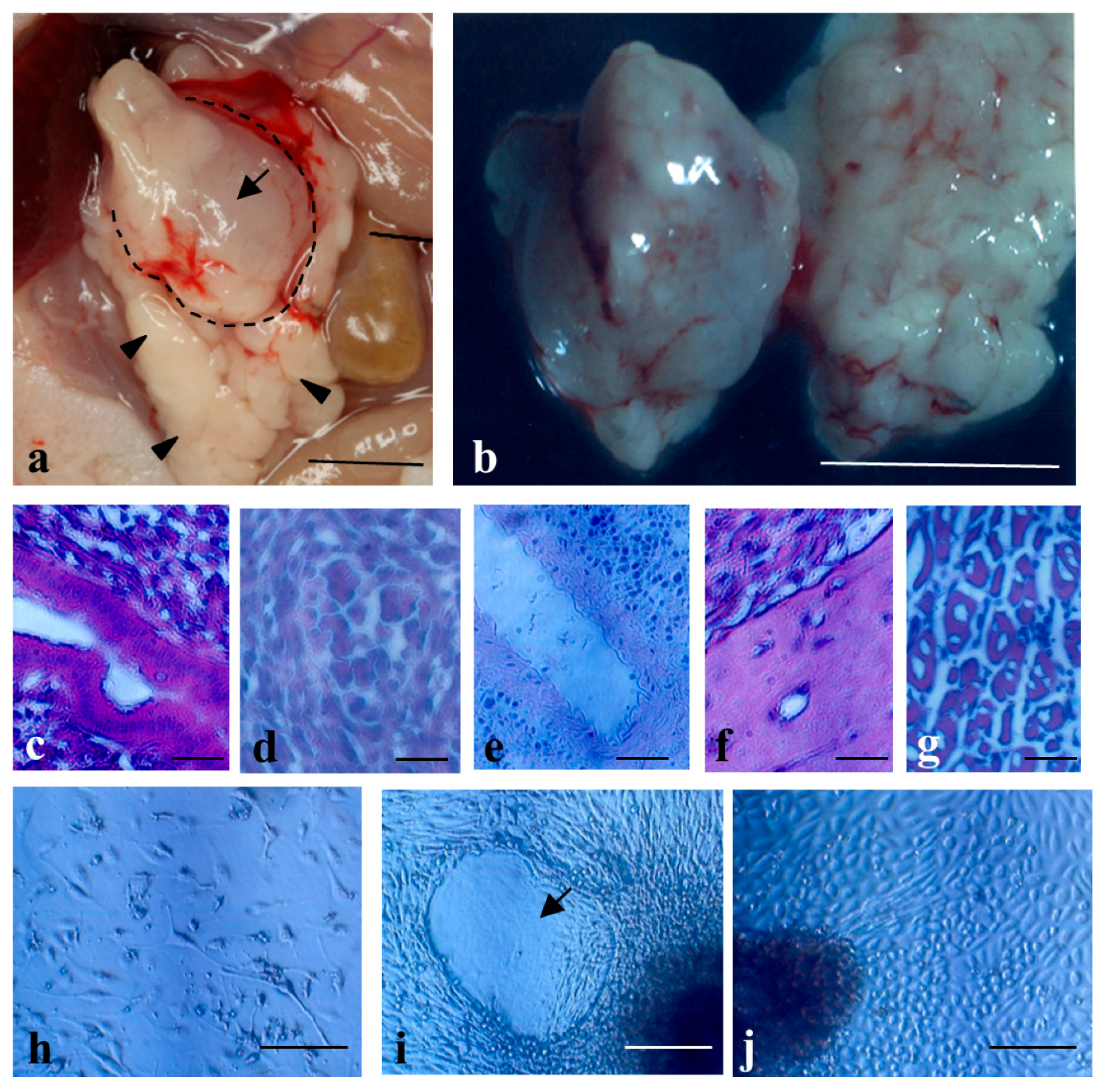
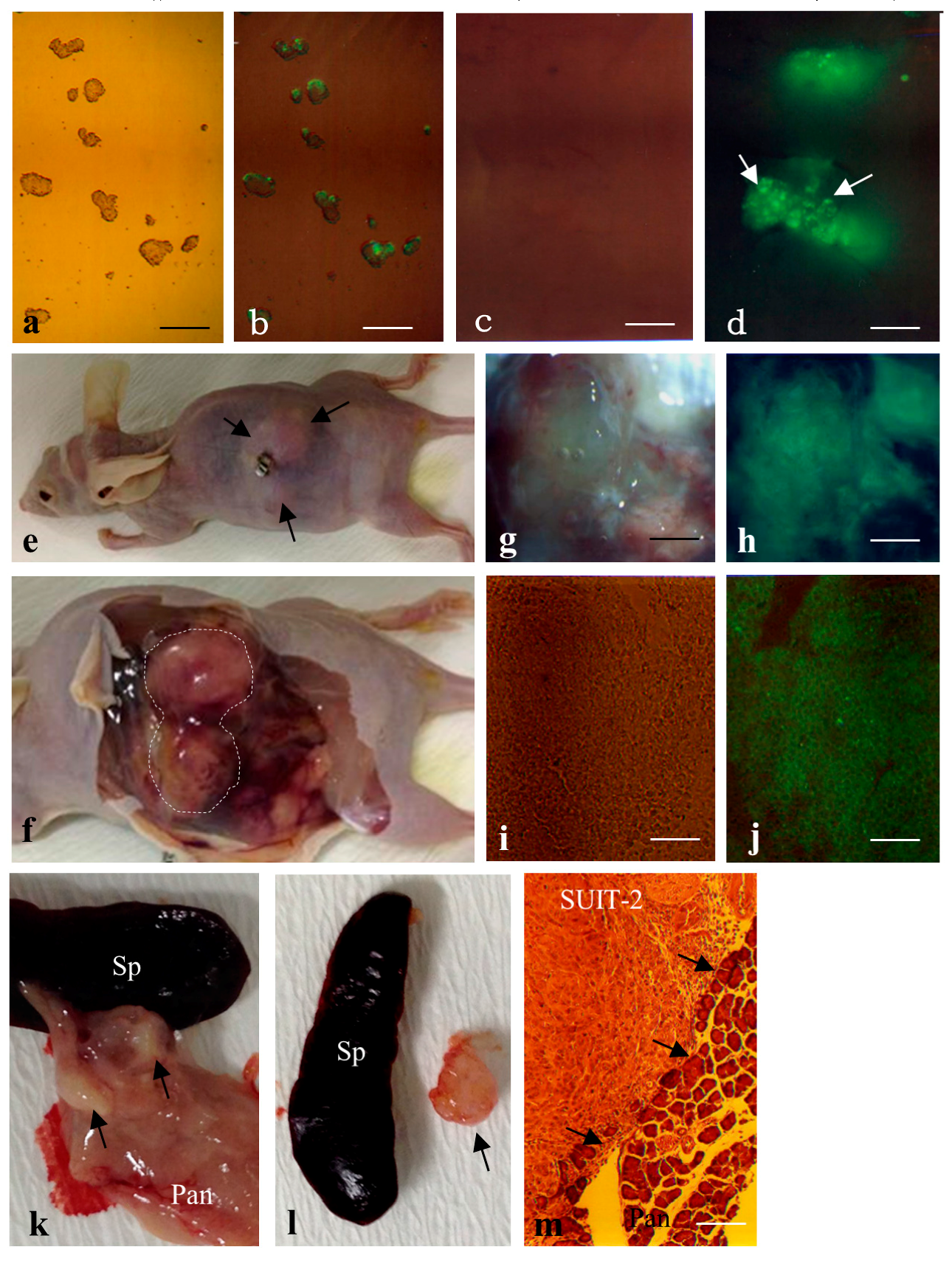
2.2. Formation of Solid Tumors after Intrapancreatic Inoculation of Proliferative Cells in Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
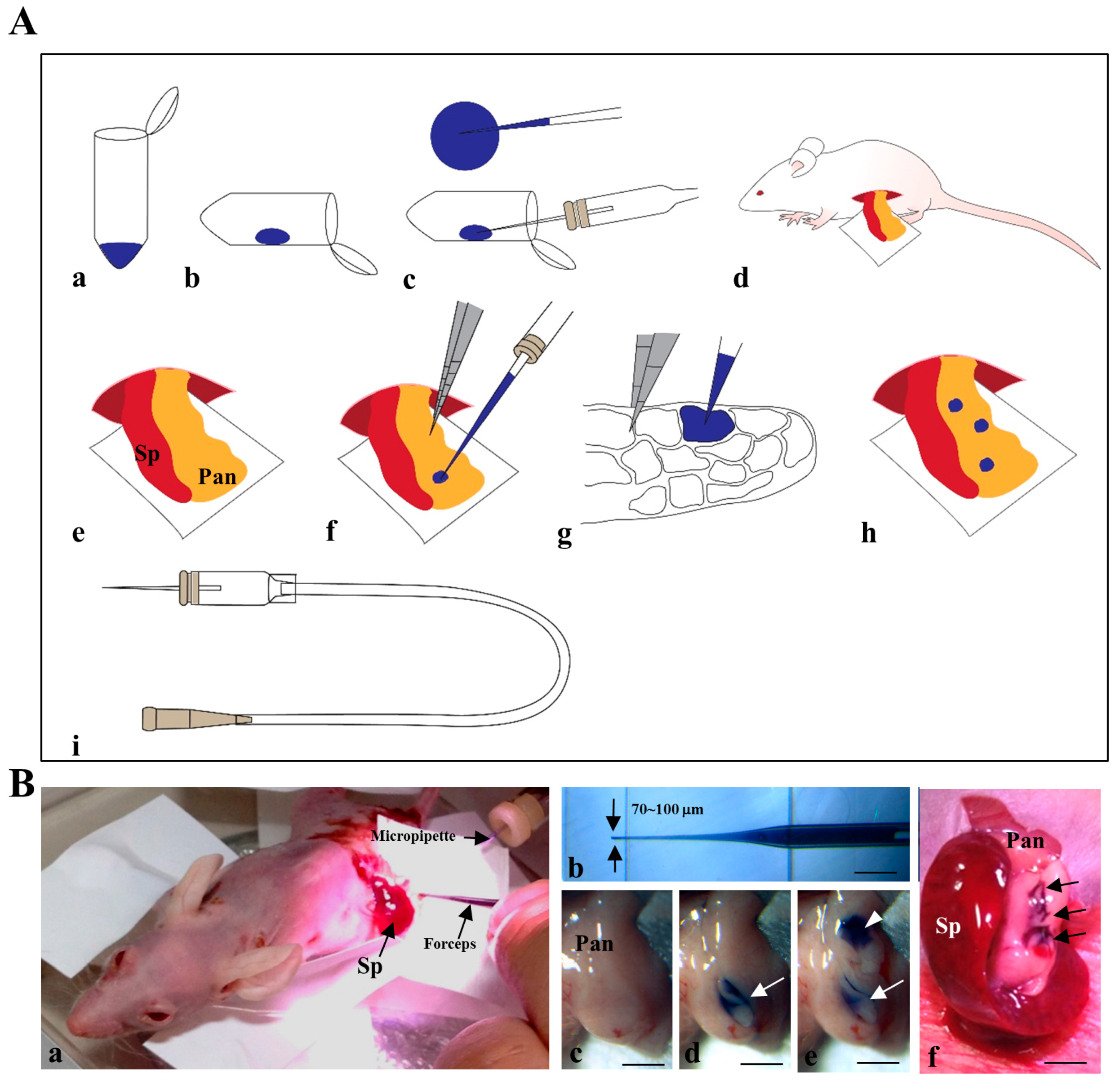
4.3. Intrapancreatic Parenchymal Gene Transfer (IPPGT)
4.4. IPPCT
4.5. Histological Analysis
4.6. Fluorescence Observation
4.7. Quantification of Teratoma Formation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Solter, D. From teratocarcinomas to embryonic stem cells and beyond: A history of embryonic stem cell research. Nat. Rev. Genet. 2006, 7, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Damjanov, I.; Andrews, P.W. The terminology of teratocarcinomas and teratomas. Nature Biotechnol. 2007, 25, 1212. [Google Scholar] [CrossRef] [PubMed]
- Leea, M.; Moonb, S.H.; Jeonga, H.; Yic, J.; Lee, T.H.; Shim, S.H.; Rhee, Y.H.; Lee, S.H.; Oh, S.J.; Lee, M.Y.; et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, E3281–E3290. [Google Scholar] [CrossRef] [PubMed]
- Brivanlou, A.H.; Gage, F.H.; Jaenisch, R.; Jessell, T.; Douglas, M.; Janet, R. Setting standards for human embryonic stem cells. Science 2003, 300, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Gertow, K.; Przyborski, S.; Loring, J.F.; Auerbach, J.M.; Epifano, O.; Otonkoski, T.; Damjanov, I.; Ahrlund-Richter, L. Isolation of human embryonic stem cell-derived teratomas for the assessment of pluripotency. Curr. Protoc. Stem Cell Biol. 2007. [Google Scholar] [CrossRef]
- International Stem Cell Initiative; Adewumi, O.; Aflatoonian, B.; Ahrlund-Richter, L.; Amit, M.; Andrews, P.W.; Beighton, G.; Bello, P.A.; Benvenisty, N.; Berry, L.S.; Bevan, S.; et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007, 25, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.J.; Goldmann, J.; Loser, P.; Loring, J.F. A call to standardize teratoma assays used to define human pluripotent cell lines. Cell Stem Cell 2010, 6, 412–414. [Google Scholar] [CrossRef] [PubMed]
- De Coppi, P.; Bartsch, G., Jr.; Siddiqui, M.M.; Xu, T.; Cesar, C.S.; Laura, P.; Gustavo, M.; Angéline, C.S.; Evan, Y.S.; James, J.Y.; et al. Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 2007, 25, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, L.J.; Tian, S.; Nie, J.; Jonsdottir, A.G.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.J.; Stojkovic, M.; Przyborski, S.A. Growth of teratomas derived from human pluripotent stem cells is influenced by the graft site. Stem Cells Dev. 2006, 15, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Prokhorova, T.A.; Harkness, L.M.; Frandsen, U.; Ditzel, N.; Schrøder, H.D.; Burns, J.S.; Kassem, M. Teratoma formation by human embryonic stem cells is site dependent and enhanced by the presence of Matrigel. Stem Cells Dev. 2009, 18, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Blum, B.; Benvenisty, N. Clonal analysis of human embryonic stem cell differentiation into teratomas. Stem Cells 2007, 258, 1924–1930. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Aranda, I.; Ramos-Mejia, V.; Bueno, C. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem Cells 2010, 28, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Hirose, M.; Hatori, M.; Matoba, S.; Miyoshi, H.; Inoue, K.; Ogura, A. Generation of induced pluripotent stem cells in rabbits: Potential experimental models for human regenerative medicine. J. Biol. Chem. 2010, 285, 31362–31369. [Google Scholar] [CrossRef] [PubMed]
- Ritner, C.; Bernstein, H.S. Fate mapping of human embryonic stem cells by teratoma formation. J. Vis. Exp. 2010, 42, 2036. [Google Scholar] [CrossRef] [PubMed]
- Gropp, M.; Shilo, V.; Vainer, G.; Miri Gov, M.; Gil, Y.; Khaner, H.; Matzrafi, L.; Idelson, M.; Kopolovic, J.; Zak, B.N.; et al. Standardization of the teratoma assay for analysis of pluripotency of human ES cells and biosafety of their differentiated progeny. PLoS ONE 2012, 7, e45532. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tang, Y.; Lu, S.; Zhou, J.; Du, Z.Y.; Duan, C.M.; Li, Z.Y.; Wang, C.Y. The tumourigenicity of iPS cells and their differentiated derivates. J. Cell. Mol. Med. 2013, 17, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Gertow, K.; Wolbank, S.; Rozell, B.; Sugars, R.; Andäng, M.; Parish, C.L.; Imreh, M.P.; Wendel, M.; Ahrlund-Richter, L. Organized development from human embryonic stem cells after injection into immunodeficient mice. Stem Cells Dev. 2004, 13, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Przyborski, S.A. Differentiation of human embryonic stem cells after transplantation in immune-deficient mice. Stem Cells 2005, 23, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, P.; Lako, M.; Stewart, R.; Przyborski, S.; Armstrong, L.; Evans, J.; Murdoch, A.; Strachan, T.; Stojkovic, M. An autogeneic feeder cell system that efficiently supports growth of undifferentiated human embryonic stem cells. Stem Cells 2005, 23, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Werbowetski-Ogilvie, T.E.; Bosse, M.; Stewart, M.; Schnerch, A.; Ramos-Mejia, V.; Rouleau, A.; Wynder, T.; Smith, M.J.; Dingwall, S.; Carter, T.; et al. Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 2009, 27, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.E.; Tran, H.T.; Garitaonandia, I.; Han, S.; Nickey, S.K.; Leonardo, T.; Laurent, L.C.; Loring, F.J. Teratoma generation in the testis capsule. J. Vis. Exp. 2011, 57, e3177. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Tang, C.; Cao, F.; Xie, X. Effects of cell number on teratoma formation by human embryonic stem cells. Cell Cycle 2009, 8, 2608–2612. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Margulets, V.; Segev, H.; Shariki, K.; Laevsky, I.; Coleman, R.; Itskovitz-Eldor, J. Human feeder layers for human embryonic stem cells. Biol. Reprod. 2003, 68, 2150–2156. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.B.; Padmanabhan, J.; Chin, A.C.; Oh, S.K. Expansion of pluripotent human embryonic stem cells on human feeders. Biotechnol. Bioeng. 2004, 88, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Plaia, T.W.; Josephson, R.; Liu, Y.; Zeng, X.; Ording, C.; Toumadje, A.; Brimble, S.N.; Sherrer, E.S.; Uhl, E.W.; Freed, W.J.; et al. Characterization of a new NIH-registered variant human embryonic stem cell line, BG01V: A tool for human embryonic stem cell research. Stem Cells 2006, 24, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Tzukerman, M.; Rosenberg, T.; Reiter, I.; Ben-Eliezer, S.; Denkberg, G.; Coleman, R.; Reiter, Y.; Skorecki, K. The influence of a human embryonic stem cell-derived microenvironment on targeting of human solid tumor xenografts. Cancer Res. 2006, 66, 3792–3801. [Google Scholar] [CrossRef] [PubMed]
- Hentze, H.; Soong, P.L.; Wang, S.T.; Phillips, B.W.; Putti, T.C.; Dunn, N.R. Teratoma formation by human embryonic stem cells: Evaluation of essential parameters for future safety studies. Stem Cell Res. 2009, 2, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Nakagawa, T.; Sakamoto, T.; Ito, J. Fates of murine pluripotent stem cell-derived neural progenitors following transplantation into mouse cochleae. Cell Transplant 2012, 21, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Obana, A.; Usami, Y.; Sakai, M.; Nohara, K.; Egusa, H.; Sakai, T. Regenerating salivary glands in the microenvironment of induced pluripotent stem cells. BioMed Res. Int. 2015, 293570, 11. [Google Scholar] [CrossRef] [PubMed]
- Reichert, M.; Rustgi, A.K. Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Investig. 2011, 121, 4572–4578. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Inada, E.; Saitoh, I.; Ohtsuka, M.; Nakamura, S.; Sakurai, T.; Watanabe, S. Site-targeted non-viral gene delivery by direct DNA injection into the pancreatic parenchyma and subsequent in vivo electroporation in mice. Biotechnol. J. 2013, 8, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ohtsuka, M.; Miura, H.; Miyoshi, K.; Watanabe, S. Determination of the optimal concentration of several selective drugs useful for generating multi-transgenic porcine embryonic fibroblasts. Reprod. Dom. Anim. 2012, 47, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Bernstine, E.G.; Hooper, M.L.; Grandchamp, S.; Ephrussi, B. Alkaline phosphatase activity in mouse teratoma. Proc. Natl. Acad. Sci. USA 1973, 70, 3899–3903. [Google Scholar] [CrossRef]
- Iwamura, T.; Katsuki, T.; Ide, K. Establishment and characterization of a human pancreatic cancer cell line (SUIT-2) producing carcinoembryonic antigen and carbohydrate antigen 19–9. Jpn. J. Cancer Res. 1987, 78, 54–62. [Google Scholar] [PubMed]
- Saitoh, I.; Inada, E.; Iwase, Y.; Noguchi, H.; Murakami, T.; Soda, M.; Kubota, N.; Hasegawa, H.; Akasaka, E.; Matsumoto, Y.; et al. Choice of feeders is important when first establishing iPSCs derived from primarily cultured human deciduous tooth dental pulp cells. Cell Med. 2015, 8, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbrusseze, J.L.; Jason B Fleming, B.J.; Gallick, E.G. Orthotopic and heterotopic generation of murine pancreatic cancer xenografts. Nat. Protoc. 2009, 4, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.J.; Lee, C.L.; Wang, Q.; Zhou, Z.W.; Yang, F.; Jin, C.; Fu, D.L. Establishment of an orthotopic pancreatic cancer mouse model: Cells suspended and injected in Matrigel. World J. Gastroenterol. 2014, 20, 9476–9485. [Google Scholar] [PubMed]
- Hara, A.; Taguchi, A.; Aoki, H.; Hatano, Y.; Niwa, M.; Yamada, Y.; Kunisada, T. Folate antagonist, methotrexate induces neuronal differentiation of human embryonic stem cells transplanted into nude mouse retina. Neurosci. Lett. 2010, 477, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Yanai, G.; Hayashi, T.; Zhi, Q.; Yang, K.C.; Shirouzu, Y.; Shimabukuro, T.; Hiura, A.; Inoue, K.; Sumi, S. Electrofusion of mesenchymal stem cells and islet cells for diabetes therapy: A rat model. PLoS ONE 2013, 8, e64499. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, M.; Saitoh, I.; Murakami, T.; Kubota, N.; Nakamura, S.; Watanabe, S.; Inada, E. Intrapancreatic Parenchymal Injection of Cells as a Useful Tool for Allowing a Small Number of Proliferative Cells to Grow In Vivo. Int. J. Mol. Sci. 2017, 18, 1678. https://doi.org/10.3390/ijms18081678
Sato M, Saitoh I, Murakami T, Kubota N, Nakamura S, Watanabe S, Inada E. Intrapancreatic Parenchymal Injection of Cells as a Useful Tool for Allowing a Small Number of Proliferative Cells to Grow In Vivo. International Journal of Molecular Sciences. 2017; 18(8):1678. https://doi.org/10.3390/ijms18081678
Chicago/Turabian StyleSato, Masahiro, Issei Saitoh, Tomoya Murakami, Naoko Kubota, Shingo Nakamura, Satoshi Watanabe, and Emi Inada. 2017. "Intrapancreatic Parenchymal Injection of Cells as a Useful Tool for Allowing a Small Number of Proliferative Cells to Grow In Vivo" International Journal of Molecular Sciences 18, no. 8: 1678. https://doi.org/10.3390/ijms18081678